通过分区中条码珠共定位生成核苷酸序列的制作方法
相关申请的交叉引用
本申请要求2018年8月20日提交的美国临时申请第62/720,014号的权益,为所有目的其全文通过引用全文纳入本文。
背景技术:
偶联寡核苷酸的珠被用于测序样品制备应用(prepapplication),诸如具有许多不同分区(partition)(例如,液滴)的高通量单细胞分析。为了独特地鉴定各分区,可以用独特的条码序列标记珠。然而,为了确保分区仅有一个珠并因此被条码所独特地标记,通常调整珠浓度以使仅有约十分之一的分区被珠所占据。这导致分区的低利用率,样品损失并且增加检测样品所需的试剂和样品的量。增加珠浓度将导致较高的分区占用率以及更大的分区利用率。分区的利用率将增加,样品损失将减少,并且检查样品所需的试剂和样品的量将减少。相反地,较高的珠浓度将导致更多数量的分区具有超过一个珠。因此,一些划分的样品将被超过一个条码标记。在这些情况中的某些情况下,分区中的样品将在超过一个条码之间分割,从而导致各条码灵敏度的预期损失,例如,因为单个分区生成了超过一个数据点而所导致的某些数据点的过度展示(overrepresentation)。
发明概述
在一些实施方式中,提供了生成dna模板核苷酸序列的方法。一些实施方式中,所述方法包括:
(a)将样品划分成多个包含颗粒的分区,所述颗粒包含固体支持物表面,所述固体支持物表面上偶联多个寡核苷酸引物,其中所述寡核苷酸引物包含条码序列,并且其中不同颗粒之间的区别在于具有彼此不同的条码(例如,其中与固体支持物表面偶联的多个寡核苷酸引物中的至少大多数包含相同的条码序列),其中至少一些分区具有超过一个颗粒/分区,并且其中不同的颗粒偶联具有不同条码序列的引物;
(b)在所述分区中提供待测序的dna模板片段;和
(c)在所述分区中,将来自所述固体支持物的寡核苷酸引物与dna模板的至少一个片段连接,从而形成条码化dna模板片段;
(d)组合来自多个分区的条码化dna模板片段;
(d)生成所述条码化dna模板的多个测序读数;
(e)以成对的方式确定不同条码之间共有的dna模板片段的测序读数的百分比;
(f)将确定的不同条码之间共有的dna模板片段的百分比与阈值进行比较,其中如果两个条码确定的共有的相同dna模板片段的百分比高于所述阈值,那么确定所述两个条码处于同一分区中;和
(g)由所述多个测序读段生成所述dna模板的核苷酸序列,其中生成所述核苷酸序列包括将确定具有处于同一分区的不同条码的测序读数视为来自同一分区;
从而生成dna模板的核苷酸序列。
在一些实施方式中,dna模板片段包含异源性末端衔接子序列。
在一些实施方式中,所述提供包括随机切割末端dna。在一些实施方式中,所述随机切割包括使所述模板dna与转座酶接触,所述转座酶将异源性末端衔接子序列引入所述模板dna,以形成包含所述异源性末端衔接子序列的模板dna片段。在一些实施方式中,所述随机切割包括使所述转座酶与细胞核中的dna接触。在一些实施方式中,所述转座酶连接模板dna片段对,从而保留所述分区中所述模板dna的连续性。
在一些实施方式中,划分导致平均值或中值为1.0-5个颗粒/分区。
在一些实施方式中,至少10%、20%、30%、40%、50%、60%或70%的所述分区具有超过一个颗粒/分区。
在上述实施方式中的任一项中,生成所述核苷酸序列可以包括排除来自包含比泊松分布所预测的颗粒更多的颗粒的分区的测序读数。
在上述实施方式中的任一项中,生成核苷酸序列可以包括排除来自包含比物理上可能存在的颗粒更多的颗粒的分区的测序读数,由所述颗粒的大小与所述分区(例如,液滴)的大小相比确定。
一些实施方式中,所述方法包括:
(a)将样品划分成多个包含颗粒的分区,所述颗粒包含固体支持物表面,所述固体支持物表面上偶联多个寡核苷酸引物,其中所述寡核苷酸引物包含条码序列,并且其中不同颗粒之间的区别在于具有彼此不同的条码(例如,其中与固体支持物表面偶联的多个寡核苷酸引物中的至少大多数包含相同的条码序列),其中至少10%、20%、30%、40%、50%、60%或70%的分区具有超过一个颗粒/分区,并且其中不同的颗粒偶联具有不同条码序列的引物;
(b)在所述分区中提供待测序的dna模板片段;和
(c)在所述分区中,将来自所述固体支持物的寡核苷酸引物与dna模板的至少一个片段连接,从而形成条码化dna模板片段;
(d)组合来自多个分区的条码化dna模板片段;
(d)生成所述条码化dna模板的多个测序读数;
(e)当所述测序读数由同一分区产生时,将具有不同条码的测序读段去卷积至同一分区;和
(f)由所述多个测序读段生成所述dna模板的核苷酸序列,其中生成所述核苷酸序列包括将确定具有处于同一分区的不同条码的测序读数视为来自同一分区;
从而生成dna模板的核苷酸序列。
在一些实施方式中,所述去卷积包括以成对的方式将确定的不同条码之间共有的dna模板片段的百分比与阈值进行比较,其中如果两个条码确定的共有的dna模板片段的百分比高于所述阈值,那么确定所述两个条码处于同一分区中。
在一些实施方式中,dna模板片段包含异源性末端衔接子序列。
在一些实施方式中,所述提供包括随机切割末端dna。在一些实施方式中,所述随机切割包括使所述模板dna与转座酶接触,所述转座酶将异源性末端衔接子序列引入所述模板dna,以形成包含所述异源性末端衔接子序列的模板dna片段。在一些实施方式中,所述随机切割包括使所述转座酶与细胞核中的dna接触。在一些实施方式中,所述转座酶连接模板dna片段对,从而保留所述分区中所述模板dna的连续性。
在一些实施方式中,划分导致平均值或中值为1.0-5个颗粒/分区。
在一些实施方式中,至少10%、20%、30%、40%、50%、60%或70%的所述分区具有超过一个颗粒/分区。
在一些实施方式中,生成所述核苷酸序列包括排除来自包含比泊松分布所预测的颗粒更多的颗粒的分区的测序读数。
在一些实施方式中,该方法包括排除来自包含比物理上可能存在的颗粒更多的颗粒的分区的测序读数,由所述颗粒的大小与所述分区(例如,液滴)液滴的大小相比确定。
还提供了一种区分源自不同分区的差异性条码化序列读数与具有不同条码但源自同一分区的序列读数的方法:一些实施方式中,所述方法包括:
(a)将样品划分成多个包含颗粒的分区,所述颗粒包含多个寡核苷酸引物,其中所述寡核苷酸引物包含条码序列,并且其中不同颗粒之间的区别在于具有彼此不同的条码,其中至少一些分区具有超过一个颗粒/分区,并且其中不同的颗粒偶联具有不同条码序列的引物;
(b)在所述分区中提供dna片段;
(c)在所述分区中将来自所述颗粒的寡核苷酸引物与dna片段连接,从而形成条码化dna片段;
(d)组合来自多个分区的条码化dna片段;
(d)生成所述条码化dna的多个测序读数;
(e)以成对的方式确定不同条码之间共有的dna模板片段的测序读数的百分比;和
(f)将确定的不同条码之间共有的dna片段的百分比与阈值进行比较,其中如果两个条码确定的共有的相同dna模板片段的百分比高于所述阈值,那么确定所述两个条码处于同一分区中。
在一些实施方式,至少大部分与颗粒关联的所述多个寡核苷酸引物包含相同的条码序列。
在一些实施方式中,dna片段是样品dna。在一些实施方式中,在进行所述划分前,所述样品dna中的甲基胞嘧啶已经转化,用于甲基化分析。在一些实施方式中,在进行所述划分前,所述样品dna已经亚硫酸氢盐处理。
在一些实施方式中,所述分区包含样品细胞,并且所述样品dna来自所述样品细胞。在一些实施方式中,所述分区包含样品细胞,并且所述样品dna是所述样品细胞生成的edna。
在一些实施方式中,该方法还包括对分区中的所述样品细胞进行预包封(pre-encapsulate)。
在一些实施方式中,所述分区还包含待测序的样品dna,并且所述dna片段对于所述样品dna是外源的。
在一些实施方式中,该方法还包括生成所述样品dna的核苷酸序列,其中生成所述核苷酸序列包括将具有确定处于同一分区不同条码的测序读数视为来自同一分区,从而生成dna模板的核苷酸序列。
在一些实施方式中,dna片段包含异源性末端衔接子序列。
在一些实施方式中,所述提供包括随机切割dna。在一些实施方式中,所述随机切割包括使所述dna与转座酶接触,所述转座酶将异源性末端衔接子序列引入所述dna,以形成包含所述异源性末端衔接子序列的dna片段。在一些实施方式中,所述随机切割包括使所述转座酶与细胞核中的dna接触。在一些实施方式中,所述转座酶连接dna片段对,从而保留所述分区中所述模板dna的连续性。
在一些实施方式中,划分导致平均值或中值为0.1-5或1.0-5个颗粒/分区。在一些实施方式中,至少10%、20%、30%、40%、50%、60%或70%的所述分区具有超过一个颗粒/分区。
在一些实施方式中,生成所述核苷酸序列包括排除来自包含比泊松分布所预测的颗粒更多的颗粒的分区的测序读数。
在一些实施方式中,该方法包括排除来自包含比物理上可能存在的颗粒更多的颗粒的分区的测序读数,由所述颗粒的大小与所述分区的大小相比确定。
在一些实施方式中,颗粒包含包含寡核苷酸的水凝胶。在一些实施方式中,颗粒包括与寡核苷酸偶联的固体表面。在一些实施方式中,寡核苷酸由分区中的颗粒释放。
附图说明
图1显示了计算机系统。
图2显示增加用于各分区条码化的珠的数量时测序数据的改善。
图3显示了用于鉴定和使用来自同一分区中的多个分区条码的数据的示例性工作流程。
图4显示了这样的示例,其将携带条码的珠和细胞递送至分区(或由包含细胞和珠的溶液形成分区),从而得到包含多种数量的珠的分区的混合物。递送至分区的细胞的数量通常保持较低(此处由许多分区缺少任何细胞表示),以避免分区中存在多个细胞。在某些情况下,为了利用尽可能多的分区,递送至分区的珠的数量导致各分区有多个珠。
图5显示了将外源性(外来)随机序列引入分区如何生成外源性序列的分区特异性“特征(signature)”,其一旦将分区中的任何条码条码化,便可以用于关联来自同一分区的条码(因为同一分区中的条码将与外源片段的相同特征关联。
图6显示了用细胞和条码连接的珠进行划分的分布的理论分析。左上图显示了细胞(理想情况下避免包含多个细胞的分区)、珠(以比分区形成期间的细胞更高的浓度提供,从而导致多个分区中的各分区有多个珠)和异源性片段(“外来特征”)(以使不同的分区将包含不同片段混合物的浓度提供)的可能分布。该分布也在该附图的右侧说明,其显示了可以根据本文所述特征访问的可能的分区集合。
图7显示了用于生成外源性dna片段的一个实施方式。在所示实施方式中,标签化用于产生外源性dna的随机片段。标签化将衔接子序列r1和r2添加到片段的末端。所得dna混合物可以作为外源性序列的储备,可以添加到分区中。
图8显示了图7的后续部分,并且显示了单链外源性片段的形成,所述单链外源性片段包含衔接子序列以及添加的序列,其在这种情况下是多聚t序列,其随后在互补序列中被转化为多聚a。
图9显示了用于将条码引入中第一链cdna以及分区中的外源性dna片段(作为一个组形成“特征”)的方法的示意图。如图所示,外源性片段具有3′多聚a序列(例如,如图7-8)并与条码连接,所述连接通过与条码连接的3′捕获序列(多聚t)和与外源性dna片段连接的多聚t序列的杂交。然后将这些链延伸,以形成可以用作分区特征的条码化序列的双链文库。
图10显示了同时形成cdna第二链以及对含有外源性多聚a的外源性dna进行条码化以形成分区特征。
图11显示了在分区中将mrna逆转录为第一链cdna,然后使用rna酶h活性以及切口(nick)引发的多重置换进行第二链合成。可以通过桥寡核苷酸(bridge-oligonucleotide)引入与条码-寡核苷酸捕获序列互补的核苷酸序列,所述桥寡核苷酸充当引物,用于通过逆转录的第一链cdna合成。第二链cdna通过多重置换产生,因此第二链末端序列与条码-寡核苷酸的捕获序列杂交,从而形成与条码连接的cdna。
图12在附图左侧显示了阈值的鉴定。在由与λ基因组(外源性dna的来源)的比对计算出jaccard指数并在条码空间进行成对比较后,将具有高于算法定义的阈值的iaccard指数的条码合并,以生成液滴水平条码。以降序排列绘制各液滴水平条码的读取数量。推断相对背景具有更高数量的读数的液滴水平条码(即,左侧拐点的数据点)代表细胞。附图右侧显示了基于分区中的外源性dna将条码分配给分区的生物信息学流程。
图13描述了混合物种单细胞atac-seq实验。
图14描述了用于使用单细胞atac-seq数据将珠共定位至单个液滴的生物信息流程。
图15描述了合并atac-seq实验期间来自同一分区的珠数据的示例。
定义
除非另有说明,本文所用的所有科技术语具有本发明所属领域普通技术人员通常所理解的含义。通常,本文所用的命名和下述细胞培养、分子遗传学、有机化学、分析化学和核酸化学以及杂交中的实验室步骤均为本领域熟知和常用的。使用标准技术进行核酸和肽合成。按照本领域和各种通用参考文献所述的常规方法进行这些技术和步骤(通常参见,sambrook等,《分子克隆:实验室手册》(molecularcloning:alaboratorymanual),第2版(1989)冷泉港实验室出版社(coldspringharborlaboratorypress),纽约冷泉港(coldspringharbor,n.y.),其通过引用纳入本文),全文中提供这些参考文献。
“随机”包括绝对随机和准随机事件。例如,“随机切割”dna包括用tn5转座酶切割dna,所述tn5转座酶以准随机方式(例如,充分随机)切割dna,所以当切割dna在不同分区之间分布时,各分区具有独特的一组片段。
“异源性末端衔接子序列”指模板dna的末端提供共有(common)序列的异源性序列,其通常以寡核苷酸形式添加。这类衔接子序列允许容易地操纵不同的模板dna片段。
术语“扩增反应”指用于以线性或指数方式倍增核酸靶序列拷贝的任何体外方法。这些方法包括但不限于聚合酶链式反应(pcr);dna连接酶链式反应(lcr);qbetarna复制酶和基于rna转录的扩增反应(例如涉及t7,t3或sp6引发的rna聚合的扩增),例如转录扩增系统(tas),基于核酸序列的扩增(nasba),和自主维持序列复制(3sr);单引物等温扩增(spia),环介导等温扩增(lamp),链置换扩增(sda);多重置换扩增(mda);滚环扩增(rca);以及本领域技术人员已知的其他方法。参见例如,fakruddin等,j.pharmbioalliedsci.20135(4):245-252。
“扩增”指将溶液置于足以扩增多核苷酸的条件下的步骤(如果反应的所有组分是完整的)。扩增反应的组分包括,例如,引物、多核苷酸模板、聚合酶、核苷酸等。术语“扩增”通常指靶核酸的“指数型”增长。然而,本文所用的“扩增”也可指核酸的选择靶序列数量的线性增长,如由循环测序或线性扩增所得。
“聚合酶链式反应”或“pcr”是指靶双链dna的特定区段或子序列得以几何级数式扩增的一种方法。pcr是本领域技术人员所熟知的;参见例如,美国专利号4,683,195和4,683,202;和《pcr方案:方法和应用指南》,innis等编,1990。示例性pcr反应条件一般包括两步循环或三步循环。两步循环具有变性步骤和之后的杂交/延伸步骤。三步循环包括变性步骤,之后是杂交步骤,之后是独立的延伸步骤。
“引物”指与靶核酸上的序列杂交,并且任选地,用作核酸合成的起始点的多核苷酸序列。引物可以有多种长度。在一些实施方式中,引物长度小于100或50个核苷酸,例如长度为约10至约900,约15至约80,或约30-85至约30个核苷酸。用于扩增反应(例如,pcr)的引物长度和序列可根据本领域技术人员所知的原理来设计,参见例如innis等编,(1990)《pcr方案:方法和应用指南》(pcrprotocols:aguidetomethodsandapplications)。引物可以包括或完全由dna、rna或非天然核苷酸形成。在一些实施方式中,引物包含一个或多个经修饰的和/或非天然的核苷碱基。在一些实施方式中,引物包含标记物(例如,可检测标记物)。
核酸或其部分在一定条件下与另一个核酸“杂交”从而使得生理缓冲剂中一定温度下非特异性杂交最小化。一些情形中,核酸或其部分与靶核酸组的共有保守序列杂交。在一些情况中,如果包括与超过一个核苷酸伴侣互补的“通用”核苷酸在内有至少约6、8、10、12、14、16或18个连续的互补核苷酸,引物或其部分能杂交至引物结合位点。或者,如果在至少约12、14、16或18个连续的互补核苷酸中有不到1或2个互补错配,引物或其部分能杂交至引物结合位点。一些实施方式中,发生特异性杂交的温度是室温。一些实施方式中,发生特异性杂交的温度高于室温。在一些实施方式中,发生特异性杂交的限定温度为至少约37、40、42、45、50、55、60、65、70、75或80℃。
本文中,“核酸”表示dna、rna、单链、双链或聚集度更高的杂交基序及其任意化学修饰形式。修饰包括但不限于给核酸配体碱基或核酸配体整体提供化学基团的那些修饰,所述化学基团引入附加电荷、极化性、氢键、静电相互作用、连接点和官能团。这类修饰包括但不限于:肽核酸(pna)、磷酸二酯基团修饰(例如,硫代磷酸酯、甲基膦酸酯)、2′-位糖修饰、5-位嘧啶修饰、8-位嘌呤修饰、环外胺修饰、4-硫尿核苷取代、5-溴或5-碘-尿嘧啶取代、骨架修饰、甲基化、稀有碱基配对组合如异碱基(isobase)、异胞苷和异胍(isoguanidine)等。核酸还可以包含非天然碱基,如硝基吲哚。修饰还可包括3′和5′修饰,包括但不限于用荧光团(例如,量子点)或其他部分加帽。
本文所用术语“划分”或“经划分的”指将样品分为多个部分或多个“分区(partition)”。分区可以是固体或流体。在一些实施方式中,分区是固体分区,例如,微通道或孔(即,在多孔微量滴定皿)。在一些实施方式中,分区是流体分区,例如,液滴。在一些实施方式中,流体分区(例如,液滴)是不互溶的流体(例如,水和油)的混合物。在一些实施方式中,流体分区(例如,液滴)是水性液滴,其被不互溶的运载体流体(例如,油)包围。
如本文所用“条码”是鉴别其所偶联分子的短核苷酸序列(例如,长度至少约4、6、8、10、12、14、16、18、20、25、30或更多个核苷酸)。在一些实施方式中,使用条码来鉴定分区中的分子。相较于存在于其他分区中的条码,这类分区特异性条码对于该分区可以是唯一的,虽然如本文所述,在一些频率下,同一分区中可以出现两个或多个条码。在一个示例中,包含来自单个细胞的靶rna的分区可以经受逆转录条件,在大部分分区中使用包含不同分区特异性条码序列的引物,从而将独特的“细胞条码”的拷贝纳入分区经逆转录的核酸。由此,来自各细胞的核酸可藉由独有的“细胞条码”与其它细胞的核酸相区分。在一些实施方式中,条码存在于与颗粒偶联的寡核苷酸上,其中“颗粒条码”为与该颗粒偶联的全部或基本全部寡核苷酸所共有(例如,在它们之间相同或基本相同)。
发明的具体实施方式
引言
发明人发现了分析由分区中测序样品制备反应生成的测序数据的新方法,其中用于各个分区的条码用于将序列读数“标记”为来自各个分区。当一个分区(例如乳剂中的液滴)具有超过一个条码时(例如,由于条码引入的随机性,某些分区包含两个或更多个条码),一些样品核酸专门分布在(例如链接至)两个或更多个条码之间,并且一些由两个或更多个条码共享。参见,例如图4。测序是批量进行的(即,合并分区的内容物),所以相较于来自同一分区的两个不同条码的序列读取,人们无法区分来自不同分区的差异性条码化(differently-barcoded)序列读数。发明人发现了如何区分这类序列读数,这利用了先前进行的片段化(例如,在划分前批量进行)或引入分区以使分区包含不同的随机“特征”的其他随机性来源。例如,发明人发现,差异性条码化测序读段之间相同片段的重叠百分比可以用于确定序列是来自相同还是不同的分区。使用模板dna(即需要其序列的靶dna)本身作为“特征”,例如,在dna经随机片段化或以其他方式带来了随机性来源(例如,甲基化特征)的情况下,或者使用引入分区中的外源性dna来源(例如,不同于模板dna并且可与之区分的经随机片段化的dna),可以应用该方法。
例如,在两个条码处于同一分区中并且样品核酸在该分区中被片段化的情况下,每个不同的条码将连接一些相同的样品核酸片段。在条码处于不同分区中的另一种情况下,各分区中的dna具有不同的片段化dna组,共有的相同片段的百分比将会少得多(对于大多数成对比较来自不同条码的序列读数而言常见的背景水平)。因此,通过确定具有不同条码的共有片段的百分比并将确定的百分比与阈值进行比较,可以区分这两种情况。一旦确定来自两个条码读取的序列源自同一分区,那么考虑到该信息,可以为分区中的模板生成核苷酸序列。作为非限制性示例,可以选择舍弃同一分区中存在超过一个条码的所有数据,或者可以合并来自两个条码的测序读取数据,将其全部解释为来自一个分区。
此外,已经发现由包含超过一个条码的多个分区生成的数据可以改善测序数据质量。例如,已经发现将各分区条码的平均数量增加到两个或更多可以改善而不是损害测序读数的信噪比。因此,例如,可以生成这样的分区,其中至少10%、20%、30%、40%、50%、60%或70%的分区包含两个或更多个不同的条码(在该上下文中,不同意味着具有不同的条码核苷酸序列)。
图3描绘了示例性工作流程。例如,可以比对测序读数(301)。任选地,可以过滤(即去除)低质量比对(302)。可以去除具有相同条码的重复读数,从而仅提供针对特定条码的一个读数副本(303)。以成对的方式在各个珠条码之间确定相同的片段并计数(304)。可以确定各条码对的片段重叠百分比(305)。将确定的片段重叠百分比与阈值进行比较。在一些实施方式中,基于在所有成对比较之间的片段重叠百分比的平滑分布来确定阈值(306)。在片段重叠百分比超过阈值的情况下,将具有不同珠条码的读数合并并分配分区条码(307)。在一些实施方式中,可以对细胞条码内的读数进行重复数据消除(308),即,对于单个分区内不同的珠条码而言,相同的读数是共有的。所得测序读数可用于识别(call)峰(309),即鉴定基因组中富含比对的读数的区域(例如,由于执行atacseq或其他测序方法的结果)。任选地,可以计算一个或多个信噪比指标,例如tss%、frip或tss富集评分或其组合(310)。
划分样品
对样品进行划分的方法和组合物描述于,例如,公开的专利申请wo2010/036352、us2010/0173394、us2011/0092373和us2011/0092376。多个分区可以是多个乳液液滴,或多个微孔等。
在一些实施方式中,在液滴形成期间添加一种或多种试剂,或在液滴形成之后将一种或多种试剂添加到液滴中。用于将试剂递送至一个或多个分区的方法和组合物包括本领域已知的微流体方法;液滴或微胶囊合并,聚结,融合,破裂或降解(例如,如u.s.2015/0027,892;us2014/0227,684;wo2012/149,042;和wo2014/028,537中所述);液滴注入方法(例如,如wo2010/151,776中所述);及其组合。
如本文所述,分区可以是皮米孔、纳米孔或微孔。分区可以是皮米,纳米或微米反应室,例如皮米,纳米或微米胶囊。分区可以是皮米,纳米或微米通道。分区可以是液滴,例如乳液液滴。在一些实施方式中,液滴包含乳液组合物,即不互溶的流体(如水和油)的混合物。在一些实施方式中,液滴是水性液滴,其被不互溶的运载体流体(如油)包围。在一些实施方式中,液滴是油性液滴,其被不互溶的运载体流体(如水性溶液)包围。在一些实施方式中,本文所述液滴是相对稳定的并在两个或更多个液滴之间具有最小聚结。在一些实施方式中,由样品生成的液滴中少于0.0001%、0.0005%、0.001%、0.005%、0.01%、0.05%、0.1%、0.5%、1%、2%、3%、4%、5%、6%、7%、8%、9%或10%与其他液滴聚结。这些乳液还可具有有限的絮凝,一种分散相以薄片中悬浮液产生的过程。在一些情况下,这种稳定性或最小聚结可保持长达4、6、8、10、12、24或48小时或更长时间(例如,在室温下,或在约0、2、4、6、8、10或12℃下)。在一些实施方式中,使油相流过水相或试剂,从而形成液滴。
该油相可包含氟化基础油,其可通过与氟化表面活性剂(如全氟聚醚)联用而进一步稳定。在一些实施方式中,该基础油包括以下一种或多种:hfe7500、fc-40、fc-43、fc-70或其他常见氟化油。在一些实施方式中,该油相包含阴离子含氟表面活性剂。在一些实施方式中,该阴离子含氟表面活性剂是ammoniumkrytox(krytox-as)、krytoxfsh的铵盐或krytoxfsh的吗啉代衍生物。krytox-as的浓度可以是约0.1%、0.2%、0.3%、0.4%、0.5%、0.6%、0.7%、0.8%、0.9%、1.0%、2.0%、3.0%或4.0%(w/w)。在一些实施方式中,krytox-as的浓度是约1.8%。在一些实施方式中,krytox-as的浓度是约1.62%。krytoxfsh的吗啉代衍生物的浓度可以是约0.1%、0.2%、0.3%、0.4%、0.5%、0.6%、0.7%、0.8%、0.9%、1.0%、2.0%、3.0%或4.0%(w/w)。在一些实施方式中,krytoxfsh的吗啉代衍生物的浓度是约1.8%。在一些实施方式中,krytoxfsh的吗啉代衍生物的浓度是约1.62%。
在一些实施方式中,该油相还包含用于调节油性质(如蒸气压、粘度或表面张力)的添加剂。非限制性示例包括全氟辛醇和1h,1h,2h,2h-全氟癸醇。在一些实施方式中,1h,1h,2h,2h-全氟癸醇添加至约0.05%、0.06%、0.07%、0.08%、0.09%、0.1%、0.2%、0.3%、0.4%、0.5%、0.6%、0.7%、0.8%、0.9%、1.0%、1.25%、1.50%、1.75%、2.0%、2.25%、2.5%、2.75%或3.0%(w/w)的浓度。在一些实施方式中,1h,1h,2h,2h-全氟癸醇添加至约0.18%(w/w)的浓度。
在一些实施方式中,乳液可以是基本上单分散的。在其它实施方式中,乳液可以是多分散的。乳液分散性可以由乳液形成方法所引起。例如,相较于“沙拉搅拌器(saladshaker)”乳液形成(可以是高度多分散的),微流体乳液形成通常具有低的多分散性。多分散性也可在乳液形成的下游出现,诸如当乳液的液滴融合在一起时。
在一些实施方式中,该乳液配制为生成具有类液界面膜的高度单分散液滴,其可通过加热转化为具有类固界面膜的微胶囊;这类微胶囊可作为生物反应器以通过一段时间的孵育保持其内容物。转化为微胶囊可在一经加热后即发生。例如,这类转化可发生在大于约40°、50°、60°、70°、80°、90°或95℃的温度下。加热过程期间,流体或矿物油覆盖物可用于阻止蒸发。过量的连续相油可在加热前去除或留在原位。这些微胶囊可在大范围的热和机械处理下抗聚结和/或絮凝。
在将液滴转化成微胶囊之后,这些微胶囊可储存于约-70℃、-20℃、0℃、3℃、4℃、5℃、6℃、7℃、8℃、9℃、10℃、15℃、20℃、25℃、30℃、35℃或40℃下。在一些实施方式中,这些微胶囊可用于储存或运输分区混合物。例如,可在一个位置处收集样品,划分到含有酶、缓冲剂和/或引物或其它探针的液滴中,任选地可进行一个或多个聚合反应,然后可加热该分区以进行微囊化,并且可储存或运输微胶囊用于进一步分析。
在一些实施方式中,将样品划分为至少500个分区,1000个分区,2000个分区,3000个分区,4000个分区,5000个分区,6000个分区,7000个分区,8000个分区,10,000个分区,15,000个分区,20,000个分区,30,000个分区,40,000个分区,50,000个分区,60,000个分区,70,000个分区,80,000个分区,90,000个分区,100,000个分区,200,000个分区,300,000个分区,400,000个分区,500,000个分区,600,000个分区,700,000个分区,800,000个分区,900,000个分区,1,000,000个分区,2,000,000个分区,3,000,000个分区,4,000,000个分区,5,000,000个分区,10,000,000个分区,20,000,000个分区,30,000,000个分区,40,000,000个分区,50,000,000个分区,60,000,000个分区,70,000,000个分区,80,000,000个分区,90,000,000个分区,100,000,000个分区,150,000,000个分区或200,000,000个分区。
在一些实施方式中,生成的液滴在形状和/或尺寸方面基本均匀。例如,在一些实施方式中,这些液滴在平均直径方面基本均匀。在一些实施方式中,生成的液滴的平均直径为约0.001微米、约0.005微米、约0.01微米、约0.05微米、约0.1微米、约0.5微米、约1微米、约5微米、约10微米、约20微米、约30微米、约40微米、约50微米、约60微米、约70微米、约80微米、约90微米、约100微米、约150微米、约200微米、约300微米、约400微米、约500微米、约600微米、约700微米、约800微米、约900微米或约1000微米。在一些实施方式中,生成的液滴的平均直径为小于约1000微米、小于约900微米、小于约800微米、小于约700微米、小于约600微米、小于约500微米、小于约400微米、小于约300微米、小于约200微米、小于约100微米、小于约50微米,或小于约25微米。在一些实施方式中,生成的液滴在形状和/或尺寸方面是不均匀的。
在一些实施方式中,生成的液滴在体积上基本均匀。例如,液滴体积的标准偏差可以低于约1皮升、5皮升、10皮升、100皮升、1nl或低于约10nl。在一些情况中,液滴体积的标准偏差可低于平均液滴体积的约10-25%。在一些实施方式中,生成的液滴的平均体积为约0.001nl、约0.005nl、约0.01nl、约0.02nl、约0.03nl、约0.04nl、约0.05nl、约0.06nl、约0.07nl、约0.08nl、约0.09nl、约0.1nl、约0.2nl、约0.3nl、约0.4nl、约0.5nl、约0.6nl、约0.7nl、约0.8nl、约0.9nl、约1nl、约1.5nl、约2nl、约2.5nl、约3nl、约3.5nl、约4nl、约4.5nl、约5nl、约5.5nl、约6nl、约6.5nl、约7nl、约7.5nl、约8nl、约8.5nl、约9nl、约9.5nl、约10nl、约11nl、约12nl、约13nl、约14nl、约15nl、约16nl、约17nl、约18nl、约19nl、约20nl、约25nl、约30nl、约35nl、约40nl、约45nl或约50nl。
在一些实施方式中,该方法包括将包括一种或多种靶核酸的样品划分成多个分区。在一些实施方式中,包括靶核酸的样品包括dna、rna或其组合或杂合体。在一些实施方式中,包含靶核酸的样品包含来自基因组的亚组的dna或基因组dna(例如,可能包含特定群体(如易患特定类型癌症的个体)的突变的选定基因)。在一些实施方式中,样品包含相邻性保留的基因组dna,其已经片段化但是通过蛋白质(例如,tn5转座酶(标签酶))与dna片段末端的连接保留相邻性。在一些实施方式中,包含靶核酸的样品包含cdna。在一些实施方式中,包括靶核酸的样品包含外显子dna(即,富含转录序列的全基因组dna的亚组,其包含基因组中的外显子组)或转录组dna(即,细胞或细胞群中产生的所有mrna或″转录本″的组)。在一些实施方式中,包括靶核酸的样品包含长片段dna(例如,具有至少约300、400、500、600、700、800、1000或更多碱基,或者对于双链dna而言的碱基对长度的dna)。在一些实施方式中,包括靶核酸的样品包括rna,例如,mrna或lncrna。在一些实施方式中,靶核酸是双链的。在一些实施方式中,靶核酸是单链的。在一些实施方式中,样品包括分离自组织或细胞的靶核酸。在一些实施方式中,样品包括位于单细胞或单核(singlenuclei)的靶核酸。
在一些实施方式中,包括靶核酸的样品是生物样品。生物样品可获自任何生物体,例如动物、植物、真菌、病原体(例如细菌或病毒)或任何其他生物体。在一些实施方式中,该生物样品来自动物,例如哺乳动物(如人或非人灵长类动物、奶牛、马、猪、绵羊、猫、狗、小鼠或大鼠)、鸟(如鸡)或鱼。生物样品可以是获自生物体的任何组织或体液,例如血液,血液成分或血液产品(如血清、血浆、血小板、血红细胞等),痰液或唾液,组织(如肾、肺、肝、心、脑、神经组织、甲状腺、眼、骨骼肌、软骨或骨组织);培养的细胞,例如原代培养物,外植体,和转化的细胞,干细胞,粪便,尿液等。在一些实施方式中,样品是包含细胞的样品。在一些实施方式中,样品是单细胞样品。
一些实施方式中,本文所述方法用于单细胞分析。相应地,在一些实施方式中,将来自单细胞的靶核酸划分成多个分区。在一些实施方式中,单细胞被递送至各个分区。在一些实施方式中,对来自包含多个细胞或核的生物样品的靶核酸进行提取和划分,从而使各个分区包含来自少于一个、一个或多个细胞或核的核酸。只要来自细胞的核酸作为物理实体保持在一起,就不需要将细胞本身递送至分区。例如,这可以这样实现:通过固定细胞(例如,通过多聚甲醛或甲醇固定),将细胞包埋在水凝胶基质,从而使基质的孔径足够小以防止靶核酸扩散,和将靶核酸的完整互补物捕获到单一固体支持实体上(一个颗粒(例如,磁珠)或结合在一起的多个颗粒)。
条码和珠(颗粒)
在一些实施方式中,将核酸条码添加到分区,使得各分区中存在相对较少(例如,平均少于10个,少于5个、1-5个、0.5-5个)不同的条码(具有不同序列的条码)。在一些实施方式中,将条码寡核苷酸的多个拷贝(例如,至少100个,例如,100-100,000个或107个或108或109个或更多个)递送至各分区。可以例如通过具有彼此不同的条码来区分不同的颗粒。例如,这可以这样实现:通过将同一条码寡核苷酸的多个拷贝与珠或其他固体支持物关联,或者将多个拷贝包含在固体或半固体材料中,可以将其递送至分区用于后续任选地释放分区中的条码。
因此,在许多情况下,方便的是将与条码化的寡核苷酸连接的固体支持物(例如珠)递送至分区或在分区周围形成。在一些实施方式中,条码递送至分区的目的是条码或与寡核苷酸的连接的珠与分区的特定(例如,1∶1)分布。然而,由于泊松分布,一些分区将包含超过一个寡核苷酸(条码化的)珠(在本文中也称为“颗粒”)。
在一些实施方式中,条码作为更长的寡核苷酸的部分引入,例如,作为包含条码序列以及允许与靶相同序列杂交的3′捕获序列的正向寡核苷酸引物。在这些实施方式中,分区将包含正向引物,用于靶核酸的扩增或其他连接方法(例如接合)。在一些实施方式中,当在分区中提供时,正向引物与珠或其他固体支持物连接。由于统计分布,虽然许多分区将仅包含与一个珠连接的一个正向引物(或仅一个正向引物的多个拷贝),其他分区将包含具有不同序列(例如,不同条码)的正向引物,因为至少一些分区中存在多个珠。在一些实施方式中,珠包括固体支持物表面,其具有多个偶联其上的寡核苷酸引物。在一些实施方式中,珠包括至少约10、50、100、500、1000、5000、10,000、50,000、100,000、500,000、1,000,000、5,000,000、10,000,000、108、109、1010或更多个与之偶联的相同的正向引物。在一些实施方式中,正向引物是双链的。在一些实施方式中,正向引物是单链的。
在一些实施方式中,珠(颗粒)是水凝胶珠。一些情况中,水凝胶是溶胶(sol)形式。一些情况中,水凝胶是凝胶(gel)形式。示例性水凝胶是琼脂糖水凝胶。其它水凝胶包括但不限于例如下列文件中所述:美国专利号4,438,258、6,534,083、8,008,476、8,329,763;美国专利申请号2002/0,009,591、2013/0,022,569、2013/0,034,592;以及国际专利申请号wo/1997/030092和wo/2001/049240。例如,颗粒可以是包含寡核苷酸的水凝胶或其它其他凝胶珠,所述寡核苷酸随后可以通过融化或溶解颗粒而被释放。
珠的固体支持物表面可以经修饰以包括用于连接条码寡核苷酸的接头。该接头可以包括可切割部分。可切割部分非限制性的示例包括二硫键、二氧尿苷部分和限制酶识别位点。本领域已知用于共价连接寡核苷酸与一种或多种水凝胶基质的许多方法。仅举一例,醛衍生化琼脂糖可共价连接至合成寡核苷酸的5’-胺基团。在一些实施方式中,构造成将水凝胶连接至条码的寡核苷酸偶联于高分子量(例如,至少约5、10、15、20、25、30、35、40、50kda或更大)的聚合物,所述聚合物能被空间约束于gel形式的水凝胶基质内。例如,寡核苷酸可偶联于高分子量线性或支链聚丙烯酰胺。如另一示例,寡核苷酸可偶联于高分子量核酸。高分子量聚合物寡核苷酸偶联物(例如,线性聚丙烯酰胺寡核苷酸偶联物)可通过与溶胶水凝胶混合并将该水凝胶硬化成凝胶形式而纳入水凝胶基质中。一些情况中,所述多个分区含有偶联于高分子线性或支链聚丙烯酰胺的寡核苷酸、sol形式的水凝胶和含有独特分区特异性条码的双功能条码模板。其它高分子量聚合物适合与寡核苷酸偶联并包封入水凝胶。示例性聚合物包括但不限于:葡聚糖、壳聚糖、苯乙烯化明胶、透明质酸、海藻酸、明胶、聚乙二醇,及其衍生物。
各分区条码的数量
本公开示出了至少两个不同方面。首先,分区中多个序列差异性条码的出现可以这样检测:通过确定共有的相同片段的百分比,或由于分区特异性随机性而在分区之间可区分的其他特征,具有不同的条码,并将确定的百分比与阈值进行比较,由此可以检测同一分区中出现的具有不同条码的序列读数。其次,已经发现的是,在一些实施方式中,其可以改善测序数据,以由各分区平均具有超过一个序列差异性条码的分区中生成测序数据。
当如本文所述递送至珠上时,各分区序列差异性条码的平均数目可以通过控制条码化珠的数量相对于生成的分区的数量来预测。在一些实施方式中,结构上不同的条码的平均数量(例如,如所述递送至珠上)在各分区为0.5-1.5个。例如,当希望拥有尽可能多的分区仅包含一个结构上不同的条码时(虽然在分区中具有给定种类的多个条码拷贝),这可能会发生。在这些实施方式中,可以使用本文描述的方法来检测和寻址(address)来自包含超过一个结构上不同的条码的分区的数据。在其它实施方式中,结构上不同的条码的平均数量(例如,如所述递送至珠上)超过一个,例如,1-5个,或1-2个,或2-5个或更多个。在这些实施方式中,可以使用任何可用的方法(包括但不限于本文所述确定具有不同条码的共有片段百分比并将确定的百分比与阈值进行比较的方法)对具有不同条码但测序性能改善(例如,信噪比改善)的序列读数进行去卷积。
形成含有至少一种条码化的引物的分区之后,可以进行分子方法来检测分区中的靶核酸因此,在多种实施方式中,样品核酸也在分区中。示例性分子方法可以包括用于检测核酸的任何分子方法,包括但不限于基于模板的引物延伸(例如,聚合酶链反应)或通过正向引物捕获序列检测与靶核酸的特异性杂交的方法。在一些实施方式中,该方法包括连接。在分区是完整时或在分区内容物已经合并后(因此该方法“批量(inbulk)”进行),可以进行这些分子方法中的任何一种。
dna模板片段化
本文所述的一些方法包括在分区中提供dna模板片段。dna模板片段可以根据需要生成。例如,片段可以通过剪切力或其他物理力或通过酶促切割来生成。生成dna片段用于测序的越来越常规的方法涉及将dna模板与修饰的转座酶(有时称为标签酶)接触,其切割dna并将短衔接子序列引入片段的末端,从而提供可以用于下游分子生物学反应的引物结合序列,所述下游分子生物学反应涉及例如杂交和扩增和/或连接。
可以如本文所述使用载有异型衔接子的标签酶和载有同型衔接子的标签酶。载有同型衔接子的标签酶是包含只有一个序列的衔接子的标签酶,其中衔接子被添加至基因组dna中标签酶诱导的断裂点的末端。载有异型衔接子的标签酶是包含两个不同的衔接子的标签酶,从而使不同的衔接子序列被添加至dna中标签酶诱导的断裂点所产生的两个dna末端。载有衔接子的标签酶进一步描述于例如美国专利公开号:2010/0120098;2012/0301925;和2015/0291942以及美国专利号:5,965,443;美国专利6,437,109;7083980;9005935;和9,238,671,其中各自内容通过引用全文并入本文用于所有目的。
标签酶是指这样的酶,所述酶能够与含转座子末端的组合物形成功能性复合物并催化含转座子末端的组合物插入或转移到与该组合物在体外转座反应中孵育的双链靶dna中。示例性的转座酶包括但不限于相较于野生型tn5过分活跃的修饰的tn5转座酶,例如,可以具有选自e54k、m56a或l372p的一个或多个突变。野生型tn5转座子是复合型转座子,其中2个几乎相同的插入序列(is50l和is50r)侧接3个抗生素抗性基因(reznikoffws.annurevgenet42:269-286(2008))。各is50包含2个反向19-bp末端序列(es),外侧端(outsideend,oe)和内侧端(insideend,ie)。然而,野生型es的活性相对较低并且被过分活跃的镶嵌端(mosaicend,me)序列体外取代。因此,具有19-bpme的转座酶复合物是转座发生所必需的,只要间插dna足够长以使这些序列中的两个靠近在一起形成活性tn5转座酶同二聚体(reznikoffws.,molmicrobiol47:1199-1206(2003))。转座在体内是非常罕见的事件,并且过分活跃的突变体历史上源自tn5蛋白的476个残基中导入三个错义突变(e54k、m56a、l372p),其由is50r编码(goryshiniy,reznikoffws.1998.jbiolchem273:7367-7374(1998))。转座通过“剪切-和-粘贴”机制起作用,其中tn5将其从供体dna中切除并插入靶序列,产生靶标的9-bp重复(schallerh.coldspringharbsympquantbiol43:401-408(1979);reznikoffws.,annurevgenet42:269-286(2008))。在当前的商业解决方案(nexteratmdna试剂盒,亿明达公司(illumina))中,游离的合成me衔接子与靶dna的5′-端通过转座酶(标签酶)末端连接。在一些实施方式中,标记酶连接固体支持物(例如,与连接正向引物的珠不同的珠)。商购的珠连接的标签酶的实例是nexteratmdnaflex(亿明达公司)。
在一些实施方式中,衔接子长度为至少19个核苷酸,例如,19-100个核苷酸。在一些实施方式中,衔接子是具有5′端突出端的双链,其中5′突出端序列在异型衔接子间不同,但是双链部分(通常为19bp)是相同的。在一些实施方式中,衔接子包含tcgtcggcagcgtc(seqidno:1)或gtctcgtgggctcgg(seqidno:2)。在涉及载有异型衔接子的标签酶的一些实施方式中,标签酶载有包含tcgtcggcagcgtc(seqidno:1)的第一衔接子和包含gtctcgtgggctcgg(seqidno:2)的第二衔接子。在一些实施方式中,衔接子包含agatgtgtataagagacag(seqidno:3)和其互补物(也就是镶嵌端,并且这是tn5转座唯一特别需要的顺式活性序列)。在一些实施方式中,衔接子包含tcgtcggcagcgtcagatgtgtataagagacag(seqidno:4)与agatgtgtataagagacag(seqidno:3)的互补物或gtctcgtgggctcggagatgtgtataagagacag(seqidno:5)与agatgtgtataagagacag(seqidno:3)的互补物。在涉及载有异型衔接子的标签酶的一些实施方式中,标签酶载有第一衔接子,所述第一衔接子包含tcgtcggcagcgtcagatgtgtataagagacag(seqidno:4)与agatgtgtataagagacag(seqidno:3)的互补物和gtctcgtgggctcggagatgtgtataagagacag(seqidno:5)与agatgtgtataagagacag(seqidno:3)的互补物。
在一些实施方式中,dna是保留相邻性的标签化的多核苷酸(例如dna)序列。在相邻性保留的转座或标签化(tagmentation)中,转座酶(例如,tn5转座酶)被用于修饰具有衔接子序列的dna,同时维持dna区段的相邻关系。制备相邻性保留的标签化的多核苷序列的条件是本领域已知的。参见例如,amini等,naturegenetics,2014,46:1343-1349;wo2016/061517;和美国临时专利申请号62/436,288;各自通过引用纳入本文。已经观察到标签酶保持与dna结合,直到向反应中加入去污剂如sds(amini等naturegenetics46(12):1343-1349)。
在一些实施方式中,将转座酶应用于具有染色质的dna(例如,形成核小体的组蛋白和/或包含形成染色质的其他dna辅助因子)。在这些实施方式中,由于核小体的存在,转座酶将不能同等地触及所有dna。这些方法有时被称为“使用测序测试转座酶可及染色质的方法(assayfortransposaseaccessiblechromatinusingsequencing)”或“atac-seq”(参见,例如,美国专利公开号20160060691;buenrostro等,(2015)currprotocmolbiol.109:21.29.1-21.29.9),并且可以用于,例如,确定染色质在不同条件下的变化。在一些实施方式中,dna包含在其天然细胞内。例如,天然细胞可以被固定和透化,使得转座酶可以进入细胞核并在染色质结构允许的情况下切割dna。这可以被认为是染色质的转座酶可及性的一种测定方法。因此,在一些实施方式中,dna为染色质形式,例如,在细胞或分离的核内。
在其他实施方式中,dna基本上不含蛋白质。例如,dna样品已用苯酚提取以去除dna结合蛋白。
可以在片段化之前或在片段化或片段化之后划分dna,或者片段化和分区可以同时发生。例如,在一些实施方式中,一旦已用转座酶处理了dna样品,dna就可以形成多个单独的分区(例如液滴)。
在一些实施方式中,替代模板dna片段化或与模板dna片段化联用,可以用允许检测胞嘧啶甲基化的生物化学处理分区中的模板dna。在一个实施方式中,用于检测胞嘧啶甲基化的生物化学使用亚硫酸氢盐处理,其中亚硫酸氢盐改变(“转化”)未甲基化的胞嘧啶,但是不改变甲基化的胞嘧啶。在另一实施方式中,用于检测胞嘧啶甲基化的生物化学使用tet-氧化甲基化的胞嘧啶和apobec转化未氧化的未保护的胞嘧啶。甲基化(例如,通过亚硫酸氢盐转化检测到的)可以用作分区特异性随机性的来源,其中不同的细胞被递送至不同的分区。在这种情况下,大多数包含细胞的分区将只包含一个细胞。因为各个单独的细胞将具有不同的甲基化模式,所以导致亚硫酸氢盐处理或tet氧化的apobec转化的dna在分区之间会发生变化,这与细胞之间的甲基化差异相符。可以将待递送到分区中的细胞预先包封于第一分区中,如液滴或水凝胶珠,以使来自单个细胞的dna在甲基转化处理过程中保持物理联系。此外,缺少100%效率的甲基转化处理可以用于通过在不同分区中生成不同的dna序列(例如,即使分区中的起始dna是相同的,由于亚硫酸氢盐转化缺少完美效率)来生成分区特异性随机性。
在其他实施方式中,分区特异性随机性的来源可以归因于通过添加到分区或形成分区时的外源分子(在本文中也称为“异外来”分子)。例如,在一些实施方式中,外源性dna(即,来自除模板dna之外的来源的dna)可以被随机切割,以生成多个随机切割的片段,其中片段的部分被添加到分区。这将导致每个分区外源性dna片段的独特组合。通过允许这些片段以及模板片段参与条码化过程(例如,通过将部分珠特异性条码与外源性片段连接或延伸),可以生成分区特异性的外源性条码化dna片段组。通过比较与不同条码相关联的片段组,如下文进一步解释,可以确定分区是否包含超过一个珠特异性条码。参见,例如图5。
外源性dna的来源可以是任何核酸来源。在一些实施方式中,例如,外源性dna来自与模板dna不同物种的细胞。外源性dna的随机片段化可以通过任何片段化方法来实现。在一些实施方式中,外源性dna通过酶切割或机械剪切。示例性的酶包括,例如,任何具有dna酶或dna切口活性的酶。在一些实施方式中,酶是将衔接子序列引入所得片段末端的转座酶,诸如上述那些(例如,tn5转座酶,包括但不限于用于标签化的那些)。参见,例如图7。在另一替代方案中,可以通过基因合成随机序列来生成独特的外源性dna片段,任选地联合在片段的一端或两端添加已知衔接子。
在一个实施方式中,各细胞的rna长度分布提供了分区中随机性的来源。这可能是任何给定细胞中rna的内源性特征,或者也可能是通过外源驱动的rna片段化所引入的。rna片段化可发生在划分之前或分区中。一个可能的实施方式是可以通过模板转换将衔接子序列添加到rna分子的5′末端。在另一个实施方式中,rna-cdna杂交体在逆转录反应期间在提供分区特异性cdna分子的分区中被标签化。然后,珠条码寡核苷酸通过引物模板化(primertemplated)线性扩增或pcr标记衔接的cdna。如果液滴中存在多个珠,那么可以使用分区特异性独特的rna或cdna长度模式来确定同一分区中的珠。
在其中将衔接子序列引入dna模板片段末端(通过标签酶或其他方法引入)的实施方式中,与条码(例如,在珠或其他固体支持物上递送的)连接的3′捕获序列可以选择杂交衔接子序列,从而允许条码寡核苷酸与dna模板片段连接。来自条码寡核苷酸或连接的基于模板的延伸(例如,基于聚合酶的扩增)可以例如用于将条码共价连接dna模板片段的拷贝。
在一些实施方式中,待测序的模板dna是cdna序列,其中cdna由分区中的rna生成。在这些方面的一些中,mrna被多聚t序列(例如5或10或更多个连续的t)或者(可替代地)与条码序列连接的3′捕获序列处的靶标特异性序列捕获。可以将外源性dna片段纳入该过程中以协助条码的去卷积。在其中外源性dna被用作分区特异性随机性来源的实施方式中,可以将多聚t序列或靶标特异性序列添加到外源性dna片段的5′末端(任选地在5′端、3′端或两者处具有已知的衔接子序列)。在一个实施方式中,一旦外源性dna片段具有衔接子序列,就可以在具有多聚t或靶标特异性序列的引物存在下的情况下扩增片段。参见,例如图8。引物上可以包括5′磷酸,使得一个链具有5′磷酸。通过扩增产生的双链dna产物的另一链将包含多聚a(或与靶标特异性互补的)序列。可以例如通过λ外切核酸酶消化含5′磷酸的链,从而留下含多聚a(或与靶标特异性互补的)单链。参见,例如图8。该序列后续可以用于第一链cdna合成过程。参见,例如图9。连接条码序列的3′捕获序列可以用于引发第一链cdna,并且还可经延伸以复制外源性(外来)dna片段,从而将珠特异性条码连接至外源性片段序列。在分区中得到的产物是条码化的第一链cdna和条码化的外源性dna片段。参见图9。可以合成cdna的第二链以及外源性dna片段。例如,在一些实施例中,可以使用图11中所述的方法。
在一些实施方式中,分区还可以包含第二寡核苷酸引物,其可以任选地与珠(颗粒)连接或不与之连接。该引物可以用作第一寡核苷酸引物的反向引物,使得两个寡核苷酸在pcr中生成扩增子。第二寡核苷酸引物将具有与衔接子序列互补的3′末端,即相较于第一寡核苷酸引物靶向的衔接子序列位于dna区段相对端的衔接子序列。在一些实施方式中,3’端将与整个衔接子序列互补。在一些实施方式中,寡核苷酸的最3′的6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个与衔接子中的序列互补。第二寡核苷酸引物还可包含通用序列或其他附加序列,以辅助下游操作或扩增子测序。例如,当使用基于illumina的测序时,第二寡核苷酸引物可以具有5’p5或p7序列用于结合illumina流动室(任选地具有第一寡核苷酸引物,所述第一寡核苷酸引物具有两个序列中的另一个)。
扩增前,可以将一种或多种寡核苷酸引物从珠去除、释放或切割。这可以通过所需的任何方法来实现。切割的方法包括但不限于,改变ph或用udg/apei或限制性内切核酸酶接触寡核苷酸。在一些实施方式中,寡核苷酸通过一个或多个尿嘧啶(u)与珠连接,并且user酶(例如,来自neb)被用于切割纳入寡核苷酸主链中的u。user有2种酶:udg和内切核酸酶viii。在一些情况下,寡核苷酸通过二硫键(通过固体支持物的硫化物和共价附接于寡核苷酸的5′或3′末端,或插入核酸的硫化物之间的二硫键)与固体支持物连接。这类情况中,可通过使固体支持物接触还原剂来切下所述寡核苷酸,所述还原剂例如硫醇或膦试剂,包括但不限于β-巯基乙醇(bme)、二硫苏糖醇(dtt)或三(2-羧基乙基)膦(tcep)。由于多种原因,由珠释放寡核苷酸引物可以是有利的。例如,dna相互作用的动力学将大幅增强。颗粒也可以如上所述融化或溶解,以释放寡核苷酸。
扩增可以在分区内实现(在合并内含物至分区之前)。各种扩增方法是已知的并且可以使用。扩增后,将分区的内容物合并,如所需进行进一步处理,即通过进一步扩增和/或dna片段大小选择并进行批量测序。可以使用所需的的任何核苷酸测序方法,只要能确定至少一些dna区段序列和条码序列。高通量测序和基因分型的方法是本领域已知的。例如,此类测序技术包括但不限于:焦磷酸测序、连接法测序、单分子测序、合成法测序(sbs)、大量同步克隆法、大量同步单分子sbs、大量同步单分子实时法,大量同步单分子纳米孔技术等。morozova和marra提供对一些此类技术的综述,见genomics,92:255(2008),该文在此通过引用全文纳入本文。
示例性的dna测序技术包括基于荧光的测序技术(参见如birren等,genomeanalysis:analyzingdna,1(基因组分析:dna分析,第1卷),纽约冷泉港,该文在此通过引用全文纳入本文)。在一些实施方式中,使用本领域已理解的自动化测序技术。在一些实施方式中,本技术提供经划分的扩增子的同步测序(pct申请号wo2006/0841,32,该文在此通过引用全文纳入本文)。在一些实施方式中,dna测序的实现是通过同步寡核苷酸延伸(参见如美国专利号5,750,341和6,306,597,两者在此通过引用全文纳入本文)。测序技术的补充示例包括:church多克隆技术(mitra等,2003,analyticalbiochemistry320,55-65;shendure等,2005science309,1728-1732;和美国专利号6,432,360,6,485,944,6,511,803;在此通过引用全文纳入本文),454皮升焦磷酸测序技术(picotiterpyrosequencingtechnology,margulies等,2005nature437,376-380;美国公开号2005/0130173;在此通过引用全文纳入本文),solexa单碱基添加技术(bennett等,2005,pharmacogenomics,6,373-382;美国专利号6,787,308和6,833,246;在此通过引用全文纳入本文),lynx大量同步极好测序技术(brenner等,(2000).nat.biotechnol.18:630-634;美国专利号5,695,934,5,714,330;在此通过引用全文纳入本文)和adessipcr克隆技术(adessi等(2000).nucleicacidres.28,e87;wo2000/018957;在此通过引用全文纳入本文)。
通常,高通量测序都具有大量同步这一共同特征,高通量策略的目的是使成本比较早的测序方法低(参见如voelkerding等,clinicalchem.,55:641-658,2009;maclean等,naturerev.microbiol.,7:287-296;两者在此都通过引用全文纳入本文)。此类方法可大致分成通常用和不用模板扩增两大类。需要扩增的方法包括罗氏公司以454技术平台商业化的焦磷酸测序(例如,gs20和gsflx),illumina销售的solexa平台,和应用生物系统公司(appliedbiosystems)销售的支持态寡核苷酸连接和检测(supportedoligonucleotideligationanddetection,solid)平台。非扩增方法也称为单分子测序,其示例有螺旋生物科学公司(helicosbiosciences)销售的heliscope平台,visigen公司、牛津纳米孔技术公司(oxfordnanoporetechnologies)、生命技术公司(lifetechnologies)/离子流(iontorrent)和太平洋生物科学公司销售的平台。
焦磷酸测序(voelkerding等,clinicalchem.,55:641-658,2009;maclean等,naturerev.microbial.,7:287-296;美国专利号6,210,891和6,258,568;其各自通过引用全文纳入本文)中,模板dna被片段化、末端修复、连接衔接子、并用珠捕获单模板分子来进行原位克隆性扩增,珠上载有与衔接子互补的寡核苷酸。载有单模板类型的各珠被分入油包水微泡中,模板被克隆性扩增,所用技术被称作乳液pcr。扩增后破乳,珠被置入皮升微孔板(picotitreplate)的各孔内,孔在测序反应中作为流动室。在测序酶和发光报告物如萤光酶的存在下,流动室中发生四种dntp试剂各自的有序迭代引入。合适的dntp被加到测序引物的3′末端时,所产生的atp导致孔内发光脉冲,用ccd相机予以记录。能够实现大于或等于400个碱基的读数长度,且能够实现106个序列读数,得到最多达5亿碱基对(mb)的序列。
在solexa/illumina平台中(voelkerding等,clinicalchem.,55.641-658,2009;maclean等,naturerev.microbial.,7:287-296;美国专利号6,833,246,7,115,400和6,969,488;其各自通过引用全文纳入本文),以较短的读数形式产生测序数据。该方法中,单链的片段化dna末端修复产生5′-磷酸化钝端,然后由klenow介导添加单一a碱基至这些片段的3′末端。添加a便于添加t-突端衔接子寡核苷酸,后者将被用来捕获流动室表面上模板-衔接子分子,流动室中插有寡核苷酸锚。锚被用作pcr引物,但由于模板的长度且其靠近其它邻近的锚寡核苷酸,pcr延伸导致分子“拱跨(archingover)”杂交邻近的锚寡核苷酸在流动室表面形成桥式结构。这些dna环被变性并切割。正链随后通过可逆染料终止子来测序。通过检测纳入后荧光来确定所纳入核苷酸的序列,在下一轮dntp添加前除去各荧光团和阻断。序列读数长度从36个核苷酸到超过50个核苷酸,总体输出为每次运行分析超过10亿个核苷酸对。
用solid技术(voelkerding等,clinicalchem.,55:641-658,2009;maclean等,naturerev.microbial.,7:287-296;美国专利号5,912,148;和6,130,073;其各自通过引用全文纳入本文)对核酸分子进行测序还包括片段化模板,连接寡核苷酸衔接子,连接珠,以及乳液pcr克隆性扩增。此后,载有模板的珠被固定化在玻璃流动室的衍生化表面,与衔接子寡核苷酸互补的引物发生退火。但该引物并不用作3′延伸,而是用来提供5′磷酸基团供连接至问询探针,这些探针含有两个探针特异性碱基及其后6个简并碱基和四种荧光标记其一。solid系统中,问询探针中每个探针3′的两个碱基有16种可能的组合而在5′末端是四种荧光标记之一。荧光颜色,及由此辨识的各探针对应于指定的颜色-空间编码方案。多轮(通常7轮)探针退火、连接和荧光检测后变性,然后用相对初始引物错开一位碱基的引物进行第二轮的测序。以此方式,模板序列可通过计算得以重建,而且模板碱基问询两次,得到更高的精确度。序列读数长度平均为35个核苷酸,总体输出为每次测序运行超过40亿个碱基。
某些实施方式中,采用纳米孔测序(参见如astier等,j.am.chem.soc.2006年2月8日;128(5)1705-10,通过引用纳入本文)。纳米孔测序的原理涉及纳米孔浸入传导液并跨纳米孔施加电压(伏特)时所发生的现象。这些条件下,可观察到由于离子传导有微弱电流通过纳米孔,而电流的量对纳米孔的大小极度敏感。随着核酸的每个碱基通过该纳米孔,就会导致通过纳米孔的电流幅度有变化,这种变化对于四种碱基的每一种是不同的,从而允许确定dna分子的序列。
某些实施方式中,采用螺旋生物科学公司(helicosbiosciencescorporation)的heliscope(voelkerding等,clinicalchem.,55.641-658,2009;maclean等,naturerev.microbial,7:287-296;美国专利号7,169,560,7,282,337,7,482,120,7,501,245,6,818,395,6,911,345和7,501,245;其各自通过引用全文纳入本文)。模板dna被片段化并在3′末端多腺苷化,最后的腺苷载有荧光素标记。变性的多腺苷化模板片段连接到流动室表面上的聚(dt)寡核苷酸上。由ccd相机记录被捕获模板的初始物理位置,然后切下并洗去标记。通过添加聚合酶并系列添加带荧光标记的dntp试剂来实现测序。纳入事件产生对应于dntp的荧光信号,而ccd相机在每轮dntp添加前捕捉信号。序列读数长度在25-50个核苷酸,总体输出为每次运行分析超过10亿个核苷酸对。
离子激流技术是基于对dna聚合所释放氢离子的检测的dna测序方法(参见如science327(5970):1190(2010);美国专利申请号2009/0026082;2009/0127589;2010/0301398;2010/0197507;2010/0188073和2010/0137143;全部通过引用全文纳入本文用于所有目的)。微孔含有待测序的模板dna链。微孔层下方是超敏isfet离子传感器。所有层都包含在cmos半导体芯片内,该芯片与电子工业中所用的类似。在dntp被纳入生长中的互补链时释放氢离子,触发超敏离子传感器。若模板系列中存在均聚重复系列,单次循环中会纳入多个dntp分子。这导致对应数量的氢释放,和成比例的更高电子信号。这一技术与其它测序技术的区别之处在于不适用带修饰核苷酸和光学元件。离子流测序仪的单碱基精确度为每50碱基读数约99.6%,每次运行产生约100mb。读数长度是100个碱基对。5个重复的均聚重复序列的精确度是约98%。离子半导体测序的优势在于测序速度快且前期和运行成本低。
可适用于本发明的另一示例性核酸测序方法是由stratosgenomics公司开发并用到xpandomer分子的测序方法。该测序方法通常包括提供由模板引导的合成产生的子链。该子链通常包括按对应于靶核酸全部或部分的连续核苷酸序列偶联的多个亚单元,各亚单元含有系连物(tether)、至少一个探针或核碱基残基和至少一个选择性可切割的键。选择性可切割的键是被切割来得到xpandomer,其长度大于子链的所述多个亚单元的长度。xpandomer通常包括系连物和报告物元件,报告物元件用以解析序列中对应于靶核酸的全部或部分的连续核苷酸序列的遗传信息。xpandomer的报告物元件随后被测得。对基于xpandomer的方法的补充细节在文献中有记载,例如美国专利公开号2009/0035777,其通过引用全文纳入本文。
其它单分子测序方法包括利用visigen平台通过合成来实时测序(voelkerding等,clinicalchem.,55:641-58,2009;美国专利号7,329,492,美国专利申请序列号11/671,956和11/781,166;其各自通过引用全文纳入本文),其中,固定化的带引物dna模板用带荧光素修饰的聚合酶和荧光素受体分子来进行链延伸,在核苷酸添加时产生可测的荧光共振能量转移(fret)。
另一由太平洋生物科学公司(pacificbiosciences)开发的实时单分子测序系统(voelkerding等,clinicalchem.,55.641-658,2009;maclean等,naturerev.microbiol.,7:287-296;美国专利号7,170,050,7,302,146,7,313,308和7,476,503;其各自通过引用全文纳入本文)利用直径50-100nm含有约20仄升(10-21l)反应体积的反应孔。利用固定化模板、改良的
在某些实施方式中,单分子实时(smrt)dna测序方法采用太平洋生物科学公司(pacificbiosciences)开发的零级波导(zero-modewaveguide,zmw)或类似方法。用此技术,dna测序在smrt芯片上进行,这些芯片各自含有数千个零级波导(zmw)。zmw是孔,直径是纳米的几十分之一,制造在100nm金属膜中,该膜置于二氧化硅底物上。每个zmw成为提供检测体积仅20仄升(10-21l)的纳米光子可视化室。以此体积,可在数千个标记的核苷酸背景中检测出单个分子的活性。zmw通过合成进行测序,为观察dna聚合酶提供了窗口。各zmw室内,单个dna聚合酶分子结合在底面从而永久保持在检测体积内。磷酸连接的(phospholinked)核苷酸每种标记有不同颜色的荧光团,这些核苷酸随后以高浓度引入反应溶液中,这些浓度提高酶速度、精确性和处理能力(processivity)。由于zmw体积小,即使在这些高浓度下,检测体积被众核苷酸占据的时间占比很小。此外,由于转运核苷酸的扩散距离很短,对检测体积的经停很快,仅持续几微秒。结果就是背景很低。
可调试用于本发明的用于此类实时测序的方法和系统记载于,例如,美国专利号7,405,281、7,315,019、7,313,308、7,302,146和7,170,050;美国专利公开号2008/0212960、2008/0206764、2008/0199932、2008/0199874、2008/0176769、2008/0176316、2008/0176241、2008/0165346、2008/0160531、2008/0157005、2008/0153100、2008/0153095、2008/0152281、2008/0152280、2008/0145278、2008/0128627、2008/0108082、2008/0095488、2008/0080059、2008/0050747、2008/0032301、2008/0030628、2008/0009007、2007/0238679、2007/0231804、2007/0206187、2007/0196846、2007/0188750、2007/0161017、2007/0141598、2007/0134128、2007/0128133、2007/0077564、2007/0072196和2007/0036511,以及korlach等(2008)“选择性铝钝化用于将单个dna聚合酶分子靶向固定在零级波导纳米结构中(selectivealuminumpassivationfortargetedimmobilizationofsinglednapolymerasemolecules1nzero-modewaveguidenanostructures)”pnas105(4):1176-81,其全部在此通过引用全文纳入本文。
完成测序后,可以通过相同的条码对序列进行分组。可以在具有不同条码的模板片段的序列读数之间进行成对比较。确定针对两个条码生成的片段读数总数中共有的相同模板片段读数的百分比。相同片段可以这样确定,例如,通过比较来自分区内包含的所有片段的不同测序读数的dna模板片段部分的起始和终止序列(即,末端部分),其中两个dna模板测序读数相同的起始序列和终止序列表明两个原始dna片段是相同的。
作为示例来说明,考虑下述预示性示例,其显示了不同片段的读数数量(为了简化说明,大幅简化了下述示例):
上述示例中,共有的片段的百分比是25个片段w加上35个片段x除以具有条码a和b的片段总数(150),即60/150=40%。
对于条码之间的各个成对比较,可以生成百分比值(共有序列除以针对进行比较的条码的序列的总数乘以100)并将其与阈值进行比较。阈值可以表示预期将会随机共同出现的片段百分比,或者可以是另一个值,其允许区分因两个条码处于同一分区相比随机出现所导致的条码之间共有的dna片段百分比。在一些实施方式中,阈值这样确定:以成对的方式绘制所有条码中共同的百分比,然后观察该百分比何时下降到随机条码之间所同有的背景百分比。参见,例如图12。在一些实施例中,如果两个条码之间共有片段的百分比大于0.5%、1%、2%或大于5%,那么将两个条码视为源自同一分区。
可以基于测序读数确定dna模板序列的核苷酸序列。例如,对于任何特定分区,如果存在两个条码珠,那么将生成针对各条码珠的序列数据集。通过应用此处所述方法,一旦达到了两个条码之间共有序列的基础阈值,就可以将两个序列数据集合并以创建新的核苷酸序列。参见示例图3,其描绘了示例性工作流程。
在又一个实施方式中,基于分区的平均体积和递送至分区的珠体积可以排除这样的测序数据,所述数据中源自分区的条码数量多于物理上可能的数量。表示如此之高数量的条码来自单个分区的数据表明液滴完整性受到损害,因此可以排除来自任何这类条码的测序读数,以提高测序分辨率。
在另一方面中,已经发现与常规做法相反,其对于改善测序中的信噪比可能是有益的。发明人令人惊奇地发现,以比1∶1平均值高的珠浓度进行加载分区导致更好的信噪比。当用多个条码化珠(具有连接的条码寡核苷酸的拷贝的珠)加载分区时,各项指标显示出了改善。例如,在atac-seq分析中,当增加引入各个液滴的条码化珠的数量时,包括转录起始位点(tss)%、峰读数分数(frip)%和tss评分在内的各种指标都会得到改善。
因此,在一些实施方式中,方法包括:提供分区,其中至少10%、20%、30%、40%、50%、60%或70%的分区具有超过一个条码化颗粒(例如,珠)/分区,将来自颗粒的条码与分区中的样品dna连接,然后对条码化样品dna进行测序(例如,如上所述进行批量反应)。一旦生成了测序读数,就可以使用任何方法对数据进行去卷积,以汇集具有来自同一分区的不同条码的测序读数。因此,在一些实施方式中,可以使用上述方法,包括确定共有的相同片段的百分比,并合并样品读数,以假定在相同片段的百分比超过如上文所述阈值时,所有这类读数来自同一分区。用于对测序读数进行去卷积的其他方法(确定读数是否来自相同还是不同的分区)可以包括,例如,pctwo2017/120531中描述的那些方法。例如,在一些实施方式中,该方法可以包括在分区中提供包含条码序列或重复克隆条码序列的底物;并且在该分区中,将与包含第一条码序列的寡核苷酸引物偶联的第一颗粒和与包含第二条码序列的寡核苷酸引物偶联的第二颗粒与来自底物的条码序列关联;从而为该分区中的颗粒生成核酸特征,其可以用于与同一分区相比区分分开分区中的条码(请参见pctwo2017/120531)。在其它方面中,为了对测序读数进行去卷积(确定读数是否来自相同还是不同分区),该方法可以包括:形成包含正向引物的分区,所述正向引物包含条码和与靶核酸3′序列互补的捕获序列或其反向互补物,其中不同分区包含具有不同条码序列的不同正向引物,以及分区id标签寡核苷酸,其包含捕获序列的反向互补序列和可变分区id标签序列;在所述分区中,使至少一种正向引物与分区id标签寡核苷酸杂交以形成杂交产物;对杂交产物进行扩增以形成扩增子,其中至少一些扩增子由正向引物和分区id标签寡核苷酸形成;和对扩增子进行测序,其中如果不同的正向引物形成具有相同可变分区id标签序列的扩增子,那么认为不同的正向引物来自同一分区。在那些实施方式中的一些中,正向引物和分区id标签寡核苷酸在递送至分区时与同一底物连接;或者分区id标签寡核苷酸具有封闭的3′末端,使得聚合酶不能在扩增过程中延伸封闭的3′末端;或者分区id标签寡核苷酸包含双链可变分区id标签序列和一个或两个包含捕获序列的反向互补物的单链3′端。参见例如,美国临时专利申请号62/624,400。
还提供了这样的系统,用于接收来自本文所述条码化dna模板的测序读数,以成对的方式确定不同条码之间共有的dna模板片段测序读数的频率百分比;将确定的不同条码之间共有的dna模板片段的频率百分比与阈值进行比较,其中如果两个条码确定的共有的dna模板片段的频率百分比高于阈值,那么确定两个条码处于同一分区中;和由多个测序读数生成dna模板的核苷酸序列,其中生成核苷酸序列包括将具有确定处于同一分区的不同条码的测序读数视为来自同一分区。
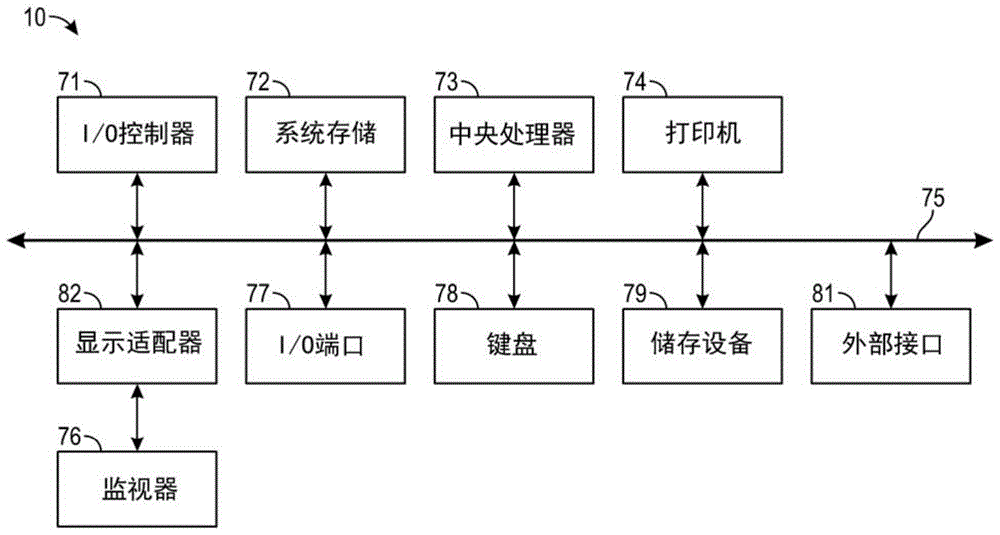
上述分析可以在如上所述的计算机软件中或系统中进行。本发明提及的任何计算机系统都可利用任何适当数量的子系统。这类子系统的示例如图1中计算机设备10所示。在一些实施方式中,计算机系统包括单个计算机设备,其中子系统可以是该计算机设备的组件。在其他实施方式中,计算机系统可包括多个计算机设备,其各是子系统,具有内部组件。
图1所示的子系统经由系统总线75互联。显示了其他子系统,如打印机74、键盘78、储存装置79、与显示适配器76偶联的监视器82等。与输入/输出(i/o)控制器71偶联的周边和输入/输出(i/o)装置可通过任何数量的本领域已知方式如输入/输出(i/o)端口977(例如,usbfire
计算机系统可包括多种相同的组件或子系统,例如通过外部接口81或通过内部接口连接在一起。在一些实施方式中,计算机系统、子系统或设备可通过网络连通。在这种情况下,可将一台计算机作为客户端并将另一台计算机作为服务器,其中各计算机都可以是同一计算机系统的部分。客户端和服务器可各包括多个系统、子系统或组件。
应理解,本发明的任意实施方式都可使用硬件(例如专用集成电路或现场可编程门阵列)以控制逻辑的形式和/或通过通常可编程的处理器使用计算机软件以模块化或集成化的方式来实施。本文中,处理器包括同一集成芯片上的多核处理器或者单个电路板上或网络连接的多个处理单元。基于本发明的公开和教导,本领域普通技术人员应知晓并理解使用硬件以及硬件和软件的组合来实施本发明的实施方式的其他方式和/或方法。
本申请中描述的任何软件组件或函数都可作为软件代码使用,以由处理器使用任何适当的计算机语言(如java、c、c++、c#)或脚本语言(如perl或python),使用例如常规或面向对象的技术来执行。软件代码可作为一系列指令或命令储存于计算机可读介质上用于储存和/或传输,合适的介质包括随机存取存储器(ram)、只读存储器(rom)、磁性介质(如硬盘或软盘)、光学介质(如光盘(cd)或dvd(数字多功能光盘)、闪速存储器等)。计算机可读介质可以是这里储存或传输装置的任意组合。
也可使用适用于传输的载波信号经由遵循多种协议的有线、光纤和/或无线网络(包括因特网)编码和传输这类程序。同样地,可使用这类程序编码的数据信号来建立本发明的一个实施方式所述的计算机可读介质。程序代码编码的计算机可读介质可与兼容性装置打包或由其他装置单独提供(例如通过因特网下载)。任何这类计算机可读介质可存在于单个计算机产品(例如硬盘、cd或整个计算机系统)之上或之内,且可存在于系统或网络中不同计算机产品之上或之内。计算机系统可包括监视器、打印机或将本发明所述任何结果提供给用户的其他合适显示装置。
本文所述方法的分析(非物理)步骤均可全部或部分地使用包括一个或多个处理器的计算机系统进行,可对其进行配置以进行完成所述方法的步骤。因此,实施方式可针对经设置以进行本文所述任意方法的步骤的计算机系统,其中不同组分可能完成相应步骤或相应步骤组合。虽然以编号的步骤形式显示,但本文中方法的步骤可同时或以不同顺序进行。此外,这些步骤的部分可与来自其他方法的其他步骤的部分联用。同样,步骤的全部或部分可以是任选的。此外,任何方法的任何步骤都可使用模块、循环或用于执行这些步骤的其他手段执行。
实施例
实施例1
用于珠条码去卷积的预示性示例(参见图3)
由培养物收获人k562细胞系细胞,监测其活力,并通过确立的方法洗涤以由细胞悬浮液去除培养基。将细胞计数为约50,000细胞/μl的浓度并裂解以产生核,并根据已公开的omni方案(corces等,naturemethods,2017年)进行标签化。完成后,将标签化的核保持在冰上。然后将标签化的核与试剂封装在一起,所述试剂支持使用条码化凝胶珠在液滴中进行pcr条码化。选择凝胶珠浓度,使得各液滴递送平均2-4个珠。进行热循环以条码化靶核酸片段。将产物经ampure珠纯化,以去除非特异性产物,并且如果需要,进行第二pcr,以增加产物浓度。第二次纯化在第二pcr条件性发生。该文库经过qced测序(qcedforsequencing)和测序。数据的生物信息处理如图3所示,以根据本文所述方法鉴定共享分区的条码珠以及生成atac特定指标。
1.比对读数:使用burrows-wheeler比对器(aligner)(bwa)比对配对末端的读数与其物种的基因组。参见例如,lih.和durbinr.(2009)使用burrows-wheeler转换快速准确的短读比对(fastandaccurateshortreadalignmentwithburrows-wheelertransform).bioinformatics,25:1754-60。
2.过滤低质量比对:去除映射质量(mappingquality)(mapq)<30的比对。剩余的所有片段正确映射的可能性≥90%。
3.对各珠条码相同片段进行删除重复数据:对具有相同起始和终止位置和相同条码的片段进行删除重复数据,以便在下游仅使用具有这些特征的单个片段。记录各珠的唯一片段计数。
4.对各珠条码之间相同片段对进行计数:对于每对珠,对具有相同起始和终止位点的片段数量进行计数。
5.确定各珠条码之间片段重叠百分比:使用下式计算在具有至少一个相同片段的各条码对之间重叠的片段的百分比:
·n条码1=与条码1关联的片段的数量
·n条码2=与条码2关联的片段的数量
·n共有=条码1和条码2之间共有的相同片段的数量
6.在珠对片段重叠百分比的平滑分布中找到阈值点:绘制并平滑各珠条码对片段重叠百分比的分布。该分布中的拐点(其中两个高点围绕一个低点)被用于将有效珠合并与无效珠合并分开。
7.合并片段重叠百分比>阈值的珠并分配细胞条码:将片段重叠百分比大于或等于拐点阈值的所有珠合并。为合并的珠分配独特的细胞/液滴/分区水平条码。
8.在细胞/液滴/分区条码内进行删除重复数据:对于合并的珠,进行第二轮相同片段的重复数据删除,以生成单个片段,以代替用于生成合并的相同片段。输出经过滤的包含细胞/液滴/分区条码的比对组,例如以.bam文件。
9.识别(call)峰:使用“峰识别(peakcalling)”程序macs2(zhangy,等.(2008)chip-seq基于模型的分析(model-basedanalysisofchip-seq)(macs),genomebiology,2008;9(9):r137)在完成珠合并(步骤8)时生成的.bam文件上识别峰。
10.计算伪批量(pseudobulk)(忽略的细胞条码)数据的信噪比指标:
·tss%:步骤8中生成的.bam文件“相交(intersect)”.bed文件,所述.bed文件是读数与之比对的基因组经注解的转录起始位点的。tss%是这样的读数的分数,其与经注释的转录起始位点上游或下游2kb内的至少一个碱基重叠。
·frip:步骤8中生成的.bam文件“相交”macs2生成的峰的.bed文件。
峰中读数的分数是与峰重叠至少一个碱基的读数的百分比。
·tss富集评分:只使用落入围绕经注释的转录起始位点周围+/-2kb窗口内的读数,如下所示计算经标准化评分:
a.计算侧接+/-2kbtss窗口的100bp区域中的平均读数深度
b.计算转录起始位点(+/-2kbtss窗口中心)的读数深度
c.(b)除以(a)
实施例2
图12描述了来自液滴中外源性片段化dna进行条码化的分析。标签化的λ基因组dna用作单细胞去卷积过程中外源性片段化dna的示例。简言之,将λ基因组随机标签化并用特定序列标记,该序列与通过转座酶的条码-寡核苷酸的捕获序列互补。在单细胞划分步骤中,将标签化的λ片段添加到反应中,从而通过聚合酶活性利用同一液滴中独特的条码-寡核苷酸来捕获外源性λdna片段和单细胞的内源性转录本。在液滴中进行条码化步骤后,破坏所有液滴,并按照生产商的说明通过下一代测序对条码标记的dna片段的单细胞文库进行测序。测序的结果以fastq文件格式捕获,用于使用下文所述且如图12所示的生物信息学管道进行后续序列分析。
下文说明了图12中流程图中的各个步骤。
a-使用伯乐公司(bio-rad)atac-seq工具包对输入fastq文件(原始读取)进行去条码化(debarcode)。将具有有效条码的读数传递到工作流程中的下一步。将具有无效条码(无条码、不正确的条码结构,不明确的条码)的读数由分析中丢弃。具有有效条码的读数具有这样的条码序列,所述条码序列由读数的dna序列移出并移入读数名(readname)。
b-首先,使用bwa-mem算法将具有有效条码的读数与λ噬菌体基因组(可在互联网上找到,网址为neb.com/products/n3011-lambda-dna#product%20information)比对。与λ基因组比对的读数以bam文件的形式传递至步骤x,并注释比对的珠条码为xb标签。与λ基因组不比对的读数传递给步骤c。
c-使用剪接转录本比对参照(splicedtranscriptsalignmenttoareference,star)比对器将未比对λ基因组的读数(大概是源自实验中细胞的读数)与其物种基因组比对。该步骤中比对的读数以bam文件的形式传递至步骤d和g,并且注释为比对的珠条码为xb标签。丢弃该步骤中不比对的读数(未图示)。
d-使用伯乐公司atac-seq工具箱对珠进行“拐点识别(kneecall)”。对各珠条码的独特基因组片段的数量进行计数,然后进行log10转换。通过珠条码分布生成转化的基因组片段的高斯核密度估算。最后,确定最有可能代表暴露于细胞的珠相对空液滴中的珠之间区别的拐点。低于该阈值的所有珠排除在进一步分析之外。将条码的“白名单”传递至步骤e。
e-通过珠去卷积对步骤d中生成的白名单上的珠进行λ比对。计算各对珠之间相同和独特比对数量的计数。计算各对珠的片段重叠指数(=相同与独特片段的比例),并传递至步骤f。高片段重叠指数表示两个珠之间存在大量相同片段,而低概率在当两个珠处于不同液滴中发生。
f-使用步骤d中概述的相同方法在该分布上进行“拐点识别”,其中拐点是jaccard指数,两个珠在此处“看见(seeing)”来自同一细胞的片段,相对不同液滴中珠上相同片段的机会水平观察。合并jaccard指数高于该阈值的所有珠条码。这将生成一个新的套件“液滴”条码。该步骤的主要输出被传递至步骤g,作为液滴条码翻译表的珠条码,其中捕获了多个珠对一个液滴条码关系。
g-将来自步骤f的珠对液滴翻译表和来自步骤c的bam文件组合,以根据步骤e和f中的计算将液滴条码注释到来自确定处于同一液滴中的珠的所有比对。该“细胞条码化的”bam文件传递至步骤h。
h-对与同一细胞(与“液滴”互换使用)条码精确比对到同一位置的读数进行重复数据删除,在液滴中>1珠与给定基因组位置比对的情况中仅保留一个比对。间该重复数据删除的bam文件传递至步骤i,用于进行细胞识别和计数。
i-使用与步骤d和f中相同的方法,基于各细胞条码独特的基因读数的数量进行拐点识别。
实施例3
图13-15显示示例性工作流程。图13显示了混合物种单细胞atac-seq实验。合并相等数量的小鼠和人细胞系,然后进行下述工作流程。从裂解的细胞中制备核,并用tn5转座酶批量标签化(试管)。使标签化的核与条码和pcr试剂一起流入微流体盒(cartridge),并在产生液滴的位置合并。将液滴收集到试管中并进行第一轮pcr。然后将液滴破碎并进行第二轮pcr。然后对衔接的dna片段进行测序。
图14描述了用于使用单细胞atac-seq数据将珠共定位至单个液滴的生物信息流程。a)质量检查。计算各碱基的序列质量,衔接子污染和丰富的序列,并决定是否应由fastq读数修剪碱基。b)分块和去条码化。提取并过滤条码序列,准备进行比对。c)fastq比对。用star比对仪比对读数与基因组。比对的读数以bam文件的形式传递至下一步,该文件的xb标签中注释了条码。d)珠过滤。对各珠条码的独特基因组片段的数量进行计数,然后进行logl0转换。通过珠条码分布生成转化的基因组片段的高斯核密度估算。最后,识别最有可能代表暴露于细胞的珠相对空液滴中的珠之间区别的拐点。低于该阈值的所有珠排除在进一步分析之外。将条码的“白名单”向前传递。e)珠合并。f-使用步骤d中概述的相同方法在该分布上进行“拐点识别”,其中拐点是jaccard指数,两个珠在此处“看见”来自同一细胞的片段,相对分开的液滴中珠上相同片段的机会水平观察。合并jaccard指数高于该阈值的所有珠条码。这将生成代表细胞的新的套件“液滴”条码。该步骤的主要输出被传递至步骤f,作为液滴/细胞条码翻译表的珠条码,其中捕获了针对一个液滴条码关系的多个珠。f)细胞过滤。使用与步骤d和e中相同的方法,基于各细胞条码独特的基因读数的数量进行拐点识别。然后,通过查找富含比对的读数的基因组区域来识别峰。
图15描述了atac-seq实验期间珠合并的示例。在使用结合小鼠和人细胞的大约5000个细胞输入运行图13中所述实验并运行图14中的生物信息管线后,可以生成下述atac-seq数据,包括将共定位后的珠合并为单个液滴。在(a)去条码化和(b)比对后,将具有高信号的珠鉴定为拐点左侧的数据点,用垂直实线勾勒,如珠过滤拐点图(c)所示。计算条码之间共有的片段%,并将珠合并为珠合并拐点图(d)中垂直蓝线左侧的点。细胞数据点通过(e)中垂直线左侧的高度独特基因组片段鉴定。在该实验中,鉴定出3974个细胞具有超过25000个平均独特片段/细胞。(f)中显示了各液滴计算的珠的直方图。
来自图15:
a)
b)
c)
d)
应理解,本文所述的实施例和实施方式仅用于说明目的,本领域技术人员应了解据此作出的各种修饰或改变,且它们包括在本申请的主旨和权益以及所附权利要求书的范围内。本文引用的所有发表物、专利和专利申请通过引用全文纳入本文以用于所有目的。
- 还没有人留言评论。精彩留言会获得点赞!