用于工程合成顺式调控DNA的方法与流程
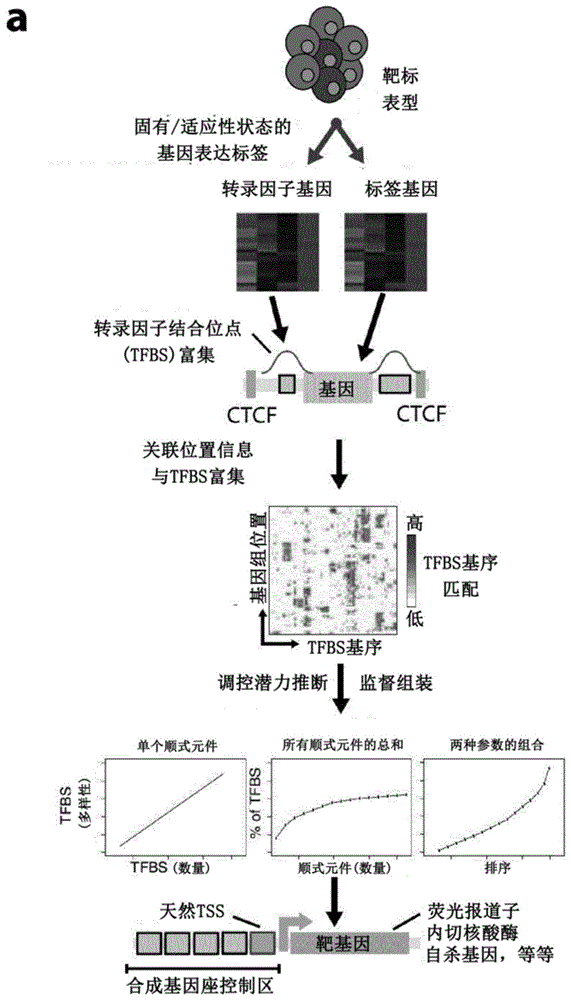
本发明涉及用于生成细胞类型特异性表达盒和报道载体的方法,以及可以通过这样的方法生成的核酸构建体。细胞类型特异性表达盒和报道载体的特征在于合成顺式调控dna,也称为合成基因座区(slcr)。slcr允许报道基因或效应基因的细胞类型特异性表达。本发明进一步涉及报道载体的各种用途,包括在基因和病毒治疗、药物发现或验证中确定细胞的特性,优选细胞类型、状态或命运转变(fatetransition)。
背景技术:
:表达盒和报道载体在基础研究、药物筛选诊断或基因治疗中具有广泛的应用。选择性鉴定细胞类型特异性的标识对于理解在其中多样化的细胞类型有助于组织内稳态的生物学过程至关重要。理想地,这种方法在涉及包括代谢、免疫、神经或精神疾患以及炎症和癌症的组织内稳态的改变的疾病环境中也将是有益的。在开发环境中,这传统上是使用谱系追踪1实现的。在最众所周知的实例中,fbx15表达的谱系追踪导致发现了能够将成纤维细胞重编程为多能细胞的定义的因子49,以及lgr5表达的谱系追踪使得能够鉴定真正的微结肠和小肠干细胞2,其后来被证明可以标记许多其他成体组织干细胞3。庞杂的报道子策略的平行开发允许在分析多个谱系中的单细胞分辨。传统上,已经利用了多种遗传追踪方法来生成用于细胞类型特异性遗传操纵和细胞标记的报道小鼠(例如lacz、mgmt、brainbow和confetti体系、双标记嵌合分析-madm等)。这些策略可以揭示复杂的神经元连接模式4,并解决悬而未决的问题,诸如活生物体中肿瘤的起源细胞5。最近,光遗传技术和基于crisrp/cas9的策略在获得更多定量读出中增加了进一步的灵活性。基于成体干细胞生物学的报道子策略的使用可以同时告知组织的起源及其异常的内稳态678。反映出特征明确的通路的遗传报道子可以导致对复杂的信号传导二分法的更深入的了解,诸如在毛囊内稳态期间转化生长因子抵消骨形态发生蛋白质(bmp)信号传导9。在癌症中,这种方法严格地揭示了异常的内稳态可能是治疗抗性的原因10,或者再生潜力和肿瘤易感性可能在某些器官之间共享,而在其他器官中则明显不同11。可以通过基于转录因子结合位点设计合成报道子来揭示定量时空模式动力学47。如从这些和许多其他研究中推断的,遗传报道子的选择是最终解决庞杂和复杂生物学问题的关键因素。这在受多种因素和复杂相互作用支配的发育或疾病环境中尤其有效12。在这些环境中,灵活设计在单个基因盒中拦截多个通路的合成报道子的能力无疑将证明是一项重要资产,然而目前的方法仍然受到限制。例如,目前采用的用于遗传追踪载体的方法依赖于使用与报道基因或功能效应子偶联的细胞类型、通路特异性或合成启动子或增强子。细胞类型特异性启动子的使用基于将报道基因或功能效应子置于目的细胞类型的标签基因(signaturegene)的最小启动子之后。因此,它允许作为给定基因的启动子介导的给定报道子或效应子的特异性转录激活。细胞类型特异性载体提供了使用一个给定基因作为细胞状态或发育阶段的代表(proxy)的可能性。一个实例是使用nestin启动子以便标记神经祖细胞。这种方法被广泛使用,并且允许研究人员指导未分化细胞中特异性报道子或效应子的激活。这些方法的显著限制是必须有标签基因的先验知识,并且假定所述基因的调控元件是已知的并且紧邻转录起始位点。此外,这些方法受限于用以描述复杂调控系统的单个基因的特异性不足。对于该问题的繁琐解决方案需要对任何给定的目的细胞类型的所有特异性增强子进行细胞类型特异性鉴定,随后选择这样的元件之一并且将其克隆至最小病毒启动子上游。然而,这种方法在技术上要求很高,并且确实依赖于监督选择48。这两个限制确实将这种方法的应用局限于在非常受选择的环境。替代方法使用通路特异性启动子,以便将报道子或效应子置于特异于给定通路的人工组装的转录因子结合位点之后。因此,可以通过已知对于所述通路必不可少的调控元件的介导来控制特异性转录激活。一个实例是特异于smad1/5/8的核活性的bmp应答元件(bre),它描述了bmp通路的激活。虽然bmp应答元件(bre)可靠地描述了经典通路激活,但它忽略了非经典激活,并且提供了对反馈回路不够敏感的报道系统。使用通路特异性启动子的限制包括需要依赖于假设所使用的调控元件的最小集足以告知通路激活。此外,这样的调控元件和其广泛的特征以及与其自然环境的隔离的先验知识是必要的,并且妨碍了它们对于复杂和不太具特征的细胞类型的应用。作为进一步的方法,已经提出了合成增强子或启动子,通过将目的报道子置于多个人工组装的转录因子结合位点之后最小启动子之前。然而,这些方法也依赖于已知与细胞类型或发育阶段相关的转录因子结合位点的先验知识。所有方法都受限于它们对先验知识的依赖或者对特异于目的细胞类型或阶段的调控元件的准确发现和验证。此外,由于在许多情况下并未覆盖所有调控元件,因此不得不使用多种标志物以便确保可靠的细胞类型表征,从而使报道子的构建和任何实验结果的评估变得复杂。经由流式细胞术的基于细胞特异性表面分子的表达的细胞表征也已经在本领域中描述。这是一种常见的做法,但在某种意义上是受限的,因为相应的标志物必须事先知道,并且并非所有细胞类型都具有特征性的表面蛋白。此外,使用这样的方法不可能在体内追踪细胞类型或非常具有挑战性。为了尝试采用多个转录因子结合位点以调控报道基因的表达,已经开发了替代的基因表达报道载体。wo2001/49868a1(韩国生命科学与生物技术研究院)公开了癌症特异性基因表达载体,其包括具有在癌基因中表达的e2f转录因子的结合位点(ef2bs)以及其他转录因子的额外的结合位点(例如sp1、ap1、nf1或c/efb)的启动子。然而,该方法仍然依赖于先前鉴定为与特定类型的癌症相关的tf结合位点(例如,ef2bs)的先验知识。wo2015/110449a1(布鲁塞尔大学/根特大学)公开了用于鉴定具有转录因子结合位点(tfbs)的富集的心脏和骨骼肌特异性调控元件的计算方法,其中,公开了300-500bp的长度的不同的调控区(csk-sh1-6;sk-sh1),每个包含多个(3-10)保守的tfbs。然而,该技术专注于采用进化的保守的tfbs,从而依赖于调控序列的基因组保守性,以便增强在肌肉中的表达。wo2008/107725a1公开了用于鉴定在目的细胞中有活性的转录因子调控元件(tfre)的计算方法,其中,tfre具有至少6至100bp的长度,其中,6个或更多个tfre可以组合在表达载体的启动子元件中。然而,该技术采用相同的预选最小启动子的融合,在任何给定条件下鉴定出额外的tfre,即具有已知功能的顺式元件的监督合并。guo等人(trendsinmol.medicine,14:410-418)回顾了几种病毒载体以及转录调控元件。gargiulo等人(mechanismsofdevelopment,35:193-203)公开了对果蝇(drospholia)的卵泡上皮中的卵黄膜蛋白基因32(vmpe)的细胞特异性表达的顺式作用元件的鉴定,其中,表达载体包括调控基因组区的不同片段。尽管在该领域中取得了这些进步,但这样的替代方法依赖于对生成报道载体不利的策略,诸如依赖于相关启动子的先验知识,专注于tfbs的遗传/进化保守性,或者使用由具有已知功能的顺式元件修饰的单一启动子。因此,在合成报道子领域中需要基于无偏倚从头方法的替代或改进的方法和构建体,用于解码和重建用于任何给定细胞类型或状态的调控信息。技术实现要素:鉴于现有技术,本发明基于的技术问题是提供替代和/或改进的方法,用于生成基于合成顺式调控dna的遗传追踪盒或载体,其允许报道基因或功能效应子的细胞类型或发育阶段特异性表达。该问题通过独立权利要求的特征解决。本发明的优选实施方式由从属权利要求提供。因此,本发明涉及用于生成细胞类型特异性表达盒的方法,所述方法包括以下步骤:a)提供目的细胞类型的基因表达谱,b)提供所述目的细胞类型的基因组序列数据,c)从所述基因表达谱中选择标签基因的集合,所述标签基因(i)与参考细胞类型相比受到差异调控,或(ii)根据基因表达水平选择,d)在c)中选择的所述标签基因的集合中鉴定编码转录因子的基因,e)从所述基因组序列数据中确定基因组区的集合,其中,每个基因组区包括编码在c)中鉴定的标签基因的序列和与编码所述标签基因的序列相邻并且位于编码所述标签基因的序列的侧翼的额外的基因组序列,f)鉴定在e)中确定的所述基因组区的集合内的具有相当和有限大小,优选相等大小的多个基因组亚区,其中,所述基因组亚区包括在d)中鉴定的一个或多个转录因子的一个或多个结合位点,g)从f)中确定的基因组亚区中选择基因组亚区的最小集合,优选2至10个之间,其中,选择所述基因组亚区的集合以包括在d)中鉴定的所有转录因子的预定百分比的转录因子结合位点,以及h)生成包括与报道基因或效应基因可操作地偶联的在步骤g)中选择的所述基因组亚区的集合的细胞类型特异性表达盒,其中,将所述基因组亚区配置为调控所述报道基因或效应基因的表达。所述方法允许生成表达盒,当将所述表达盒引入目的细胞中时,会以对特定实体或状态诸如细胞类型或状态高度特异性的方式产生报道基因或效应基因的表达,其中,报道子已被设计为描述在所述目的实体或状态中基因表达的调控而不需要先验知识。与现有技术相反,本发明的方法和构建体基于用于解码和重建任何给定细胞类型/状态的调控信息的无偏倚从头方法。本发明代表了基本上基于在细胞类型/状态特异性标签基因上的细胞类型/状态特异性tfbs的聚类的完全新颖的方法。本发明的特征还在于对于任何给定的细胞类型/状态的相关tfbs采用定量和/或统计富集的优点。在一些实施方式中,所述方法基本上采用系统生物学方法,通过从目的细胞类型的给定转录标签中鉴定内源性发生的顺式调控元件的集合,并将这些顺式调控元件置于报道基因或效应基因之前,来生成表达盒。此方法独立于目的细胞类型的特定特征的预想信息,从而允许标准化、无偏倚和直接地产生针对任何给定细胞类型的报道构建体。为此,所述方法鉴定了包括细胞类型特征性的转录因子结合位点的基因组亚区,并将它们组装到包括目的细胞类型内的转录调控序列信息的相关部分的基因组亚区的集合中。所述基因组亚区的集合也可以被称为“合成顺式调控dna”、“合成调控区”或“合成基因座控制区(slcr)”。当引入细胞中时,所述报道基因或效应基因的表达将会发生,因为在所述细胞类型中,存在与特征性转录因子结合位点相对应的转录因子,并启动所述报道基因或效应基因的表达。因此表达水平与特定细胞类型有关。每种细胞类型将根据标签基因集合本质上产生不同的基因集合,并且每种细胞类型将根据存在的转录因子和slcr中组装的调控区的组合示出不同的报道子表达水平。有利地,所述方法不限于某些细胞类型,而是可以实际上应用于任何细胞类型,并且甚至区分某个细胞类型内的细胞状态或命运转变。为此,不需要目的细胞类型中基因调控的先验知识。相反,所述方法仅依赖于给定细胞类型的基因表达谱和基因组序列数据的提供,这可以使用标准生物分子技术或访问公共数据库获得。基因表达谱反映了目的细胞类型内的基因表达的水平。为此,例如,可以使用rna-seq或其他测序或基于微阵列的技术来定量目的细胞类型中rna转录物的水平。然而,也可以使用蛋白质组学可能地推导出基因表达谱,例如通过量化存在于目的细胞类型中的表达的蛋白质或肽,可以将其平方到基因表达谱。从基因表达谱中,选择目的细胞类型、细胞状态或实体特征性的标签基因。标签基因的选择可以适应于期望的应用。例如,可以根据标签基因的基因表达水平来选择标签基因,通过将目的细胞类型的基因根据其基因表达水平进行排序,并选择高于或低于某个阈值的基因,或者选择预定数量的最高或最低表达的基因。对于这样的标签基因的选择,将目的细胞类型的基因的绝对表达水平用作参考。因此得到的表达盒可以独立于待探测的细胞而忠实地报道在各种测定中的目的细胞类型的存在。然而,对于某些应用,可能期望生成一种表达盒,其以特定的高特异性将目的细胞类型与参考细胞或参考细胞状态相区分。对于这样的应用,通过鉴定与参考细胞类型中的表达水平相比上调或下调的基因来选择差异调控的标签基因。在这些实施方式中,提供目的细胞类型和参考细胞类型的基因表达谱。通过选择差异调控的基因,可以将表达盒微调用于需要将目的细胞类型(或状态或命运)与某一参考类型(或状态或命运)相区分的测定。从选择的标签基因中,鉴定出标签基因的集合中的编码转录因子的所有基因。为此,所述方法可依赖于公开可访问的带注释的数据库,诸如encode、mencode(encode项目的鼠标版本)、jaspar、ensemble、entrezgene、genebank等。因此,鉴定了特征性表达的目的细胞类型的转录因子的集合。转录因子可由技术人员通过常用数据库中的功能的注释来鉴定。此外,每个转录因子的靶序列,即转录因子结合位点通常是技术人员已知的和/或使用适当注释的数据库诸如上述那些可获得的。优选地,在一些实施方式中,所述方法涉及转录因子的使用,这些转录因子的结合位点(以dna序列或序列基序的形式)是已知的和/或优选地在公共数据库中注释的。此外,使用选择的基因的集合以确定来自目的细胞类型的基因组序列数据的基因组区的集合,其中,每个基因组区包括编码标签基因的序列和与编码所述标签基因的序列相邻(优选位于编码所述标签基因的序列的紧邻的侧翼)的额外的基因组序列。此基因组序列例如非编码参考dna(尽管顺式调控元件可能存在于编码区中)旨在涵盖调控序列,该序列可位于编码区的上游、下游或内部,更通常紧邻转录起始位点但不仅限于此。与标签基因相邻的额外的基因组序列的大小可以变化,因为所述方法有利地对额外的基因组序列的附加部分的存在不过度敏感。因此,额外的基因组序列应足够大以涵盖调控标签基因的表达的顺式调控元件(尤其是转录因子结合位点,或增强子或沉默子)。众所周知,这样的顺式调控元件在结构上可以与编码区非常接近,但是——考虑到基因组在核仁中的3d结构分布——就线性基因组序列而言,顺式调控元件可以位于相当远的距离。在优选的实施方式中,通过使用拓扑相关结构域作为边界,基于细胞类型中染色质内dna的折叠的三维状态,选择调控基因组序列。优选地,在一些实施方式中,所述方法假定细胞类型特异性的非编码ctcf结合位点作为拓扑相关结构域的代表。ctcf结合位点(以dna序列或序列基序的形式)通常是技术人员已知的和/或通常在公共数据库中注释的。在优选的实施方式中,在确定了基因组区的集合之后,所述方法搜索相似或相当大小(例如相等大小)的多个基因组亚区,所述基因组亚区包括由标签基因编码的转录因子的一个或多个,优选数个结合位点。因此,在所述方法的步骤f)中鉴定的所有基因组亚区均包括在目的细胞类型中特征性表达的转录因子的dna结合位点。当将所述基因组亚区组装在slcr中并且将所述slcr引入目的细胞中时,特征性表达的标签转录因子可以与所述slcr结合并且调控下游报道基因或效应基因的表达。通常,鉴定出的许多基因组亚区比组成slcr的基因组亚区更大,这些基因组亚区就特征性转录因子的结合位点而言是冗余的。有限数量的所有已鉴定的基因组亚区的组装足以代表整个调控复杂性,并且包括所有元件不会导致特异性增加,而是产生不必要的大表达盒。因此,所述方法进一步涵盖以下步骤,选择包括由选择的标签基因编码的所有转录因子的预定百分比的转录因子结合位点的基因组亚区的最小集合。举例来说,可以假设在标签基因的集合内可以鉴定100个转录因子,其中100个转录因子结合位点是已知的。然而,在一些实施方式中,由选择的标签基因编码的转录因子的数量不一定等于转录因子结合位点的数量。在一些选定的实施方式中,不是所有的转录因子都可以具有已知的结合位点,或者多个转录因子结合位点矩阵可以与一些转录因子相关。为了在slcr的组装中使用尽可能少的基因组亚区的数量,例如为了保持所得调控序列的紧凑,则所述方法优选地除了根据转录因子结合位点的多样性之外,还根据转录因子结合位点的数量将基因组亚区进行排序。例如,排序最高的基因组亚区可包含用于步骤d)的转录因子的35个转录因子结合位点,其中这些结合位点中的3个在同一基因组亚区中重复出现5次,而其余结合位点仅出现一次。那么此排序最高的基因组亚区将包括23个不同的(独特的)转录因子结合位点,其代表标签基因的23个转录因子的结合位点。因此,此排序最高的基因组亚区将覆盖23%的步骤d)的特征性转录因子。如果例如将预定百分比设置为50%,则将搜索优选涵盖尚未包含在第一基因组亚区的23个结合位点中的转录因子结合位点的第二(并且潜在地第三)基因组亚区,等等,使得进一步的一个或多个基因组亚区将包括尚未被第一排序最高的基因组亚区所覆盖的转录因子的至少7个结合位点。通常,2-10个基因组亚区的最小集合将包括是由标签基因编码的转录因子的至少50%的结合靶标的转录因子结合位点。当将表达盒引入目的细胞类型中时,基因组亚区的最小集合充当特征性转录因子可以结合的合成顺式调控dna。因此,在本文的所述方法的步骤g)中选择的基因组亚区的最小集合因此称为合成基因座控制区(slcr)。在一些实施方式中,所述盒因此包括富集了被转录因子(所述转录因子例如在目的细胞类型中表达或高度表达)结合的调控序列的调控区(slcr)。因此,此调控区是对于该特定细胞类型特有的/定制的,并且导致该细胞类型特有的报道基因的表达水平。考虑到在d)中鉴定的特征性转录因子的总量反映了目的细胞类型的调控机制,转录因子的覆盖的预定百分比可以被视为被基因组亚区的最小集合覆盖的“调控信息的百分比”。理论上,所覆盖的调控信息的量越高,报道基因或效应基因的表达对细胞类型就更具特异性。然而,有利的是,如通过实验验证所测量的,就细胞类型特异性表达谱而言,覆盖至少30%的调控信息、优选至少40%或50%的百分比产生优异的结果。在所述方法的步骤h)中,通过以下生成细胞类型特异性表达盒:将在步骤g)中选择的基因组亚区最小的集合与报道子或效应子组装,使得它们可操作地偶联,即包括作为顺式调控元件的转录因子结合位点的基因组亚区被配置为调控报道基因或效应基因的表达。借助于组装的基因组亚区对调控信息的高覆盖率无需先验信息,为本文所述的方法和构建体提供了巨大的应用潜力。所述表达盒作为报道载体的一部分,可以在体外和体内被利用作为固有细胞状态的报道子,用于对外部信号传导或化学输入、细胞命运转变、重编程、正向和化学遗传筛查的适应性应答。此外,当细胞类型特异性slcr与内切核酸酶或自杀基因组合时,所述载体可在基因治疗或其他遗传修饰环境中用于使细胞类型、发育阶段或疾病特异性群体缺失。在这些其他遗传修饰设置中,slcr可以驱动溶瘤病毒和/或共刺激分子的结构组分的肿瘤特异性表达,旨在提高溶瘤治疗的特异性和有效性。在本发明的优选的实施方式中,所述方法的特征在于所述基因表达谱包括目的细胞类型中基因的表达水平,以及o根据步骤c)(i)提供参考细胞类型的基因表达谱,所述基因表达谱包括参考细胞类型中基因的表达水平,并且通过鉴定与所述参考细胞类型中的表达水平相比被上调或下调的基因来选择差异调控的标签基因,优选选择在所述目的细胞类型中上调3至10倍或更多的基因,或o根据步骤c)(ii)根据所述目的细胞类型的基因的基因表达水平将所述目的细胞类型的基因排序,并且基于标签基因的预定水平或预定数量的表达,诸如所述目的细胞类型中100至1000个最高表达的或者100至1000个最低表达的基因,选择标签基因。第二种替代允许基于可从基因表达谱得到的所述细胞类型的基因的表达水平的比较来选择标签基因。这样的实施方式尤其很好地适于生成在不同的实验设置中代表目的细胞类型的表达盒。为此,选择相比平均表达水平上调3至10倍或更多的基因已产生了优异的结果。第一种替代允许定制表达盒以区分目的细胞类型相比于参考细胞类型。举例来说,目的细胞类型可以是某种肿瘤细胞,而参考细胞类型是指通常被肿瘤侵袭的组织类型的正常细胞或肿瘤细胞所起源的细胞类型。然而,参考细胞类型也可以指相同类型的但是处于不同的细胞状态或在命运转变之前或之后的细胞。目的细胞类型的基因表达谱可以是指在上皮至间充质转化(emt)后处于间充质状态的癌细胞的基因表达谱,而参考细胞类型的基因表达谱可以指相同类型的但是处于其上皮状态,即在上皮-至-间充质转化(etm)之前的癌细胞的基因表达谱。在此情况下,表达盒将能够区分经历过emt的细胞与未经历过emt的细胞。通过基于与参考细胞类型相比的相对调控选择标签基因而可得到的表达盒的特征在于,具有尤其高的特异性,允许在不需要任何额外标志物下区分参考细胞类型与目的细胞类型。在本发明的优选实施方式中,所述方法的特征在于所覆盖的转录因子的预定百分比为30%或更多、优选40%或更多、最优选50%或更多。在本发明的进一步优选的实施方式中,所述方法的特征在于,在e)中确定的基因组区对应于包含差异调控基因的拓扑相关结构域的基因组序列,其中,优选地拓扑相关结构域对应于两个ctfc结合位点之间的基因组序列。通过基于拓扑相关结构域选择基因组区的大小,可以实现控制所述标签基因的转录的潜在顺式调控元件的最优覆盖。在拓扑相关结构域内,dna序列物理上的相互作用比与在拓扑相关结构域外的序列更加频繁,从而形成可用于转录机制的三维染色体结构。通过选择两个ctfc结合位点之间的基因组序列,可以获得尤其好的结果。这样的实施方式在计算能力资源、非编码顺式调控dna对它们最有可能调控的基因的特异性以及覆盖特征性转录因子结合位点的侧翼dna的大小之间产生最优的平衡。在所述方法的优选的实施方式中,通过在e)中确定的所述基因组区的滑动窗口算法来进行步骤f)中的对相当例如相等大小的基因组亚区的鉴定,其中,优选地所述窗口具有的长度为500bp至5000bp、优选700bp至2000bp、更优选800bp至1200bp、最优选1000bp,以及所述滑动步长具有的长度为100bp至1000bp、优选120bp至300bp、更优选130bp至170bp、最优选150bp。在一种实施方式中,所述滑动窗口固定为1000bp大小,以150bp步长滑动,尽管扫描产生的基因组亚区大小可能会在大小上变化,因为它取决于tfbs的统计得分和分布。进一步优选地,所述滑动窗口算法从限于与步骤d)中鉴定的转录因子相对应的转录因子结合位点的相关数据库(例如,jaspar)计算转录因子结合位点基序的统计富集。因此,生成了特异性区内的特征性转录因子结合位点的显著富集的列表,并用于鉴定相当的、优选相等大小的基因组亚区,所述基因组亚区包括由标签基因编码的至少一个特征性转录因子的至少一个转录因子结合位点。优选地并且最可能地,在相当大小的基因组亚区中包括数十个(10至200,优选在20至180之间)tfbs。根据本发明,在e)中确定的基因组区的集合内的相当和有限大小优选相等大小的多个基因组亚区(根据步骤f)通常具有相同的大小,但是可以变化。在本文中相当的是指优选展现出500bp至5000bp的任何窗口大小的多个基因组亚区。在本发明进一步优选的实施方式中,所述基因组亚区具有的长度为100bp至1000bp、优选120bp至300bp、更优选130bp至170bp、最优选150bp。如果使用滑动窗口算法,则基因组亚区的长度将优选与滑动步长相关。在其他实施方式中,滑动窗口方法可以使用任何给定的步长大小,从1bp至最高对于上述窗口大小所指定的那些步长大小。优选的长度已经通过采用对不同细胞类型和测定系统的方法确定,并且反映在表达特异性和表达盒总大小方面的最优结果。在本发明的进一步优选的实施方式中,所述方法的特征在于g)中的基因组亚区的集合的选择通过计算f)中鉴定的每个基因组亚区来进行:-在基因组序列数据中根据d)的转录因子的结合位点的富集,以及-存在结合位点的转录因子的多样性的得分,-其中,根据存在结合位点的转录因子的累计百分比将所述基因组亚区排序,以及-其中,选择基因组亚区的最小集合以包括d)中鉴定的所有转录因子的预定百分比的结合位点。例如,在c)中选择的标签基因的集合内鉴定编码转录因子的基因之后,已经生成了转录因子结合位点的数量和类型。此外,提供了在步骤f)中生成的基因组亚区的列表。用此信息,可以计算每个基因组亚区(例如,tfbs=35)的转录因子结合位点(tfbs)的数量,其代表了在基因组序列数据中根据d)的转录因子的结合位点的富集。此外,优选地,计算每个基因组亚区的转录因子结合位点的多样性。例如,在35个tfbs中,3个tfbs可以存在5次,而剩余的tfbs仅存在一次,得到所述基因组亚区为35个tfbs数量,具有多样性得分为23。在进一步的步骤中,优选的方法基于tfbs的最高数量和最佳多样性得分将基因组亚区排序。作为排序第一的实例,在基因组基因座chr10:6019558-6019708中,所述方法与间充质gbm状态相关联存在20个tfbs,其中一些重复2至6次。一旦确定了最佳排序的基因组亚区,就可以计算所有剩余基因组亚区中的第二最佳,其中,将存在于第一基因组亚区中的tfbs排除在排序之外。通过迭代可以计算出需要多少不同的基因组亚区来覆盖转录因子结合位点的整个集合或预定的百分比。当需要所有调控潜力(tfbsnxtfbsd)的百分比时,可以生成两个独立的lcr。通常,4-5个元件足以达到最高50%的调控潜力,并且这已被验证为足以生成应答相同的信号传导的两个独立的slcr(参见实施例)。在本发明的进一步优选的实施方式中,所述方法的特征在于,h)中的基因组亚区的配置使得包括转录起始位点的基因组亚区组装在编码报道基因的序列的相邻处和上游,并且不包括转录起始位点的基因组亚区优选地组装在从最近的转录起始位点的进一步上游。在此种情况下,尤其优选的,所述方法可以注释包含天然转录起始位点的所有基因组亚区元件(例如150bp元件),以及那些不包含的,并且将从包含转录起始位点的基因组亚区开始排序。在选择了包含转录起始位点的最佳排序的基因组亚区之后,可以独立于无论那些基因组亚区是否包含转录起始位点而进行额外的基因组亚区的排序。根据本发明,在一些实施方式中,术语“生成细胞类型特异性表达盒”涉及核酸分子的设计和物理产生。在一些实施方式中,术语“生成细胞类型特异性表达盒”涉及在无物理产生相应的核酸分子下设计细胞类型特异性表达盒,例如所述方法可以是计算机实现的方法或可以在所述方法中包括一个或多个计算机实现的步骤。在一些实施方式中,所述方法为或包括计算机实现的元件和产品,作为所述方法的输出,为所述构建体的计算机(insilico)设计、产品、模拟和/或计算机表示。因此,在一些实施方式中,盒或构建体的“生成”可以发生在计算机中,即在计算机软件中,例如,输出可以是核酸序列、核酸序列信息,即以计算机可读格式。在一些实施方式中,本发明的方法还可以涉及计算机程序产品,诸如软件产品。该软件可以配置为在通用计算设备上执行,并且配置为实施本文描述的方法的步骤a)至h)中的一个或多个。因此,本发明的计算机程序产品还涵盖并且直接涉及针对本文提供的方法所描述的特征。优选的基于计算机的方法的进一步细节在如本文所述的实例和相关参考文献中提供。如果所述方法是在计算机程序中实施的,例如借助于发明的盒的模拟或计算机设计,则所述序列可以在一些实施方式中随后通过实验室技术人员已知的方法合成并在期望的体外或体内应用中使用。本发明还涉及用于实施本文所描述的方法的系统,包括一个或多个计算设备、数据存储设备和/或软件作为系统组件,其中,所述组件优选地可以彼此紧邻连接或经由数据连接,例如通过互联网,并且被配置为与所述组件中的一个或多个交互和/或实施本文所述的方法。所述系统可以包括计算设备、数据存储设备和/或适当的软件,例如,单独的软件模块,它们彼此交互以实施本文所描述的方法。关于计算机实现:步骤a),关于提供目的细胞类型的基因表达谱,可以是计算机实现的,即,用于目的细胞类型的基因表达谱的信息优选地以计算机可读格式呈现,配置为在所述方法的进一步的步骤中处理。步骤b),关于提供所述目的细胞类型的基因组序列数据,可以是计算机实现的,即,用于基因组序列数据的信息优选地以计算机可读格式呈现,配置为在所述方法的进一步的步骤中处理。步骤c),关于从基因表达谱中选择标签基因的集合,其中所述标签基因(i)与参考细胞类型相比受到差异调控或(ii)根据基因表达水平选择,优选地是计算机实现的。在优选的实施方式中,基因及其表达谱以配置为由计算设备处理的格式的信息表示,使得可以基于此信息选择基因的特定组。根据所采用/需要的选择特征或用户的技能,此步骤可以自动化或手动执行。步骤d),关于鉴定在c)中选择的标签基因的集合中的编码转录因子的基因,优选地以计算机实现的方法实施,由此用功能注释基因,使得在任何一个或多个鉴定的标签基因中可以(任选地)自动询问转录因子功能。如由本文的实例所提及的,可以采用适当的数据库。步骤e),关于确定来自所述基因组序列数据的基因组区的集合,其中,每个基因组区包括编码在c)中鉴定的标签基因的序列和与编码所述标签基因的序列相邻的额外的基因组序列,优选地以计算机实现的方法实施。技术人员可以基于基因组序列,即可从数据库中获得的,通过使用自动选择准则,或通过手动评估和选择相邻序列,来实施与目的基因相邻的基因组序列的评估和选择。步骤f),关于鉴定在e)中确定的所述基因组区的集合内的具有相等大小的多个基因组亚区,其中,所述基因组亚区包括在d)中鉴定的一个或多个转录因子的一个或多个结合位点,优选地使用计算机实现的方法来实施。一个或多个转录因子的结合位点的鉴定可以使用本领域建立的方法来实施,例如,搜索和/或询问任何给定的序列是否存在由特定序列或序列基序定义的已知结合位点。被配置为筛选序列是否存在这样的已知序列的软件对于本领域技术人员是可获得的。步骤g),关于从f)中确定的基因组亚区中选择基因组亚区的最小集合,优选2至10个之间,其中,选择所述基因组亚区的集合以包括在d)中鉴定的所有转录因子的预定百分比的转录因子结合位点,优选地使用(任选地)自动化计算机算法来实施。以上提供了确定基因组亚区的细节。适用于选择期望的基因组亚区的软件方案有多种选项,或者该选择可以通过熟练的用户评估各种亚区并将它们编译为包括步骤d)中鉴定的相关转录因子的一定百分比的结合位点来手动实施。技术人员可以使用已建立的编程、编码和生物信息学技术来设计和/或配置软件,以评估基因组亚区中转录因子结合位点的存在,将这些结合位点与鉴定为标签基因的转录因子进行比较,并选择基因组亚区的编译以覆盖预定百分比的相关转录因子。根据所述方法的步骤h),生成了包括与报道基因或效应基因可操作地偶联的在步骤g)中选择的基因组亚区的集合的细胞类型特异性表达盒。如上所述,所述“生成”可以涉及计算机可读形式的核酸序列信息的计算机实现的产生和/或涉及基于和/或包括所述序列的物理核酸分子的合成。本发明因此进一步涉及用于设计和/或制造对应于、包括或基于从步骤a)至g)获得的产物dna序列信息的核酸分子的方法。所述方法优选地包括包括实施本文所描述的方法,和随后合成、克隆和/或分离所述核酸分子。在这样的实施方式中,术语“生成盒”可以包括用于在生成核酸分子中使用的克隆、突变、重组、pcr扩增和/或合成的任何相关分子生物学或化学技术。在优选的实施方式中,基于通过本发明的方法获得的信息,使用从头核酸合成来合成盒。在进一步优选的实施方式中,本发明涉及包括通过本文所描述的方法生成的表达盒的细胞类型特异性报道载体。在进一步的方面,本发明涉及细胞类型特异性报道载体,包括合成调控区,所述合成调控区包括2至10个100bp至1000bp的基因组亚区,所述基因组亚区位置相邻,没有接头或具有小于100bp的接头序列位于所述亚区之间,其中所述亚区源自目的细胞类型的同一基因组中的分开(非相邻)的位置,其中所述亚区累计包括至少5个、优选至少10个、最优选至少20个转录因子的结合位点,以及报道基因或效应基因,其中,所述基因组亚区与报道基因或效应基因可操作地偶联,以调控所述报道基因或效应基因的表达。尤其优选地,通过根据如本文所描述的步骤a)至g)的方法选择基因组亚区。本领域技术人员将理解,对于所述方法公开的优选实施方式同样适用于本文所描述的细胞类型特异性报道载体。本发明的方法导致载体的结构特征在该领域中是独特的。本发明的优选实施方式涉及构建体设计,其中,来自基因组亚区的转录因子结合位点具有的长度为100至1500bp或100至1250bp、优选100至1000bp、更优选120bp至300bp、更优选130bp至170bp、最优选基本上150bp,与来自相同基因组的非相邻区的基因组亚区的起源组合。通过此种组合,本发明的构建体由新的从头且无偏倚的构建定义,通过将反映出调控信息的相关大小的不同/分开的但高度相关的调控区聚集在一起,尤其是对于优选120bp至300bp、更优选130bp至170bp、最优选150bp的大小,这近似dna包裹在其上的组蛋白颗粒的大小。本发明的优选实施方式涉及构建体设计,其中,使用5个或更多个转录因子结合位点,即,通过将足够数量的tfbs聚集在一起以覆盖在任何给定细胞类型/状态中的相关tf的大的调控部分,更高数量的tfbs反应出新的从头且无偏倚的构建。基因组亚区的特征在于它们源自细胞类型的相同基因组中分开的位置,并且累计包括至少5个、优选至少10个、最优选至少20个或更多个转录因子的结合位点。在一些实施方式中,编译2-10(即2、3、4、5、6、7、8、9或10)个基因组亚区以形成包括至少5、10、15、20、25、30、35、40或更多的转录因子结合位点的slcr。因此,基因组亚区覆盖了通常足以覆盖目的细胞类型的调控信息的大量转录因子的结合位点。优选的转录因子的结合位点是指在目的细胞类型中特征性表达的转录因子。为确定在目的细胞类型中特征性表达的转录因子,例如可以采用本文所描述的方法的步骤a)至d)。使用包括2至10个具有100bp至1000bp的长度的此类基因组亚区的合成调控区,已证明是就最小化载体大小,同时维持由转录因子结合位点表示的大量调控信息而言的最优方案。在此方面,使基因组亚区位置相邻而无接头或具有小于100bp的接头序列,也确保了报道载体的紧凑设计和不包括大量的调控信息下的有效转导。在本发明的尤其优选的实施方式中,所述载体的特征在于所述基因组亚区的每个具有的长度为120bp至300bp、更优选地130bp至170bp、最优选地150bp。基因组亚区的这样的长度最优地覆盖了在背景基因组区上富集统计显著性的相关转录因子结合位点。最优大小为150bp可能是由于组蛋白在其核心颗粒周围包裹着大约146个dna基因组碱基对(bp),阻止了转录因子的接近。相反,无核小体区(nfr),其通常与活性顺式调控dna相关,当展开dna后使得转录因子可以接近,因此其最小为146pb。顺式调控dna的平均大小通常由nfr的平均大小推断——也被称为dnasei超敏感位点——其为约1000bp,并且通常在这些长度尺度上包含相关转录因子结合位点的聚类。在本发明的进一步优选的实施方式中,所述载体的特征在于与报道基因或效应基因相邻的基因组亚区包括转录起始位点。这确保了效应子和报道子处于框架中,并且可以由上游合成调控区正向调控。本文所描述的本发明的独特设计具有以下优点:根据期望的应用,可以将多种报道基因或效应基因偶联至包括基因组亚区的合成调控区。在本发明的优选实施方式中,载体的特征在于报道基因或效应基因编码选自包括以下的组的蛋白:荧光蛋白、自杀基因、荧光素酶、β-半乳糖苷酶、氯霉素乙酰转移酶、表面受体、蛋白标签包括但不限于6xhis标签、v5标签、gfp标签、自加工核酶盒、甲羟戊酸激酶及其衍生物、生物素连接酶及其衍生物包括但不限于bira、工程化过氧化物酶及其衍生物包括但不限于apex2、内切核酸酶或位点特异性重组酶及其衍生物包括但不限于限制性酶、cre、flp、tn5、spcas9、sacas9、talens、矫正单基因疾病的基因、病毒抗原诸如e1a和e1b以诱导细胞类型特异性疫苗接种、或佐剂细胞因子/趋化因子以增强免疫识别诸如gm-csf或il-12。荧光蛋白可尤其适用于对于指示报道基因的表达的信号的任何种类的光学测量。为此,所述方法可以从使用现有技术的显微和/或荧光激活的细胞分选设备和定量技术中受益。此外,本发明可以使用不同种类的载体系统而很容易采用,并且容易地适用于目的细胞。在本发明的优选的实施方式中,载体是病毒载体,优选慢病毒或腺相关病毒载体。在本发明的进一步优选的实施方式中,载体包括根据seqidno1-6的核酸序列或与seqidno1-6的任何一个具有至少80%、优选至少90%的同一性的核酸序列。如本文所描述的,本发明允许提供细胞类型特异性载体构建体,其在目的细胞类型中介导期望的报道基因或效应基因的可靠表达而无需先验知识。这样,载体构建体允许从基础研究到临床研究或治疗策略的各种不同应用。例如,载体构建体可用于鉴定细胞类型或确定细胞的固有细胞状态或发育状态。所述载体还允许研究细胞如何与外部信号或化学物质反应。此外,所述载体可用于诊断学,例如确定癌症的状态或类型,例如上皮或间充质胶质母细胞瘤是否存在,并且从而允许更有效的治疗指导。此外,所述载体本身也可以用作药物剂,例如在基因治疗方法中。在优选的实施方式中,本发明涉及载体用于在基因和病毒治疗、药物发现或验证中转化细胞和/或确定细胞特性优选细胞类型、状态或命运转变的用途。如本文所述的载体或slcr在已经转化的细胞内的存在,被本发明的实施方式所覆盖。在一种实施方式中,本发明涉及用于确定细胞特性优选细胞类型、状态或命运转变的方法,包括以下步骤a.提供如本文所描述的细胞类型特异性报道载体,b.提供细胞,c.用所述载体转导细胞,d.测量指示报道基因或效应基因的表达的信号,其中,所述信号的量对所述细胞的特性优选细胞类型、状态或命运转变有指导性。可以采用任何合适的测量技术。例如,所述报道基因或效应基因可以是荧光蛋白,在这种情况下,可以使用显微设备定量评估荧光信号,并从而定量评估所探测的细胞中报道基因或效应基因的表达。在一种实施方式中,本发明涉及用于确定固有细胞状态的方法,包括以下步骤a.提供如本文所描述的细胞类型特异性报道载体,b.提供其中固有细胞状态存在或不存在或可选地可诱导的细胞,c.用所述载体转导细胞,d.任选地诱导细胞,e.测量指示报道基因的表达的信号,其中,所述信号的量对细胞中的每一个的固有细胞状态有指导性。在一种实施方式中,本发明涉及用于确定细胞命运转变的方法,包括以下步骤:a.提供如本文所描述的细胞类型特异性报道载体,b.提供响应于外部信号传导和/或化学扰动而经历命运转变的细胞,c.用所述载体转导细胞,d.将细胞暴露于外部信号传导和/或化学扰动下,e.测量指示报道基因的表达的信号,其中,所述信号的量对细胞的命运转变有指导性。在一种实施方式中,本发明涉及用于确定细胞命运重编程因子的方法,包括以下步骤:a.提供如本文所描述的细胞类型特异性报道载体,b.提供响应于包括转录因子的重编程因子、外部信号传导和/或化学扰动而经历命运转变的细胞,c.用所述载体转导细胞,d.将细胞暴露于转录因子、外部信号传导和/或化学扰动下,e.测量指示报道基因的表达的信号,其中,所述信号的量对引入细胞的命运转变的因子有指导性。在一种实施方式中,本发明涉及用于确定预期表型的体外细胞增殖的最低要求的方法,包括以下步骤:a.提供如本文所描述的细胞类型特异性报道载体,b.提供在体内具有固有标签的细胞,c.用反映所述标签的所述载体转导细胞,d.将细胞暴露于一系列生物和化学物质下,e.测量指示预期表型的信号,其中,所述信号的量对所述表型有指导性。在一种实施方式中,本发明涉及用于疾病细胞的靶向的矫正的方法,包括以下步骤:a.提供如本文所描述的细胞类型特异性报道载体,b.提供具有固有疾病状态的细胞,所述固有疾病状态可以通过给定基因给定细胞的表达或消除来矫正c.用驱动矫正所述疾病的基因、或自杀基因、或内切核酸酶的表达的所述载体转导细胞d.将细胞暴露于矫正所述疾病的基因下、于激活自杀基因或内切核酸酶的药物下e.测量指示报道基因的表达的信号和指示疾病矫正的信号。在一种实施方式中,本发明涉及用于溶瘤病毒治疗的方法,包括含有以下步骤:a.提供如本文所描述的肿瘤细胞类型特异性报道子,b.提供编码溶瘤病毒基因组的载体,包括腺病毒、马拉巴病毒(maraba)、vsv、hsv-1、麻疹病毒(measles)、呼肠孤病毒(reovirus)、逆转录病毒和牛痘病毒,可以对所述载体进行修饰以在肿瘤slcr的表达下转基因表达肿瘤相关抗原(taa)和/或分子佐剂,c.用所述载体生成病毒颗粒,d.用所述病毒颗粒转导靶生物体以感染肿瘤细胞,e.测量肿瘤组织内且非周围组织内的病毒遗传物质。本文描述的方法,例如用于确定细胞特性优选细胞类型、状态或命运转变的那些方法,可以在各种生物学、生物技术或药物(筛选)设置中采用。本发明的进一步的实施方式涉及使用dna甲基化和/或atac-seq谱作为标签基因发现的输入。atac-seq(使用测序的转座酶可及性染色质测定)是一项用于评估全基因组染色质可及性的技术,通过用将测序衔接子(adapter)插入基因组的开放区的高活性突变体tn5转座酶探测开放染色质。突变的tn5转座酶会在称为tagmentation的过程中切割任何足够长的dna,从而通过预负载有测序衔接子的tn5转座酶进行dna的同时片段化和标记。然后将标记的dna片段纯化、通过pcr扩增并送去测序。然后可以将测序读段用于推断可及性增加的区,以及映射转录因子结合位点和核小体位置的区。几类顺式调控元件的染色质可及性是由转录因子结合的体内dna的预测标志物。染色质中所有可及位点的库(repertoire)是细胞标识(identity)的最强预测因子。事实上,在癌症中,染色质可及性是癌症类型相似性的最强预测因子,并且可用于鉴定个体癌症类型的常见二维空间内的亚型标识。为了研究由slcr描述的获得的异质性是否伴随着全基因组染色质可及性的变化,可以根据本文所描述的报道构建体的表达水平细胞分选进行atac-seq。因此,染色质可及性的差异分析可以发现许多正在经历重构的基因。在下面的实施例中描述的这些结果突显了slcr在以下中的效力:揭示例如肿瘤内异质性,并能够对肿瘤模型与主要的癌症数据一起进行深入的细胞和分子表征。本发明的进一步实施方式涉及在应激反应(例如杀死具有高er应激或炎症信号传导的细胞)和senolitic(例如杀死衰老细胞)的领域中对药物靶标的靶标发现和验证。使用本发明的方法,可以针对任何给定的细胞状态鉴定特异性调控谱,并且有效地生成报道构建体。在一些实施方式中,可以针对具有高er应激或炎症信号传导或经历衰老的细胞类型/状态生成slcr。因此,这样的报道子可用于测量是否有任何给定的药物候选,即在筛选期间应用的,导致细胞状态改变。本发明的进一步实施方式涉及在细胞标识/命运改变的领域中对药物靶标的靶标发现和验证。如本文详细描述的,可以针对任何给定的细胞标识或针对标识或命运改变之前和之后的状态,鉴定特异性调控谱,并有效地生成报道构建体。在一些实施方式中,可以在标识改变之前和之后针对细胞类型生成slcr。因此,这样的报道子可用于测量是否有任何给定的药物候选,即在筛选期间应用的,导致细胞状态改变。本发明的进一步实施方式涉及使用本文所述的方法和构建体对于合成肽的靶标发现和验证。本发明的进一步实施方式涉及使用本文所述的方法和构建体对于治疗性外体(exosome)和反义寡核苷酸的靶标发现和验证。本发明的进一步实施方式涉及在免疫治疗中药物候选的治疗潜力的发现,包括但不限于先天免疫细胞在治疗应答和抗性中的作用,以及slcr工程化治疗性适应性免疫细胞(t细胞,nk)以抵抗衰竭和主要靶标特异性的用途。在一些实施方式中,可以生成slcr作为免疫细胞活性和/或靶标特异性的读出,并且可以测试候选分子并且测量slcr读出的变化,以便评估免疫细胞(t细胞,nk)当用候选化合物增强/处理时是否可以抵抗衰竭。在进一步的实施方式中,本发明涉及用于确定合成基因座控制区(slcr)的序列的计算机实现的方法,包括如本文所描述的方法的步骤a)至g)。因此,本发明还涉及能够并且适于实施如本文所描述的方法步骤a)至g)的计算机软件产品,以及用于本文所述的方法的包括指令的计算机程序,其中当计算机执行该程序时,这些指令使得计算机实施本文所述方法的步骤a)至g)。具体实施方式本发明涉及用于生成细胞类型特异性表达盒的方法、使用这样的表达盒的细胞类型特异性载体以及这样的载体的应用。在关于实施例描述本发明之前,应当理解,本文使用的术语仅是为了描述特定实施方式的目的,并不旨在限制本发明的范围。所有引用的专利和非专利文献的文件,其全部内容通过援引并入本文。除非本文另有描述,否则所有术语均应具有其通常的技术含义。如本文所用,术语“表达盒”是指包括足以表达基因产物的核酸元件的核酸构建体。如本文所述,表达盒还涵盖表达盒的电子表示。典型地,表达盒包括编码为基因产物的核酸(序列),报道基因或功能效应子可操作地连接至包括转录结合位点的所选基因组亚区,所述转录结合位点充当基因产物的表达的调控元件。如本文所用,术语“合成顺式调控dna”、“合成调控区”或“合成基因座控制区(slcr)”是指包括以非天然存在的顺序(即在天然存在的基因组中不以这种顺序或排列存在)相邻(有或没有间隔子)排列的经验证的和/或潜在的(假定的/预测的)顺式调控序列的多个基因组亚区的排列。顺式调控序列的实例是转录因子结合位点(tfbs)、启动子、增强子、沉默子或能够顺式作用于编码区的表达的其他调控序列。当将这些调控区排列到合成调控区内时,这些调控区典型地是细胞类型的特征。本文所描述的方法优选地将这些调控区组装到包括目的细胞类型内的转录调控序列信息的相关部分的基因组亚区的集合中。如本文所用,术语“报道载体”是指包括表达盒和允许将表达盒在体外或体内引入细胞中的进一步的核酸元件的核酸构建体。术语“报道载体”、“载体”和“效应子载体”可以互换使用。“载体”可以具有一个或多个限制性内切核酸酶识别位点(i、ii或iis型),在该位点可以以可确定的方式切割序列而不会损失载体的基本生物学功能,并且在该位点中可以剪接或插入核酸片段以实现其复制和克隆。载体还可包括允许在两个核酸分子之间交换核酸序列的一个或多个重组位点。载体可以进一步提供引物位点,例如用于pcr、转录和/或翻译起始和/或调控位点、重组信号、复制子、选择标志物等。载体可以进一步包含适合用于鉴定用载体转化的细胞的一种或多种选择标志物。本领域已知的载体和可商购的那些(及其变体或衍生物)可以与本文所描述的表达盒一起使用。此类载体可以从以下获得,例如vectorlaboratoriesinc.、invitrogen、promega、novagen、neb、clontech、boehringermannheim、pharmacia、epicenter、origenestechnologiesinc.、stratagene、perkinelmer、pharmingen和researchgenetics,或者可以通过addgene在科学家之间自由分配。如本文所用,术语“病毒载体”是指包括病毒起源的至少一个元件并且具有被包装至病毒载体颗粒中的能力,编码至少一种外源核酸的核酸载体构建体。载体和/或颗粒可以用于在体外或体内将任何核酸转移到细胞中的目的。病毒载体的多种形式是本领域已知的。术语病毒粒子用于指单个感染性病毒颗粒。“病毒载体”、“病毒载体颗粒”和“病毒颗粒”还指具有其dna或rna核心和蛋白外壳(因为其存在于细胞外部)的完整病毒颗粒。术语“转染”优选是指将dna递送至真核(例如哺乳动物)细胞中。术语“转化”优选是指将dna递送至原核(例如大肠杆菌)细胞中。术语“转导”优选是指用病毒颗粒感染细胞。核酸分子可以稳定地整合至本领域通常已知的基因组中。然而,术语“转导”、“转染”和“转化”在本文中可以互换使用,并且是指将包括表达盒的载体引入细胞的过程。如本文所用,术语“细胞类型特异性”涉及当将如本文所描述的表达盒引入与其他(例如参考细胞)相比的目的细胞中时,报道基因或效应基因的表达的特异性。术语细胞类型特异性涵盖特异于目的细胞的细胞类型及其细胞状态或命运的表达(水平)。术语细胞类型特异性表达盒或载体因此涵盖细胞状态特异性以及细胞命运特异性表达盒或载体。如本文所用,术语“报道子”、“效应子”或“报道基因或效应基因”是指由包括在如本文提供的表达构建体中的核酸编码的基因产物,其可以通过本领域已知的测定或方法来检测,因此“报道”构建体的表达和/或“效应”它们在其中表达的细胞的状态或命运。报道子和效应子以及编码报道子的核酸序列是本领域众所周知的。报道子或效应子包括例如荧光蛋白,诸如绿色荧光蛋白(gfp)、蓝色荧光蛋白(bfp)、黄色荧光蛋白(yfp)、红色荧光蛋白(rfp)、增强的荧光蛋白衍生物(例如egfp、eyfp、mvenus、erfp、mcherry等)、酶(例如催化产生可检测产物的反应的酶,诸如荧光素酶、β-葡糖醛酸糖苷酶、氯霉素乙酰转移酶、氨基糖苷磷酸转移酶、氨基环醇磷酸转移酶或嘌呤霉素n-乙酰基-转移酶)和表面抗原。适当的报道子或效应子对于相关领域的技术人员将是显而易见的。优选的蛋白选自包括以下的组:荧光蛋白、自杀基因包括但不限于胸苷激酶、荧光素酶、β-半乳糖苷酶、氯霉素乙酰转移酶、表面受体、蛋白标签包括但不限于6xhis标签、v5标签、gfp标签、自加工核酶盒、甲羟戊酸激酶及其衍生物、生物素连接酶及其衍生物包括但不限于bira、工程化过氧化物酶及其衍生物包括但不限于apex2、内切核酸酶或位点特异性重组酶及其衍生物包括但不限于限制性酶、cre、flp、tn5、spcas9、sacas9、talens、矫正单基因疾病的基因、肿瘤相关的抗原或编码免疫调节剂以促进免疫治疗的基因包括但不限于magea3mgm-csf、ifnγ、ifnβ、cxcl-9-10-11。术语“基因”基本上意指当可操作地连接至合适的调控序列时,在体外或体内被转录(dna)以及被翻译(mrna)成多肽的编码核酸序列。该基因可以包括或可以不包括在编码区之前和之后的区,例如5'非翻译(5'utr)或“前导”序列和3'utr或“尾随(trailer)”序列,以及各个编码片段(外显子)之间的插入序列(内含子)。如本文所用,“基因表达”是指基因的绝对或相对表达水平和/或表达模式。可以在dna、cdna、rna、mrna、蛋白质或其组合的水平上测量基因的表达。基因表达也可以从蛋白表达中推断。“基因表达谱”是指针对目的细胞类型测量的多个不同基因的表达水平。基因表达谱可以在样本中,诸如包括各种细胞类型、不同组织、不同器官或体液(例如血液、尿、脊髓液、汗、唾液或血清)的样本中,通过以下各种方法测量:包括但不限于通过大规模平行标签测序(mpss)的rna-seq、基因表达系列分析(sage)技术、微阵列技术、微流控技术、原位杂交方法、定量和半定量rt-pcr技术或质谱技术。本文涵盖了本领域中可用于检测基因表达的任何方法。“检测表达”旨在确定rna转录物或其表达产物例如在蛋白水平上的量或存在。如本文所用,应用于基因的术语“表达水平”是指基因产物的归一化水平,例如对于基因的rna表达水平或对于基因的多肽表达水平所确定的归一化值。本文使用的术语“基因产物”或“表达产物”是指基因的rna转录产物(转录物),包括mrna,以及此类rna转录物的多肽翻译产物。基因产物可以是例如未剪接的rna、mrna、剪接变体mrna、微rna、片段化rna、多肽、翻译后修饰的多肽、剪接变体多肽等。如本文所用,术语“rna转录物”是指基因的rna转录产物,包括例如mrna、未剪接的rna、剪接变体mrna、微rna和片段化rna。用于检测本发明基因的表达的方法,即基因表达谱分析,包括基于多核苷酸的杂交分析的方法、基于多核苷酸的测序的方法、免疫组织化学法和基于蛋白质组学的方法。这些方法通常检测基因的表达产物(例如,mrna)。许多表达检测方法使用分离的rna。起始材料典型地是从生物学样本诸如分别为目的细胞类型和参考细胞类型中分离的总rna。用于rna提取的一般方法是本领域众所周知的,并且公开在分子生物学的标准教科书中,包括ausubeletal.,ed.,currentprotocolsinmolecularbiology,johnwiley&sons,newyork1987-1999。用于从石蜡包埋的组织中提取rna的方法公开于例如rupp和locker(labinvest.56:a67,1987)以及deandresetal.(biotechniques18:42-44,1995)中。特别地,可以使用来自商业生产商诸如qiagen(瓦伦西亚,加利福尼亚州)的纯化试剂盒、缓冲液组和蛋白酶,根据生产商的说明书进行rna分离。分离的rna可以用于杂交或扩增测定中,包括但不限于pcr分析和探针阵列。一种用于检测rna水平的方法涉及使分离的rna与可以杂交至由被检测的基因编码的mrna的核酸分子(探针)接触。核酸探针可以是例如全长cdna或其部分,诸如至少7、15、30、60、100、250或500个核苷酸长度并且足以在严格条件下特异性杂交至本发明的固有基因的寡核苷酸,或任何衍生的dna或rna。mrna与探针的杂交表明所讨论的固有基因正在被表达。替代地,在目的细胞类型中基因表达的水平涉及核酸扩增的过程,例如,通过rt-pcr(美国专利no.4,683,202)、连接酶链反应(barany,proc.natl.acad.sci.usa88:189-93,1991)、自我持续序列复制(guatellietal.,proc.natl.acad.sci.usa87:1874-78,1990)、转录扩增系统(kwohetal.,proc.natl.acad.sci.usa86:1173-77,1989)、q-beta复制酶(lizardietal.,bio/technology6:1197,1988)、滚环复制(美国专利no.5,854,033)或任何其他核酸扩增方法,随后是使用本领域技术人员众所周知的技术检测扩增的分子。如果核酸分子以非常低的数量存在,那么这些检测方案特别适用于这类分子的检测。特别地,可以通过定量rt-pcr评估基因表达。许多不同的pcr或qpcr方案是本领域已知的。通常,在pcr中,通过与至少一种寡核苷酸引物或寡核苷酸引物对反应来扩增靶多核苷酸序列。一种或多种引物与靶核酸的互补区杂交,并且dna聚合酶延伸一种或多种引物以扩增靶序列。在足以提供基于聚合酶的核酸扩增产物的条件下,一种大小的核酸片段在反应产物(作为扩增产物的靶多核苷酸序列)中占主导。重复扩增循环以增加单个靶多核苷酸序列的浓度。该反应可以在通常用于pcr的任何热循环仪中进行。然而,优选的是具有实时荧光测量功能的循环仪。在一些情况下,优选定量pcr(qpcr)(也称为实时pcr),因为它不仅提供定量测量,而且减少时间和污染。如本文所用,“定量pcr”(或“实时qpcr”)是指在其发生时直接监测pcr扩增的进程,而无需重复采样反应产物。在定量pcr中,可以在信号升高到高于背景水平之后但在反应达到平稳之前,随着反应产物被生成并被跟踪而经由信号传导机制(例如,荧光)监测反应产物。在pcr过程开始时,达到可检测的或“阈值”水平的荧光所需的循环数量直接随可扩增靶的浓度而变化,从而能够测量信号传导强度,以提供对样本中靶核酸量的实时测量。此外,微阵列可用于基因表达谱分析。“微阵列”意指可杂交的阵列元件诸如例如多核苷酸探针在基质上的有序排列。术语“探针”是指能够选择性结合至特定的靶生物分子的任何分子,例如,由固有基因编码或与固有基因相对应的核苷酸转录物或蛋白。探针可以由本领域技术人员合成,或衍生自适当的生物制剂。探针可以特别设计为有标记的。可用作探针的分子的实例包括但不限于rna、dna、蛋白、抗体和有机分子。dna微阵列提供了一种用于同时测量大量基因的表达水平的方法。每个阵列由附接在固体承载体上的捕获探针的可重复模式组成。标记的rna或dna与阵列上的互补探针杂交,并然后通过激光扫描检测。确定阵列上每个探针的杂交强度,并将其转换为表示相对基因表达水平的定量值。参见,例如,美国专利nos.6,040,138、5,800,992和6,020,135、6,033,860和6,344,316。高密度寡核苷酸阵列对于确定样本中大量rna的基因表达谱尤其有用。基因表达的系列分析(sage)是一种允许对大量基因转录物进行同时和定量分析的方法,而无需为每个转录物提供单独的杂交探针。首先,生成包含足够的信息以唯一地鉴定转录物的短序列标签(约10-14bp),前提是该标签是从每个转录物内的唯一位置获得的。然后,许多转录物连接在一起以形成可以被测序的长系列分子,同时揭示多个标签的鉴定。通过确定单个标签的丰度并鉴定与每个标签相对应的基因,可以定量评估任何转录物群体的表达模式。更多详细信息请参见velculescuetal.,science270:484-487(1995);以及velculescuetal.,cell88:243-51(1997)。核酸测序技术是分析基因表达的合适方法。这些方法潜在的原理是在样本中检测到cdna序列的次数与对应于该序列的mrna的相对表达直接相关。这些方法有时被称为术语数字基因表达(dge),以反映所得数据的离散数值属性。应用此原理的早期方法是基因表达的系列分析(sage)和大规模平行标签测序(mpss)。参见,例如s.brenner,etal.,naturebiotechnology18(6):630-634(2000)。“下一代”测序技术的出现已使dge变得更简单、更高通量和更实惠。结果,与以往相比,更多的实验室能够利用dge在更多目的细胞类型中筛选更多基因的表达。参见例如j.marioni,genomeresearch18(9):1509-1517(2008);r.morin,genomeresearch18(4):610621(2008);a.mortazavi,naturemethods5(7):621-628(2008):n.cloonan,naturemethods5(7):613-619(2008)。下一代测序通常允许比传统的sanger方法显著更高的通量。参见schuster,next-generationsequencingtransformstoday'sbiology,naturemethods5:16-18(2008);metzker,sequencingtechnologiesthenextgeneration.natrevgenet.2010january;11(1):31-46。这些平台可以允许对核酸片段的克隆扩张的或未扩增的单分子进行测序。某些平台涉及,例如,通过染料修饰探针的连接(包括环状连接和切割)测序、焦磷酸测序和单分子测序。核苷酸序列种类、扩增核酸种类和由此生成的可检测产物可以通过这样的序列分析平台进行分析。下一代测序可以在本发明的方法中使用,例如,以确定目的细胞类型的基因表达谱或基因组序列数据。rna测序(rna-seq)使用大规模平行测序以允许例如对基因组进行转录物组分析,其分辨率通常远高于基于sanger测序和微阵列的方法获得的分辨率。在rna-seq方法中,使用下一代测序技术对从目的rna生成的互补dna(cdna)进行直接测序。rna-seq已成功用于精确定量转录物水平,确认或修改基因的先前注释的5'和3'末端,以及映射外显子/内含子边界(eminagaetal.,2013.quantificationofmicrornaexpressionwithnext-generationsequencing.currentprotocolsinmolecularbiology.103:4.17.1-4.17.14)。如本文所用,“测序”因此是指允许鉴定核酸的至少一部分的连续核苷酸的本领域已知的任何技术。示例性测序技术包括illuminatm测序、直接测序、随机鸟枪法测序、sanger双脱氧终止测序、全基因组测序、大规模平行标签测序(mpss)、rna-seq(也称为全转录物组测序)、通过杂交的测序、焦磷酸测序、毛细管电泳、凝胶电泳、双重(duplex)测序、循环测序、单碱基延伸测序、固相测序、高通量测序、大规模平行标签测序、乳液pcr、通过可逆染料终止子测序、双末端测序、短期(near-term)测序、外切核酸酶测序、通过连接测序、短读段测序、单分子测序、通过合成测序、实时测序、反向终止子测序、纳米孔测序、454测序、solexa基因组分析仪测序、solidtm测序、illuminahiseq4000、illuminanextseq500、illuminamiseq和miniseq、ms-pet测序、质谱及其组合。基因表达谱也可以从蛋白质组的信息中推导出来。术语“蛋白质组”在本文中定义为在一定时间点存在于细胞类型中的蛋白的全体。蛋白质组学除了其他方面还包括研究样本中蛋白表达的整体变化(也称为“表达蛋白质组学”)。蛋白质组学通常包括以下步骤:(1)通过2-d凝胶电泳(2-dpage)分离样本中的单个蛋白;(2)鉴定从凝胶中回收的单个蛋白,例如my质谱或n端测序,以及(3)使用生物信息学对数据进行分析。如本文所用,术语“基因组”通常是指以一个或多个核酸序列的形式的遗传信息的完整集合,包括其文本或计算机表示。基因组可以包括dna或rna,取决于其起源的生物体。大多数生物体具有dna基因组,而一些病毒具有rna基因组。如本文所用,术语“基因组”不必包括遗传信息的完整集合。该术语还可以指基因组的至少大部分,诸如整个基因组的至少50%至100%或它们之间的任何整数或分数百分比。术语“基因组序列数据”是指基因组上的数据,包括其文本或计算机表示,其中基因组序列数据还可以涉及优选大部分基因组诸如整个基因组的至少50%至100%或其中间的任何整数或分数百分比的基因组。提供基因组序列数据可以包括对目的细胞类型的基因组的实际测序,或者依赖于基因组序列数据的公共可用的数据库,诸如由国家基因组资源中心(nationalcenterforgenomeresources,ncgr)操作的注释的基因组序列数据库(gsdb)。提供大量物种的基因组序列数据可通过由ucsantacruz(ca,usa)的ucsc基因组浏览器组(genomebrowsergroup)创建的ucsc基因组浏览器公开获得。如本文所用,术语“基因组区”通常是指基因组的区。典型地,基因组区是指包括至少一个基因的目的细胞类型的基因组的连续核酸序列延伸。术语“基因组亚区”是指基因组区的一部分,其如本文所描述被鉴定为包括已经基于一个或多个基因表达谱被鉴定为标签基因的一个或多个转录因子的一个或多个结合位点。术语“核酸”是指任何核酸分子,包括但不限于单链或双链形式的dna、rna及其杂交体或修饰的变体和聚合物(“多核苷酸”)。除非特别限制,否则该术语涵盖包含天然核苷酸的已知类似物的核酸,其具有与参考核酸相似的结合特性并且以与天然存在的核苷酸相似的方式代谢。除非另有说明,否则特定的核酸分子/多核苷酸还隐含地涵盖其保守修饰的变体(例如简并密码子替换)和互补序列以及明确指出的序列。具体而言,简并密码子替换可以通过生成在其中将一个或多个所选(或全部)密码子的第三位置用混合碱基和/或脱氧肌苷残基替换的序列来实现(batzeretal.,nucleicacidres.19:5081(1991);ohtsukaetal.,j.biol.chem.260:2605-2608(1985);rossolinietal.,mol.cell.probes8:91-98(1994))。核苷酸通过以下标准缩写由其碱基表示:腺嘌呤(a)、胞嘧啶(c)、胸腺嘧啶(t)和鸟嘌呤(g)。“外源核酸”或“外源遗传元件”涉及引入细胞的任何核酸,其不是细胞“原始”或“天然”基因组的成分。外源核酸可以是整合的或非整合的,或涉及稳定转染的核酸。“功能变体”或“功能类似物”优选是指分别具有与参考序列“相同”、“基本上相同”、“大体上相同”、“同源”或“相似”的核苷酸序列或氨基酸序列的核酸或蛋白,通过非限制性实例,该参考序列可以是分离的核酸或蛋白的序列,或者是通过比较两种或更多种相关的核酸或蛋白而获得的共有序列,或者是给定核酸或蛋白的同种型的组。同种型类型的非限制性实例包括由例如交替的rna剪接或蛋白水解切割产生的不同分子量的同种型;并且同种型具有不同的翻译后修饰,诸如糖基化;等等。如本文所用,术语“变体”或“类似物”是指不同于参考核酸或多肽但保留其基本特性的核酸或多肽。通常,变体总体上非常相似,并且在许多区中与参考核酸或多肽相同。因此,转录因子的“变体”形式总体上非常相似,并且能够结合dna并激活基因转录。如本文所用,术语“有义链”是指被翻译或可翻译成蛋白的基因的dna链。当基因相对于核酸序列中的启动子沿“有义方向”定向时,“有义链”位于启动子下游的5’末端,蛋白的第一个密码子位于启动子的近端,并且最后一个密码子位于启动子的远端。相反的被称为“反义”链。如本文所用,术语“可操作地连接”是指在核酸构建体中的调控元件被配置为使得能够在调控元件和基因之间功能性偶联,从而导致基因的表达,即,调控元件优选地与编码蛋白或肽的核酸在框内。如本文所用,术语“包括(comprising)”或“包括(comprises)”用于指表达盒、报道载体及其各自的一个或多个组分,其开放式包括未指定的元素。术语“由……组成”是指本文所描述的表达盒、报道载体及其各自的一个或多个组分,其排除了在该实施方式的描述中未列举的任何元素。术语“标签基因”涉及选自目的基因细胞类型的基因的基因,其特征在于所述目的细胞类型的表达谱。差异调控的标签基因可以例如通过鉴定与参考细胞类型中的表达水平相比被上调或下调的基因来选择,或通过对目的细胞类型的基因表达水平进行排序并且基于基因的阈值水平或预定数量(例如,最高或最低表达的)来选择标签基因。如本文所用,术语“转录因子”是指结合至特定dna序列并且由此控制遗传信息从dna至mrna的转移(或转录)的蛋白。转录因子的功能主要是调控基因的表达。转录因子可以通过促进(作为激活因子)或阻止(作为阻遏因子)rna聚合酶募集至特定基因而单独或与其他蛋白在复合物中组合来发挥作用。转录因子包含至少dna结合结构域,该结构域附接在典型地与它们调控的基因相邻的dna的特定序列(“结合位点”)上。术语“显微设备”涉及包括用于细胞的显微分析的工具的设备。显微分析可以通过但不限于以下进行:光学显微镜、双目立体显微镜、明场显微镜、偏光显微镜、相差显微镜、微分干涉相差显微镜、自动显微镜、荧光显微镜、共聚焦显微镜、全内反射荧光显微镜、激光显微镜(激光扫描共聚焦显微镜)、多光子激发显微镜、结构照明显微镜、透射电子显微镜(tem)、扫描电子显微镜(sem)、原子力显微镜(afm)、扫描近场光学显微镜(snom)、x-射线显微镜、超声显微镜。显微设备可以额外地包括用于记录细胞图片的照相机和/或检测器,例如以及用于控制显微设备的计算机系统。由报道基因产生的信号的存在和/或强度可以借助于显微设备确定,但也可以通过可以检测由报道基因生成的信号的其他设备来确定,诸如但不限于流式细胞仪、光度计(luminometer)、光谱仪、测光仪(photometer)或色度计。如本文所用,术语“拓扑相关结构域”优选是指自相互作用的基因组区,意指拓扑相关结构域内的dna序列在物理上彼此相互作用比拓扑相关结构域之外的序列更为频繁,从而形成三维染色体结构。拓扑相关结构域的大小范围可以从数千个到数百万个dna碱基。已知许多蛋白与拓扑相关结构域形成有关,包括蛋白ctcf和蛋白复合体黏连蛋白。在优选的实施方式中,拓扑相关结构域是指两个ctfc或黏连蛋白结合位点之间的基因组序列。如本文所用,术语“生成细胞类型特异性表达盒”在一些实施方式中涉及在无物理产生相应的核酸分子下设计细胞类型特异性表达盒,例如所述方法可以是计算机实现的方法或可以在所述方法中包括一个或多个计算机实现的步骤。如本文所用,术语“生成细胞类型特异性表达盒”在一些实施方式中涉及核酸分子的设计和物理产生,优选通过核酸分子的从头合成。人工基因合成(或从头合成)是生成本发明的盒的优选方法,并且涉及在合成生物学中使用以创建任何给定的核酸序列的方法。在一些基于固相dna合成的情况下,人工合成与分子克隆和聚合酶链反应(pcr)有所不同,因为用户不必从预先存在的dna序列开始。因此,有可能制造完全合成的双链dna分子,而对核苷酸序列或大小没有大的限制。基因合成方法可以基于有机化学和分子生物学技术的组合,并且整个基因可以“从头”合成,而不需要前体模板dna。该方法已用于生成包含大约一百万个碱基对的功能性细菌染色体。基因合成在重组dna技术的许多领域中已成为重要工具,包括异源基因表达、疫苗开发、基因治疗、载体构建和各种形式的分子工程。核酸序列的合成通常比典型的克隆和诱变程序更经济。多种技术已被良好建立并且是技术人员已知的。术语“基因疗法”优选是指将dna转移至受试者中以治疗疾病。本领域技术人员知道使用基因治疗载体进行基因治疗的策略。优化这样的基因治疗载体以将外源dna递送至受试者的宿主细胞中。在优选的实施方式中,基因治疗载体可以是病毒载体。病毒具有天然发展的策略以将dna整合到宿主细胞的基因组中,并且可以因此被有利地使用。优选的病毒基因治疗载体可包括但不限于逆转录病毒载体诸如莫洛尼(moloney)鼠白血病病毒(mmlv)、腺病毒载体、慢病毒、腺病毒相关的病毒(aav)载体、痘病毒载体、单纯疱疹病毒载体或人免疫缺陷病毒载体(hiv-1)。然而,非病毒载体也可以优选用于基因治疗,诸如由真核启动子驱动的质粒dna表达载体或者包含与宿主基因组的同源性的质粒dna序列,以便将表达盒直接整合到目的基因组中的优选位置。dna转移也可以使用脂质体或相似的细胞外囊泡进行。此外,优选的基因治疗载体还可以指转移dna的方法,诸如电穿孔或将核酸直接注射至受试者中。本领域技术人员知道如何根据应用的需要选择优选的基因治疗载体以及如何实现核酸构建体诸如本文所描述的表达盒至基因治疗载体中的方法。(p.sethetal.,2005,n.koostraet,al.2009.,w.waltheretal.2000,waehleretal.2007)。本发明的方法、系统或其他计算机实现的方面可以在一些实施方式中包括和/或采用具有以下的一种或多种常规计算设备:处理器,输入设备诸如键盘或鼠标、存储器诸如硬盘驱动器和易失性或非易失性存储器,以及用于运行本发明的计算机代码(软件)。该系统可以包括一个或多个预装有所需的计算机代码或软件的常规计算设备,或者它可以包括定制设计的软件和/或硬件。该系统可以包括进行本发明的步骤的多个计算设备。在某些实施方式中,多个客户端诸如台式机、笔记本或平板计算机可以连接至服务器,使得例如多个用户可以在本方法的不同步骤处提供数据或进行计算。计算机系统还可以通过局域网(lan)连接或经由因特网连接与其他计算机或必要的数据库诸如基因组数据库联网。该系统还可以包括保留通过本发明获得的数据的副本的备份系统。可以经由用于数据传输的任何合适的方式来执行或配置本方法的各种步骤之间必需的数据连接,诸如以有线或无线通过局域网(lan)连接或经由因特网连接。客户端或用户计算机可以具有其自己的处理器、输入工具诸如键盘、鼠标或触摸屏以及存储器,或者它可以是不具有其自己的独立处理能力而是依赖于另一计算机诸如与其连接或联网的服务器的计算资源的终端。取决于本发明的特定实施方式,如果出现这样的需求,则客户端系统可以包含必要的计算机代码以承担对系统的控制。在一种实施方式中,客户端系统是平板或笔记本电脑。尽管可以为每个特定实现定制配置系统,但是用于执行本方法的计算机系统的组件可以是常规的。计算机实现的方法步骤或系统可以在任何特定架构,例如个人/微型计算机、小型计算机或大型机系统上运行。示例性的操作系统包括applemacosx和ios、microsoftwindows和unix/linux;sparc、power和基于itanium的系统;以及z/architecture。可以用任何编程语言或基于模型的开发环境编写计算机代码以执行本发明,诸如但不限于c/c++、c#、objective-c、java、basic/visualbasic、matlab、r、simulink、stateflow、labview或汇编程序。计算机代码可以包括用专属于结合本发明使用的电路板、控制器或其他计算机硬件组件的制造商的专用计算机语言编写的子程序。通过本方法处理和/或产生的信息,即作为核酸序列、基因表达谱、基因列表和/或特定序列元件诸如tf结合位点的数字表示可以采用行业中使用的任何种类的文件格式。例如,数字表示可以以专用格式、dxf格式、xml格式或本发明使用的其他格式存储。可以利用任何合适的计算机可读介质。计算机可用或计算机可读介质可以是例如但不限于电子、磁、光学、电磁、红外或半导体系统、装置、设备或传播介质。计算机可读介质的更具体实例(非详尽列表)将包括以下:具有一根或多根电线的电气连接、便携式计算机磁盘、硬盘、随机存取存储器(ram)、只读存储器(rom)、可擦可编程只读存储器(eprom或闪存)、光纤、便携式小型光盘只读存储器(cd-rom)、光学存储设备、传输介质诸如支持因特网或内部网的那些、云存储或磁存储设备。在表1中列出了细胞类型特异性报道载体(即合成基因座区)的基因组亚区的最小集合的优选实施方式的核苷酸序列。表1:细胞类型特异性报道子的优选合成基因座区的核苷酸序列:在一种实施方式中,本发明因此涵盖包括选自由以下组成的组的核酸分子的载体:a)包括根据seqidno1-6的核苷酸序列或由根据seqidno1-6的核苷酸序列组成的核酸分子b)与根据a)的核苷酸序列互补的核酸分子;c)包括具有足够的序列同一性以与根据a)或b)的核苷酸序列在功能上类似/等同的核苷酸序列的核酸分子,包括优选与根据a)或b)的核苷酸序列的序列同一性为至少70%、80%,优选90%,更优选95%。d)根据a)至c)的核苷酸序列的核酸分子,其通过缺失、添加、替换、易位、倒位和/或插入修饰,并且与根据a)至c)的核苷酸序列在功能上类似/等价。功能上类似的序列优选地是指合成调控区在目的细胞类型中促进可操作偶联的报道基因或效应基因的转录的能力。在一种实施方式中,本发明涵盖包括选自由以下组成的组的核酸分子的溶瘤病毒治疗的载体:a)包括根据seqidno1-6的核苷酸序列或由根据seqidno1-6的核苷酸序列组成的核酸分子b)与根据a)的核苷酸序列互补的核酸分子;c)包括具有足够的序列同一性以与根据a)或b)的核苷酸序列在功能上类似/等同的核苷酸序列的核酸分子,包括优选与根据a)或b)的核苷酸序列的序列同一性为至少70%、80%,优选90%,更优选95%。d)根据a)至c)的核苷酸序列的核酸分子,其通过缺失、添加、替换、易位、倒位和/或插入修饰,并且与根据a)至c)的核苷酸序列在功能上类似/等价。e)根据本方法生成的核酸分子。功能上类似的序列优选地是指合成调控区在目的疾病靶细胞中并且不在非疾病细胞中促进病毒必需基因和/或效应基因诸如共刺激分子(例如细胞因子/趋化因子)的转录的能力。附图本发明由以下附图进一步描述。这些并非旨在限制本发明的范围,而是表示本发明方面的优选实施方式,以便提供对本文所述的本发明的更好说明。附图的简要说明图1:合成基因座控制区(slcr)的生成和验证图2:由slcr揭示的mes-和pn-gic中的固有和适应性应答。图3:使用slcr的gbm亚型化和重编程。图4:由slcr揭示的不依赖组织的上皮-间充质内稳态。图5:由体内slcr揭示的异质性间充质转分化。图6:mesgbm-亚型亚型特异性基因的选择。图7:自动合成基因座控制区(slcr)生成。图8:由slcr揭示的mes-和pn-gic中的固有和适应性应答。图9:结合至mgt#1顺式调控dna的转录因子。图10:乳腺癌细胞中mgt#1表达的内稳态维持。图11:mgt#1反映了tgfb和gsk126对emt的单个和组合贡献。图12:mgt#1能够筛选由外部信号传导和/或化学扰动驱动的细胞命运转变。图13:由扩张的slcr揭示的mes-和pn-gic中的固有和适应性应答。图14:由体内扩张的slcr揭示的异质性间充质转分化。图15:slcr有助于发现肿瘤和免疫细胞之间的非细胞自主性串扰(crosstalk)的治疗意义。图16:合成基因座控制区(slcr)的延伸表征。图17:由slcr揭示的适应性应答的进一步实例。图18:由gic中的slcr测量的mes-gbm状态诱导是特异性的且可逆的。图19:mes-slcr以剖析mes-gbm中电离辐射和nfkb信号传导的作用。图20:支持在表型crispr/cas9正向遗传筛选中使用slcr的进一步证据。图21:支持hmg细胞诱导hgic中mgt#1表达以及对治疗剂和hmg细胞的差异敏感性的进一步证据。图22:支持在表型crispri筛选中使用slcr的进一步证据。附图的详细说明图1:合成基因座控制区(slcr)的生成和验证。a)从差异调控的基因(drg)开始的slcr生成的示意图。b)在基因组gbm亚型特异性基因座上检测到具有显著性的tfbs基序的两两相关热图。每个图上方指定了分析中tfbs和drg的数量。c)slcr以及生成报道的胶质瘤起始细胞(gic)的实验步骤的示意图。d)左;mgt#1转染的293t或(右)-慢病毒转导的经冰冻切片的mes-hgic神经球的共聚焦成像。尺度=10μme)分选h2b-cfp的用slcr和facs修饰的mes-hgic和pn-hgic的代表性mvenusfacs谱。mes-hgic表达更高水平的mgt#1(箭头)f)在指定的gic中对肿瘤坏死因子α(tnfa)治疗的应答的代表性量化。mes-hgic表达更高水平的mgt#1(箭头)。mes=间充质;pn=原神经;cl=经典。mgt#1-2=mes遗传追踪#1-2。tmd=pdrgfra跨膜结构域。g)双重if和smrna-fish。示出了合并的通道(左)和单独的通道(右)的图像。黄色的重叠信号和箭头表示med1和mgt#1驱动的mvenus之间的共定位。h)分选h2b-cfp的用指定的slcr和facs转导的mes-hgic和pn-hgic的mvenusfacs谱。选通(gating)和箭头示出mes-hgic比pn-hgic表达更高水平的mgt#1。图2:由slcr揭示的mes-和pn-gic中的固有和适应性应答。a)tfna是促成间充质gbm表型的主要信号传导。左,使用指定的细胞因子最长48小时通过适应性应答筛选,tnfa鉴定为最高调控子作为mes-hgic中两个独立设计的mes-gbm报道子(mgt#1-mgt#2)的激活因子。相对于对照归一化数据。与pn-hgic相比,mes-hgic表达更高基础水平的mgt#1。b)在mgt#1诱导中il-6和小胶质细胞之间的协同。在指定治疗后,mes-hgic中mgt#1表达的活细胞成像。c)指定条件和抗体的免疫印迹。d-e)差异mgt#1激活告知了对tnfa的差异适应性应答。通过rna-seq和分层样本聚类测量的mes-hgic或pn-hgic中的通过tnfa调控的基因的表达变化。f)在指定的gic中,响应于肿瘤坏死因子α(tnfa)处理的指定基因的rt-qpcr验证。n=3个生物学独立的样本,anova测试;****p<0.0001;g)由mgt#1表达揭示的tnfa和治疗诱导的间充质定型(commitment)之间的协同性。在指定的刺激后间充质转分化的facs定量。h)指定条件和抗体的免疫印迹。mes=间充质;pn=原神经;cl=经典。mgt#1-2=mes遗传追踪#1-2。fbs=胎牛血清,cbd=大麻二醇。irr=电离辐射。图3:使用slcr的gbm亚型化和重编程。a)在使用细胞重编程或外部信号传导下使用gbm亚型特异性slcr确定固有gbm亚型以及增强亚型鉴定的示意图。b)增强常规胶质瘤细胞系中的原神经鉴定。将t98细胞用原神经slcr或间充质slcr驱动mcherry作为报道子转导,并且用pn亚型鉴定50的指定的主调控子转染或空转染。t98细胞的代表性显微照片(左)和facs图(右)示出了t98细胞中pngt#2(但不是mgt#2)报道子的更高的高固有和tf诱导的表达,尺度=100μm。图4:由slcr揭示的不依赖组织的上皮-间充质内稳态。a)mgt#1揭示了乳腺癌细胞中的固有细胞命运差异。左,转导至上皮(上)和间充质(下)乳腺癌细胞中的mes-gbm报道子mgt#1的代表性表达。facs图示出了mda-231中报告子比mcf7细胞中的报告子具有更高的高固有表达。注意,报道子表达独立于间充质诱导剂10pmtgfβ2。尺度=100μm。b)mgt#1揭示了对肺癌细胞中化学物质/形态发生的适应性应答。左,在96孔中接种的a549细胞中代表性mgt#1表达,并在指定的时间内增殖。300,000个细胞/板在rpmi培养基中增殖。在0和48小时时补充10pmtgfβ1+2和5μmgsk126。测量荧光,以及右,通过incucyte成像系统拍摄的代表性显微照片(右)。误差棒代表独立孔的标准偏差(n=3)。c)crispri和mgt#1揭示了肺癌emt的机械调控子。描绘筛选的示意图。dox,多西环素。d)crisrpi筛选的代表性中间时间点的免疫印迹。在裂解之前拍摄mgt#1-荧光显微照片。e)用于纯化mgt#1高和低群体的facs分选选通策略。f)示出了mgt#1高mgt#1crispri筛选中grna的相对富集的ma图。注意,两个脱落(dropout)的grna鉴定了emt的已知和新型调控子。g)使用两个独立的grna进行crispr介导的arid1a和cnksr2的敲除,并且随后是对mgt#1表达的facs验证。h)在野生型以及arid1a和cnksr2ko细胞中的emt标志物的免疫印迹。图5:由体内slcr揭示的异质性间充质转分化。a)mes-hgic的代表性冠状前脑图像;nsg小鼠(n=10)中在人道终点的mgt#1-mvenus微弱异种移植物。左,he染色;右渐进性插图,显示gfp、微管蛋白和dapi复染色组织的放大。注意,侵袭性胶质瘤前端是同质地mgt#1-mvenus高。b)代表性的混合mgt#1-mvenus高/mgt#1-mvenus阴性病变。c-d)分别在mgt#1阳性和阴性病变中的代表性h2b-cfp表达(箭头)。e)代表性的流式细胞术图,显示了mes-hgic中的cd133和mgt#1-mvenus表达;在nsg小鼠或体外的mgt#1-mvenus微弱异种移植物(左)。各个组件示出在右侧。注意proprole从体外转移至体内。f)a-e中显示的数据的示意图。图6:mesgbm-亚型亚型特异性基因的选择。a)热图,表示从tcga排序的微阵列的显著性分析(sam)列表中选择的基因的倍数变化,用于所指定的两两比较。下方,颜色代码指示了与元数据相关的gbm亚型表达谱。b)热图,表示所选基因的表达水平,说明了它们在原发活检和衍生自它们的胶质瘤干样细胞(gsc)中的表达和倍数变化。下方,颜色代码指示了与元数据相关的gbm亚型表达谱。所有基因具有绝对cpm>4,并且大多数基因在gsc内示出倍数变化,表明它们的表达也以细胞自主方式起作用。将spearman排序相关性用于样本,并且将pearson相关性用于基因。图7:自动合成基因座控制区(slcr)的生成。a)左上(i),与特定基因标签相关的顺式调控元件(cre)的鉴定的示意图;右上(ii),cre对基因组位置的注释;下方(iii),基于tfbs多样性和得分[σ-log10(p值)+numtfbs)]的150bpcre的迭代选择。slcr生成涉及从最接近天然tss至最远的远端cre的n个cre的组装,最高达>50%的tfbs多样性(本实施例中为mes-gbm)。b)基于tfbs得分/多样性的单个slcr的spearman相关性。(a)注释由自动算法生成的slcr。图8:由slcr揭示的mes-和pn-gic中的固有和适应性应答。来自图2a的gic中mgt#1表达的代表性活细胞成像。图9:结合至mgt#1顺式调控dna的转录因子。a)上方,mgt#1slcr的示意图。下方,可以在使用的任何细胞系中在encode公共数据库中观察到chip-seq信号的tf的列表。图10:乳腺癌细胞中mgt#1表达的内稳态维持。a)被测试的两种hyphotes的示意图:mgt#1静态反映细胞状态或mgt#1动态反映细胞内稳态,并且在扰动后重新建立体外内稳态调控(即mgt#1微弱群体的facs纯化)。绿色虚线圆圈突显了图4a中的结果,其中mcf7和mda-231由于其细胞标识而分别示出具有固有的低或高mgt#1表达。b)基于最佳可比较的mgt#1强度对mcf7和mda231进行facs分选,并且在4a中示出的facs分析之前在体外增殖。图11:mgt#1反映了tgfb和gsk126对emt的单一和组合贡献。a)在暴露于指定处理下5天的a549细胞中mgt#1表达的facs谱。每个样本需要最少10,000个细胞。b)在暴露于指定处理下5天的a549细胞中细胞形态和mgt#1表达的facs谱。注意细胞形状中的tgfb依赖性变化以及在tgfb1+2和gsk126之间的协同性。图12:mgt#1能够筛选由外部信号传导和/或化学扰动驱动的细胞命运转变。示出的是从筛选获得的数据的主成分分析(pca)。pc1和pc2这两种成分说明了实验中最大的变化。为了生成数据,在初始上皮a549-mgt#1和gsk126处理的细胞的程序结束时,增殖a549-mgt#1细胞并且拍摄细胞图像。注意间充质转变与先前发表的数据一致。对来自增殖的a549-mgt#1细胞的归一化荧光数据进行分层聚类,并且使用sparm20mtecan读板器扫描底部读数的荧光。使用pearson相关性聚类。颜色代码指示荧光强度倍数变化(蓝色-白色-红色)和生物学复制品(黄色/橙色=媒介物,绿色=gsk126)。进行了活细胞成像,示出在gsk126处理的和对照a549-mgt#1细胞中对lps的响应。图13:由slcr揭示的mes-和pn-gic中的固有和适应性应答。a)使用slcr的表型筛选的示意图(上)和结果的气泡图可视化(下)。对于每个gic和slcr,气泡大小示出每种处理相对于对照的变化的幅度(log2倍变化),气泡颜色指示变化的迹象(红色或橙色表示富集,浅蓝色表示缺失)。b)表型筛选的facs验证。cd133和pngt#2的表面表达是细胞标识的内源性标志物。注意到相比于pn-hgic更高的mes-hgicmgt#1表达。c)在指定的刺激下间充质转分化的代表性facs定量。d)mgt#1激活的功能剥离的实验设计。e)来自d中筛选的与药物相关的sgrna的火山图(红色,正调控子;蓝色,负调控子;灰色,不显著)。相对于所有mgt#1低部分和未分选的对照(n=6),计算了所有mgt#1高部分(n=3,初始、tmz+ir、tnfa+fbs的平均值)的倍数变化。padj由deseq2计算(参见方法)。突显了选定的sgrna-化合物对。f)在用指定的处理和tnfα顺序处理下指定基因的rt-qpcr。将padj指定用于代表性比较,并且表示整体双向anova和dunnett的多次比较的结果。mes=间充质;pn=原神经;mgt#1-2=mes遗传追踪#1-2。fbs=胎牛血清,tnfa=肿瘤坏死因子-α。ir=电离辐射。tmz=替莫唑胺。图14:由体内slcr揭示的异质性间充质转分化。a)指定条件的atac-seq谱的散点图,在5e)中由黄色和蓝色框表示。突显了在tnf受体超家族(tnfrs)基因座的开放染色质。b)fadd/tnfrs6基因座的ucsc基因组浏览器视图。体外和体内mgt#1高细胞之间的可及性的变化由箭头和颜色(红色-高,灰色-中)表示。c)泛癌(pancancer)数据集和指定条件的atac-seq谱分析的无监督t-sne。当可以获得时,每个点表示给定的样本或所有技术复制品的合并。该分析包括所有样本中250,000个变化最大的峰的最高主成分。灰点是所有tcga癌症类型,但gbm/lgg与来自(parketal.,2017,cellstemcell21,209–224august3,2017)的胶质瘤干细胞以及来自该研究的gic一起着色。圆圈表示由主要gbm/lgg和gic/gsc占据的尺寸。d)atac-seq谱分析的无监督t-sne限于胶质瘤尺寸内的样本。图15:slcr有助于发现肿瘤和免疫细胞之间的非细胞自主性串扰的治疗意义。a)代表性的mes-hgic的明场视野和if,具有指定的报道子以球状体或具有永生化的人小胶质细胞(hmg)的类器官增殖(分别为上图和下图)。比例尺=50um。b)无接触hgic-hmg共培养的示意图。左,共培养中的hgic和mg的明场图像。c)单独的或用tnfa或hmg共培养刺激的mes-hgic-mgt#1高的代表性facs谱和选通策略。下方是针对指定条件的通过drg的独创通路分析(ingenuitypathwayanalysis)的nfkb相关基因的维恩图(venndiagram)。与对照gic相比,drg被富集(fc>1,padj<0.05)。d)与患者的那些重叠的hmg驱动的mesgbm标签的维恩图。注意与其他相比,与nefteletal.的重叠更高。e)指定条件的drg的热图。将rna-seq读段归一化为每百万的转录物,log2转换并且z得分。通过使用r-包limma评估统计学显著性(对照,n=3,hmg,n=3;tnfαn=2;padj<0.05)。f)所指定的比较的ma图。突显并用颜色编码显著的drg。g)与mes-hgics-mgt#1高中的tnfa相比,通过hmg共培养上调控的基因的独创性上游调控子分析。h)左,slcr高和低状态的化学敏感性谱分析测定的示意图。右,针对响应于指定药物的增加的浓度的facs分选的mes-hgics-mgt#1高和-mgt#1低部分生存力计算的logic50值。图16:合成基因座控制区(slcr)的延伸表征。mgt#1-和pgk-驱动的基因表达的单分子rnafish定量。箭头/黄色表示胞质共定位。图17:由slcr揭示的适应性应答的进一步实例。指定刺激下的代表性mgt#1激活。图18:由gic中的slcr测量的mes-gbm状态诱导是特异性的且可逆的。a-b)条形图,示出了诱导48小时后对指定的因子/slcrr的个体响应。c-d)线形图,示出了指定的因子/slcr的纵向表达。图19:mes-slcr以剖析mes-gbm中电离辐射和nfkb信号传导的作用。a)右,ir和mgt#1激活之间的剂量响应。左侧示出了实验设置的实例。b)在指定的刺激下间充质转分化的代表性facs定量。图20:支持在表型crispr/cas9正向遗传筛选中使用slcr的进一步证据。a)从全基因组crispr筛选中,在对mgt#1高和mgt#1低进行分选用于grna扩增之前指定条件的facs图。b)箱形图,示出了数据质量评估,通过比较在未分选的筛选条件下的高度信息化必需和所有非必需或非靶向grna的分布(p值=学生的t检验)。c)在brunello文库和未分选的mes-hgics+brunello条件之间的对于指定的grna集合的sgrna倍数变化值分布(参见“方法”)。d)sgrna丰度(x轴)和倍数变化(y轴)的代表性ma图。将携带brunello文库的初始mes-hgic在mgt#1高和mgt#1低中facs分选,并且将grna相对于最大数据集归一化并且log2转换(参见方法)。与mgt#1高部分相比,所指示的grna缺失。e)来自crispr/cas9ko筛选的所有命中的独创通路分析(ipa)前25种毒性类别(fc±1.5;padj<0.05)。只有“正调控子”超出了统计界限。粗体为与视黄酸受体信号传导相关的类别。来自crispr/cas9ko筛选的所有命中的ipa上游调控子分析(fc±1.5;padj<0.05)。mes-gbm表型的正调控子和负调控子分别以水蓝色和红色着色。灰色表示没有方向性富集的显著类别。f)来自e中筛选的最高调控的sgrna的火山图。相对于所有mgt#1低部分和未分选的对照(n=6)计算所有mgt#1高部分(n=3,初始,平均的tmz+ir,tnfa+fbs)的倍数变化。通过deseq2计算padj,并且突显选定的sgrna-fda批准的化合物对(参见“方法”)。图21:支持hmg细胞诱导hgic中mgt#1表达以及对治疗剂和hmg细胞的差异敏感性的进一步证据。a)图4中共培养实验的延伸的示意图;对于详细的培养基组成,参见“方法”。b)单独或与人小胶质细胞(hmg)或人cd34+衍生的髓系来源的抑制细胞(mdsc)共培养的mes-或pn-hgics-mgt#1高的facs谱。c)所指示的rna-seq谱的主成分分析。基于从gosselinetal2017获得的选定的人mg标志物的平均表达水平来计算距离。d)响应于指定药物浓度增加的facs分选的mes-hgics-mgt#1高和-mgt#1低部分生存力。e)指定基因列表的散点图和基因集合富集分析(gsea),示出hmg细胞诱导mes-gbm并且抑制dna损伤转录标签基因。图22:支持在表型crispri筛选中使用slcr的进一步证据。a)激酶组筛选中所有样本的累计曲线分布(n=42),包括技术复制品和生物学条件:质粒文库、a549-h1944输入、a549-h1944+gsk126高、中、低-对照-a549-h1944+gsk126+dox高、中、低和a549-h1944+dox高、中、低–分别用于gsk126驱动的emt和内稳态emt的筛选。将所有grna(n=6615)通过每百万读段的总计数归一化,通过百分位归一化(75百分位)对数转换,并通过转化为z得分进行转换。b-c)图3c-f中的筛选中所有grna(n=6615)的散点图以及分别对于非必需sgrna(n=483)和必需基因(n=352)的gsea。通过t检验以及柯尔莫哥罗夫-斯米尔诺夫检验(kolmogorov-smirnov),fc<-1和padj<0.001,必需基因的缺失是显著的。d-e)合并的a549+h1944+gsk126+dox筛选中所有grna(n=6615)的散点图以及分别对于非必需sgrna(n=483)和必需基因(n=352)的gsea。通过t检验以及柯尔莫哥罗夫-斯米尔诺夫检验,fc<-0.5和padj<0.001,必需基因的缺失是显著的。实施例本发明由以下实施例进一步描述。这些并非旨在限制本发明的范围,而是表示为更好地说明本文所描述的发明而提供的优选实施方式。实施例示出,本文所描述的方法和报道载体允许报道基因和效应基因在各种目的细胞类型中的细胞类型特异性表达。实施例中使用的材料和方法:slcr生成和tfbs发现:使用具有--输出-门限值(pthresh)1e-4--否-q值(qvalue)的fimo(pmid:21330290)来鉴定定义的基因组区(drg基因座;表x)中的高亲和性tf结合位点。从文献(portales-casamaretal.,2010;badisetal.,2009;bergeretal.,2008;bucher,1990;jolmaetal.,2010)中,生成了代表已知转录因子结合偏好(位置权重矩阵,pwm)的1,818个模型的数据库。pwm是基于亚型特异性tf预选择的。从ucsc基因组浏览器(hg19;2012年10月5日下载的refseq表)检索与drg相对应的区,并且以150bp的窗口和50bp步长(以下称为顺式单位)扫描。每个标签基因周围的扫描区域由距tss或tes>10kb的两个远端ctcf位点界定。使用fimo将亚型特异性pwm映射到基因组区。pwm最好显著过表达的区(adj.p值<0.01;多个背景)。对于每个窗口,只要鉴定出同一pwm的多个匹配,就将最佳匹配的p值视为该tf在该区上的亲和性的代表。给定一个区,基于所考虑的每个pwm的最佳-log10(p-值)之和来计算总体得分。通过比较基序/背景(经验p值<0.01)确定显著过表达的区(多个背景)。图1a中的tfbs两两相关热图使用根据上面定义的得分的前500个区。基因组座标与tfbs相关性热图,包括图1a中的代表图,是在得分最高的前100个区生成的。slcr生成的自动化:为了专注于细胞固有基因标签,在先导方法中,我们从我们先前的实验中滤出了在gbm干样细胞(gsc)中低表达的基因,而本方法的当前实施涉及着重于经过验证的胶质瘤固有标签20。基于pwm得分和多样性手动选择得分最高的顺式单位,从而设计第一slcr。同样,手动进行包含tss的区的选择。自动的slcr生成是以python(urlgithub/gitlab)编写的。脚本将tf、pwm和表型基因标签的列表作为输入。有了这些,就可以从定义的顺式调控区生成顺式单位(默认参数:150bp窗口/50bp步长)。通过使用基于定义的选择规则的算法,生成对于任何给定的表型最佳的顺式单元的选择。该算法首先通过应用以下公式生成排序和最佳顺式单元的选择:[总和的得分-log10(p值)*多样性(不同tfbs的数量)]。迭代地,它会移除所选顺式单元中包括的tfbs。为了增加成功转录触发的机会,该算法还基于5'cage数据将顺式单元排序。排序的列表是算法的输出。自动化程序返回与手动选择重叠的结果(图7)。图1a-b中的热图是使用来自gplotsr包的热图.2函数生成的。rna-seq生成:使用trizol(invitrogen)提取rna,使用异丙醇沉淀,并且使用rnacleanxp珠纯化。使用truseq链式总rna文库制备试剂盒构建了用于本研究生成的rna-seq文库。将基于珠的方法用于rrna缺失(ribo-zerogold;illumina),并且按照制造商的方案进行pcr扩增。在bioanalyzer或tapestation上分析最终的文库,并在具有单读段51bp或成对末端100碱基方案的illuminahiseq2500或hiseq4000平台上对条形码文库汇集和测序。用cutadapt使用来自原始读段修剪illumina衔接子,并且用tophat将原始读段与人基因组(hg19或hg38)比对。将htseq用于评估每个基因的独特分配读段的数量;然后将表达值相对于107个总读段归一化,并且log2转换以获得每百万计数(cpm)。分析:对于图2d中的热图,我们使用seqmonkv1.42。简而言之,将bam文件使用hisat2与hg38比对,并且用rna-seq管道(pipeline)定量转录物定量在转录物计数读段上在外显子上校正特征长度。图形表示使用定量、对数转换和比对假定反向链特异性文库,随后是百分位数归一化补充有匹配分布。在图15e中,使用seqmonk分析数据,并且通过标准分析管道归一化读段,采用dna污染校正(contaminationcorrection)并生成原始计数以进行deseq2差异分析。使用具有对数转换的相同管道用于可视化。使用标准seqmonk设置确定显著性:在benjamimi和hochberg校正后,采用独立强度过滤,p<0.05。如上进行定量。使用ipa确定mg与gic以及tnfa与gic中的nfkb相关基因,通过各自的出版物获得mesgbm标签,并使用venny生成图。对于mes-gbmfc>0.5倍与padj=0,对于pnfc<-0.4,padj=0以及对于srebpfc>1倍与padj=0确定gsea显著性。图15e相互作用图是使用来自ipa的功能ingenuity上游调控子生成的,用于比较mgt#1高tnfa与mgt#1高c20mg共培养。atac-seq:对来自体内实验的20-50,000个细胞以及在体外实验中的50-100,000个细胞进行facs分选的群体上的atac-seq。将细胞在pbs中离心,并且将沉淀轻轻重悬于50μl的主混合物中(25μl2×td缓冲液、2.5μl转座酶和22.5μl无核酸酶的水、nexteradna文库制备、illumina),在37℃适度振荡下孵育60min(500-800rpm)。用5ul蛋白酶k和50ul的al缓冲液(quiagen)终止转座,在56℃下孵育10min,并使用1.8xvol/volampurexp珠纯化dna,并在18ul中洗脱。每个各自样本的文库扩增的pcr循环的最佳数量使用2ul的模板,随后使用热激活的kappahifi聚合酶和evagreen1x进行qpcr扩增来确定。在50ulqpcr体积和8-12ul的模板dna中进行最终扩增。引物为先前描述的(buenrostroetal.201)。使用qubit(lifetechnologies)对文库单独定量,并使用高灵敏度d1000screentapes在tapestation(agilent)上确定适当的阶梯分布。使用v2化学试剂在illuminanextseq500上进行150个循环的测序(配对末端75nt)。使用seqmonk进行图14a中的atac-seq散布分析,通过使用tss±5kb作为探针,对ensemblmrna进行最终注释。使用读段计数定量归一化,并且对仅在每百万读段的探针中的总计数进行读段校正,对数转换snf通过大小因子归一化进一步转换。atac-seq分析使用trim-galorev0.6.2--nextera对读段移除了衔接子,然后使用bowtie2v2.3.5(参考)默认参数进行映射。使用seqmonk进行atac-seq分析,通过使用tss±5kb作为探针对ensemblmrna进行最终注释(2019assembly)。使用读段技术定量功能归一化计数,并且对于仅每百万个读段的探针中的总计数进行读段校正,对数转换并且通过大小因子归一化进一步转换。根据建立的方案生成图14c的slcratac-seq和tcgaatac-seq的集成。载体生成:slcr最初在idt合成,以及后来在genscript合成。将mgt#1-mvenus克隆至哺乳动物表达,慢病毒fugw的paci-bsrgi片段中(来自davidbaltimore的赠予;addgene#14883)。额外的修饰,诸如将mvenus换成mcherry,或者mgt#1具有使用限制酶消化或gibson克隆的所有其他slcr。slcr载体是第3代慢病毒系统,并且已与pcmv-g(addgene#8454)、prsv-rev(addgene#12253)和pmdlg/prre(addgene#12251)一起使用。从ccsb-broad慢病毒表达文库获得sall2(ccsbbroad304_11117)pou3f2(ccsbbroad304_14774)。细胞系:mes-hgic和pn-hgic由我们的实验室生成,并且将在其他地方描述。简而言之,pn-hgic是通过转化人npc生成的,借助于plenti6.2/v5-idh1-r132h、tp53r173h和tp53r273h(从ccsb-broad慢病毒表达文库引入至tp53ccsbbroad304_07088的点突变,以及prs-puro-sh-pten(#1)。通过转化人npcprspuro-sh-pten(#1)、plko.1-sh-tp53(trcn0000003754)和prs-shnf1生成mes-hgic。对于这些品系,已经进行了全面的遗传、转录和表观遗传表征,以及体内肿瘤形成和表型模拟能力。在体外,如描述的76用一种修饰增殖gic。除了使用egf(20ng/ml;r&d)、bfgf(20ng/ml;r&d)、肝素(1μg/ml;sigma)和5%青霉素和链霉素外,还将pdgf-aa(20ng/ml;r&d)补充至rhb-a(takara)中。此培养基组成将被称为rhb-a完全。hgic在37℃下在5%co2、3%o2和95%湿度培养箱中培养。将t98g和u87mg(由nki的vantellingen实验室友情提供)在emem培养基中增殖。对于图13a中的实验,将t98g替换为补充有egf(20ngml-1)、bfgf(20ngml-1)、肝素(1μgml-1)和5%青霉素和链霉素的rhb-a,并且首先在经过标准组织培养处理的塑料上增殖,然后在超低结合塑料(corning)上增殖。将mcf7、mda-231、a549和h1944细胞系(由nki的renebernards实验室友情提供)培养在rpmi培养基中。在37℃下在5%co2–95%空气培养箱中,所有细胞系均补充有10%fbs和5%青霉素和链霉素。在补充有1%fbs、2.5mm谷氨酰胺(thermofisher;35050038)、1μm地塞米松(sigma;d1756)和1%青霉素和链霉素的rhb-a培养基(takara)中于37℃下在5%co2、19%o2和95%湿度培养箱中培养永生化原代人小胶质细胞c20。在sfemii(stemcell)、scf、flt3-l、tpo、il6(全部100ng/ml;easyexperiments.com)、um171(selleck,0.035μm)、sr1(selleck,0.75μm)、19-脱氧-9-亚甲基-16,16-二甲基pge2(cayman,10μm)中增殖供体来源的cd34细胞。全基因组crispr敲除体外筛选:对于全基因组汇集的crispr敲除筛选,我们利用由靶向19,114个基因(每个基因平均4个sgrna)的77441个sgrna和1000个非靶向对照组成的brunello文库。为了获得超过100x的文库表示,我们以~0.5的moi转导了总计16x106mes-hgics-mgt#1低细胞,并在引入处理之前将细胞扩增10天。在第10天,将细胞用tnfa(10ng/ml)和fbs(0.5%);替莫唑胺(50μm)和辐照(20gy)处理或不进行处理。在gdna提取之前,我们对每种条件进行了fac分选,收集mes-hgics-mgt#1低、mes-hgics-mgt#1高和未分选的群体。通过在56℃下在补充有蛋白酶k(invitrogen)和rnasea(thermoscientific)的al缓冲液(qiagen)中裂解细胞沉淀10’,随后用ampure珠纯化并在eb缓冲液(qiagen)中洗脱,从而提取基因组dna。ngs文库是在两步pcr设置中构建的,其中,pcr1用于扩增sgrna支架并插入交错序列以增加整个流通池中的文库复杂性,而pcr2引入了具有独特p7条形码的illumina兼容衔接子,从而允许样本多重性。对于pcr1,将5μg的每个gdna样本分成5个平行反应,随后汇集在一起,并使用ampure珠纯化。通过使用kapahifihotstartreadymix(roche)和1xevagreen(biotium)进行qpcr扩增,单独确定对于1μl的每个pcr1的pcr2的最佳循环数。将10μl的每个样本的纯化的pcr1用作最终pcr2的输入。pcr1和pcr2两者均使用kapahifihotstartreadymix进行。可根据需求提供引物。使用用于定量的qubitdsdnahs试剂盒(invitrogen)和用于确定pcr片段大小的tapestation高灵敏度d1000screentapes(agilent)进行最终文库的定量控制。将条形码文库以等摩尔汇集在一起,并且使用75个循环v2化学(1x75nt单读取模式)在illuminanextseq500上测序。穿透小室(transwell)共培养:使用具有0.4μm孔径的亲水性ptfe6孔细胞培养插入物(merck)建立hgic和永生化原代人小胶质细胞c20的共培养。将人小胶质细胞以1.5x105细胞/孔接种在各自培养基中的6孔板上24h。吸出培养基,并且用pbs洗涤细胞一次,然后加入1ml的rhb-a完全培养基。将穿透小室插入物置于板中,并且将在总体积为1ml的rhb-a完全培养基中的5x105单hgic铺板在插入物表面上。共培养48h后,收集hgic和c20人小胶质细胞,用于进一步分析。转染-转导:先前详细描述了转染和转导。简而言之,将12μg的dna混合物(慢病毒载体、pcmv-g、prsv-rev、pmdlg/prre)与fugene-dmem/f12混合物在室温下孵育15min,加入到覆盖293t细胞的无抗生素培养基中,并且在转染后40h收集第一层(first-tap)病毒上清液。根据制造商的说明,使用lenti-xp24快速滴定试剂盒(takara)评估效价。我们在补充有2.5μg/ml硫酸鱼精蛋白的适当的完全培养基中将病毒颗粒应用于靶细胞。在与病毒上清液一起孵育12-14h后,用适当的完全培养基更新培养基。冷冻切片的制备:通过重力使肿瘤球体沉降,固定在新鲜制备的甲醛于pbs(1.0%)中,其用140mm甘氨酸2m阻断。用30%蔗糖冲洗,随后加入冷冻培养基(o.c.t/冷冻模具(cryomold))。通过干冰冷冻获得冷冻块,并在-80℃下保存直至使用。用leicacm1950切割块。免疫组织化学:将组织或肿瘤球体固定在4%pfa中20’。固定后,用从70%增加至100%的etoh、二甲苯和过夜石蜡孵育进行脱水。使用hm355s切片机(thermoscientific)切割石蜡包埋的样本(pes)。进行苏木精/伊红(he)标准染色,并且用自动显微镜(keyence)获得载玻片图像。免疫荧光:在室温下,细胞生长在盖玻片上或球状体旋转下来到玻璃上,随后是4%多聚甲醛(pfa,16005-sigmaaldrich)于pbs中固定10min,在pbs中洗涤5min(3x),用0.5%tritonx100于pbs中透化5min,用4%bsa(3854.4roth)阻断15min,用初级和二级抗体和20μm/ml的hoechst33258(16756-50,cayman)染色,并且使用甲油和vectashield(h1000-linaris)固定到玻璃载玻片上。在石蜡包埋的组织上,我们用标准方案进行了去石蜡化和柠檬酸抗原修复。用triton0,25%于pbs中进行透化,并且——当适当的情况下——用3%h2o2于水中阻断内源性过氧化物酶。通常,我们用5%正常山羊血清(ngs)进行阻断。初级抗体为:抗-gfp(抗-gfpab6556,1:000)、抗-med1(abcamab649651:500)、抗-tubulin(bdt5168,1:2000),以及二级抗体为:a31573、a11055和a31571alexafluor647、a21206alexafluor488、a31570alexafluor555。rnafish和双重fish-if:将细胞在70%乙醇(仅rnafish)中或用0.5%tritonx-100(对于双重if-rnafish)透化,在无rnase的pbs(1x(lifetechnologies,am9932)中洗涤,在室温下用10%去离子甲酰胺(emdmillipore,s4117)于20%stellarisrnafish洗涤缓冲液a(biosearchtechnologies,inc.,smf-wa1-60)和无rnase的pbs中固定5min。使用以31.5μm在100μl中的smf-1084-5calred635和smf-1063-5570定制fish探针(根据需求可提供寡核苷酸序列)于10%去离子甲酰胺90%stellarisrnafish杂交缓冲液(biosearchtechnologies,smf-hb1-10)中的溶液转移至盖玻片上,在37℃下在暗处杂交,来探测igk-mgt#1-mvenus和h2b-cfp。在o/n孵育后,将载玻片用无rnase的pbs洗涤5min(3x)。如果发生了初级/二级染色,则如上所述。成像:使用的显微镜为zeisslsm800,leicasp5-7-8,nikonspinningdisk。用leicasp5获得图s41中的共聚焦图像。使用ex=488nm、em=535nm获得mvenus荧光,以及对于图1d中的,使用zeisslsm800获得,对于mvenus-quasar570使用ex=558nm、em=575nm,对于brd4-或med1-af647,分别使用ex=653、em=668。对于h2b-cfp-quasar670,我们使用ex=631、em=670。使用imagej或photoshop处理图像。表型筛选:如上所述增殖肿瘤细胞直至筛选。然后,我们在补充有适当生长因子的gibcofluorobritedmem培养基中,在384孔板(corning)中以15'000/50μl/孔接种。使用spark20m注射器系统(50μl注射体积;100μl/s注射速度)将细胞以50μl悬浮液分配至每个孔中。对于非贴壁细胞(例如gic),将细胞在37℃下进一步以1500rpm离心1h30min。使用sparm20mtecan读板器在37℃下在5%co2-95%空气(对于gic为3%)中在加湿的盒中扫描底部读数荧光,对于mvenus具有以下设置:单色仪,ex505nm±20nm、em535nm±7.5nm,手动增益:198,闪烁:35,积分时间:40μs。在独立的复制子中,用在fluorobrite培养基中的0.02%alamarblue溶液测量细胞生存力,使用以下设置:荧光最高读数。单色仪,ex565nm±10nm、em592nm±10nm,手动增益:88,闪烁:30,积分时间:40μs。使用d300e自动等分dmso可溶性化合物诸如gsk126,而使用andrew移液机器人(andrewalliance)将细胞因子自动等分至每个孔中,使用以下浓度:细胞因子产品代码储备液工作浓度il6206-il;r&d系统100μg/ml15ng/mllpsalx-581;enzo200x1xtnfα210-ta;r&d系统100μg/ml20ng/mltgfb240-b;r&d系统35μg/ml5ng/mlifng285-if;r&d系统100μg/ml10ng/ml生腱蛋白cmbs230239;mybiosource100μg/ml100ng/mlhgf294-hg;r&d系统10μg/ml10ng/mligf50356.100;biomol2μg/ml2ng/mlfbs10270106;gibco100%10%gsk1265mm5μmcbd10mm4μm激活素abv-p1078;enzo50μg/ml50ng/mlnrg197642.10;biomol16μg/ml90ng/mlil1bcyt-094;biotrend100μg/ml10ng/ml将数据输入prism7(graphpad)中。从所有值中将来自对照死细胞的荧光强度作为背景减去。将各个值相对于对照的平均值归一化,并表示为倍数变化。药物剂量响应筛选:将来自穿透小室共培养实验的转导的hgic收获至单细胞悬浮液中,并使用bdfacsariaiii分选成mvenus高和低群体。计数细胞,并使用spark20m注射器系统(50μl注射体积;100μl/s注射速度)将7000细胞/50μl/孔接种到384孔黑壁板上于rhb-a完全培养基中。通常将药物溶解为10mm储备液于dmso中,并且使用d300e复合打印机(tecan)进行分配,用于具有平板随机化和dmso归一化的靶向剂量响应。孵育72h后,在用10μl的cell-titer-blu(promega)测定试剂孵育2-6h后用以下设置测量细胞生存力:荧光最高读数。单色仪,ex565nm±10nm、em592nm±10nm,增益设置:最优扫描,闪烁:30,积分时间:40μs。将数据输入prism7(graphpad)中。从所有值中将来自空孔的荧光强度作为背景减去。将浓度log10转换为log[m]标度,并将各个值相对于未处理的阳性和sds处理的阴性对照条件的平均值归一化。使用非线性回归模型(log(抑制剂)与归一化响应-可变斜率)得出剂量响应曲线和ic50值。hgic的辐照:使用配备有用于靶向辐照的225kvx射线管的xenx辐照器平台(xstrahllifesciences)传送辐照。将在6孔板或96孔板中培养的hgic置于光束线的焦平面中,并如用内部计算软件计算的根据靶剂量暴露于辐射特定的时间。基质胶(matrigel)类器官的生成:为了用c20人小胶质细胞和hgic的共培养生成类器官,使用生长因子减少的并且无酚红的基质胶(bd;734-1101)液滴作为细胞外基质承载体。收获靶细胞,并且制备具有1.5x105的c20人小胶质细胞和3.5x105的hgic在500μl的体积中的单细胞悬液。使用预冷的消耗品和移液器吸头,将30μl的基质胶在冰上融化,添加至冷的60孔minitray(thermofisher;439225)的每个孔中。使用5μl制备的细胞悬液以每液滴5000细胞注射至每个类器官中,并通过移液混合。将液滴在37℃下,在5%co2、3%o2和95%湿度培养箱中培养最长达14天,并且每2-3天更换rhb-a完全培养基。在第10天使用leicasp8共聚焦显微镜进行活细胞成像。rt-qpcr:使用20μl中的superscripttmvilotmmastermixrna(0.5-2.5μg)在25℃下孵育10',在42℃下孵育60'和在85℃下孵育5',生成cdna。以10ul/孔,在使用1xpowerupsybrgreenmastermix(appliedbiosystems)的384wviiatm7系统中,用10ngcdna/孔进行rt-qpcr。可根据需求提供引物。组织剥离和细胞表面染色:先前详细描述了脑肿瘤剥离77。简要地,将组织用解剖刀剥离,在37℃下在accutase/dnasei(947μlaccutase、50μldnasei缓冲液、3μldnasei)中消化直至需要。先通过120μm细胞过滤器,再通过40μm细胞过滤器过滤,然后rbc裂解(nh4cl,155mm;khco3,10mm;edta,ph7.4,0.1mm)。在冷pbs中洗涤后,使用tecanspark20m用0.4%锥虫蓝染色自动评估生存力和细胞计数。当评估表面标志物时,通常在15mlfalcons中使用200.000个细胞/抗体。染色体积为50μl在具有初级抗体(例如cd133-apc;miltenyi)的rhb-a培养基中,在冰上在黑暗中30’。用两次pbs洗涤去除未结合的抗体。根据是否分析或分选细胞,在bdlsrfortessa上进行数据采集,或者使用bdariaii或astriosmoflo分选细胞。根据所分析的荧光团选择合适的激光滤光片组合。通常,为了去除死细胞,首先根据形状和粒度(fsc-ssc)对事件选通,并且我们将annexinv或活/死可固定的水性死细胞染色试剂盒(live/deadfixableaquadeadcellstainkit)用作生存力染料(取决于所分析的荧光团)。用flowjo_v10进行分析。facs分析:用flowjo_v10进行分析。facs分选:将转导的hgic收获到单细胞悬液中,并重悬至冷的rhb-a完全中,并过滤至facs管中。使用bdfacsariaiii或fusion进行分选。根据要分选的荧光团选择合适的激光滤光片组合。通常,为了去除死细胞,首先根据形状和粒度(fsc-a与ssc-a)对事件选通,并排除二重态(fsc-a与fsc-h)。在pgk驱动并结构性表达的h2b-cfp作为分选报道子上建立正向选通,以对具有低至中强度的slcr依赖性荧光团表达的群体进行分选。免疫印迹:在补充有1x蛋白酶抑制剂混合物(roche)、10mmnappi、10mmnaf和1mm正钒酸钠的ripa缓冲液(20mmtris-hclph7.5,150mmnacl,1mmedta,1mmegta,1%np-40)中裂解细胞沉淀。如有必要,对裂解物进行超声处理,并且使用nupagebis-tris预制凝胶(lifetechnologies)在nupagemopssds运行缓冲液(50mmmops,50mmtris碱,0.1%sds、1mmedta)中进行电泳。在转移缓冲液(25mmtris-hclph7.5,192mm甘氨酸,20%甲醇)中以120ma将蛋白转移至硝酸纤维素膜上1h。在用tbs-t洗涤两次后,通过用丽春红(ponceaured)染色5min来评估蛋白质转移。在室温下用5%bsa于pbs中进行膜的阻断1h。在pbs+5%bsa中制备初级抗体的稀释液,并且将膜在4℃下孵育过夜。在用tbs-t洗涤三次5min后,在pbs+5%bsa中制备适当的hrp偶联的二级抗体的稀释液,并且将膜在室温下孵育45min。在用tbs-t洗涤三次5min后,采用ecl检测试剂(sigma;rpn2209)并且将膜暴露于eclhyperfilm(sgima;ge28-9068-37)以检测化学发光信号。抗体靶标产品代码制造商gfpab6556abcam黏着斑蛋白p-stat3y7059145lcellsignalingstat3sc-482xsantacruzp-nfkbp653033pcellsignalingnfkbp6586299abcamp-p38t180d3f945115cellsignalingp-p389211smillipore神经上皮干细胞蛋白611658bdbiosciencesp-yh2axser13905-636milliporek27me307-449millipore总h3(h3total)1791abcame-钙黏蛋白31950cellsignaling波形蛋白5741scellsignaling山羊抗小鼠igg(hl)-hrp626520invitrogen山羊抗兔igg(hl)-hrpg21234invitrogenincucyte:incucyte自动化纵向成像是在96孔黑壁板(greiner)中进行的。在实验结束时,每板接种300,000个细胞,以达到最优汇合。使用d300e等分gsk126,而手工等分tgfb1+2至每个孔。两者均每两天更换。最后的时间点使用读板器(bmcclariostar)独立验证。crispri筛选:对于crispri筛选,在astriosmoflo上分选a549-mgt#1±gsk126±dox细胞。我们旨在每个群体内的在10%的最低(微弱)和10%的最高(亮)的细胞中1000x(>600万个细胞)的文库表示。还将中间群体分选,并作为对照包括在筛选分析中。将细胞在56℃下在al+proteinasek缓冲液(quiagen)中裂解10’,随后dna提取是使用ampure珠(agencourt)和rnasea处理提取的。crispri文库的pcr扩增和条形码标签化基本上如所描述的进行,包括pcr缓冲液组合物77。对于每个样品,在pcr1中,我们使用分成包括来自输入对照的10个平行反应的20ug的dna,而质粒文库在pcr1中需要0.1ng的dna。将平行pcr1反应混合在一起,并将5ul用作pcr2的模板。我们在pcr1和pcr2两者中都使用了phusion聚合酶(neb)、gc缓冲液和3%dmso。可根据需求提供引物。测量文库浓度,并且汇集条形码文库,并在illuminahiseq2500测序上测序。用定制脚本(可根据需求提供)将读段映射到计算机文库中,以生成读段计数,随后将其用作seqmonk的输入。我们使用定制基因组用于seqmonk分析(可根据需求提供),并将样品相对于rpm归一化并且对数转换以生成ma图,而在padj<0.001的deseq2则以原始读段计数运行。我们在a549中运行2个独立的crispri筛选,并且在h1944中运行一个额外的筛选。crispr/cas9ko:按照说明使用cas9rnpsynthego试剂盒敲除a549-mgt#1的cnksr2和arid1a。使用bioradxcell于pbs中和使用标准脉冲对a549细胞进行电穿孔。首先使用t7e1以及tide计算(https://tide.nki.nl/)评估试剂盒中的最优grna。之后,我们使用流式细胞术以及低汇合铺板和手动克隆挑选对mgt#1荧光进行总体评估。动物实验:所有小鼠研究均根据由机构动物护理和应用委员会(institutionalanimalcareandusecommittee)批准的方案进行,并且符合欧盟的法规。原位胶质瘤异种移植物研究如先前所描述的76用修饰进行。nod-scid-il2rg/(nsg)小鼠购自thejacksonlaboratory,并保持在无特异性病原体(spf)的条件下。我们使用了7-12周龄之间的雄性和雌性小鼠。基因敲除:使用synthego基因敲除试剂盒进行基因敲除。将sgrna溶解在无核酸酶的1xte缓冲液中,至储备液浓度为30um。rnp复合物是通过将cas9核酸酶-grna以6:1的比率混合形成的。使用bioradgenepulserxcell(150伏,10ms),将每个rnp复合物电穿孔至于1xpbs中的在2mm比色皿中的250ka549-mgt1#1中。电穿孔后,在补充有10%胎牛血清和1%青霉素/链霉素的rpmi中培养细胞。电穿孔后的大约7天,使用invisorb旋转组织分离试剂盒(stratec)提取gdna,在50ul洗脱缓冲液中洗脱,并且使用以grna靶基因座为中心的800至1200bp产物对目的靶基因进行pcr(可根据需求提供引物)。使用tide(nki)和t7ei测定计算敲除效率。建立单独的克隆,或使用bdlsrfortessa和flowjo程序通过facs直接测定总体ko细胞。实施例1:包括多形性胶质母细胞瘤(gbm)肿瘤细胞的亚型特异性合成基因座控制区(slcr)的表达盒的设计。高度的细胞和分子异质性被认为促成了在实体瘤中对标准治疗的抗性,并且这为靶向方法的发展带来了障碍。多形性胶质母细胞瘤(gbm)是最常见的原发性成人脑肿瘤,它是异常异质性的并且对治疗有抗性13。gbm也是具有最高程度的基因组和表观基因(epigenomic)表征的癌症之一14-16。基于转录物组,gbm肿瘤被反复分类为三种亚型,其中间充质和原神经更经常被交叉验证52、53、54。数项研究对亚型特异性基因表达标签和对治疗的差异响应以及患者的总体存活之间的相关性进行了辩论。这表明gbm亚型标识和命运改变可能具有治疗潜力。在gbm肿瘤中,主要的亚型和具有不同亚型标识的肿瘤细胞可能共存17,18。而且,肿瘤可以在复发时改变优势表达谱19,20。谱系追踪先前对小鼠模型中我们理解的gbm生物学具有主要影响,告知了——除其他之外——单个亚型5的细胞起源,以及异常的内稳态调控如何影响体内对护理标准的响应10。在本实施例中,我们描述了系统生物学方法,以设计合成系统,以遗传标记复杂发育和疾病环境中的任何细胞状态或转变,并测试该系统以寻求人gbm的分子亚型潜在的生物学原理。首先,我们假设亚型特异性gbm基因将实质上包括特异性亚型标识所需的调控活性(即顺式调控元件)。我们进一步假设在每个亚型中表达的转录因子基因(tf)将主要负责建立和维持亚型标识。为了设计将拦截最少信号传导和调控信息的基因盒,我们确定了与tcga数据集16中所有其他亚型相比具有最高倍数变化的亚型特异性gbm基因。可以使用任意严格的切断(即>6log2fc;图6)来实现调用mes、cl和pn亚型特异性基因。同样,可以使用较不严格的切断(即>0log2fc)和标准通路分析工具(例如,独创通路分析,david等)来鉴定tf。最初,来自我们先前实验的在gbm干样细胞(gsc)中低表达的基因(例如,<每百万4个计数,cpm)被丢弃,作为专注于细胞自主调控的测量(图6)。本方法的当前实现使用单细胞rna-seq谱,如例如胶质瘤固有标签14。为了鉴定在亚型差异调控的基因(dgr)中具有高固有顺式调控潜力的基因组区,我们计算了与在每个亚型中表达的tf相关的最佳位置权重矩阵(pwm)的所有配对频率(图1a)。由于顺式调控dna通常是无核小体区(nfrs;>147bp),并且涉及平均~1000bp21,为了精确定位这些元件,我们设置了具有150bp步长的1kb滑动窗口方法。对可能调控drg的顺式单元的搜索通过由encode联盟22,23确定的两个外部ctcf结合位点来界定,距基因起始/末端的距离任意设置为>10kb。这些准则近似于拓扑相关结构域(tad)的功能定义,据信其包含给定基因座的顺式调控元件之间的绝大多数接触点,并使用ctcf作为边界蛋白24。为了使用上面描述的tfbs分析组装驱动亚型特异性表达的合成顺式调控元件,此类合成基因座控制区(slcr)理想地应包括具有最高数量(i)和多样性(ii)的顺势单元的最小集合。理想地,组成一个slcr的至少一个顺式单元还将包括自然转录起始位点(tss),并将直接置于报道元件的上游(图1a)。利用这些准则,我们生成了用于遗传追踪mes、cl和pngbm的slcr(以下称为mgt、clgt和pngt)。可以使用算法以最小化决策并自动化slcr生成(图7a)。潜在地调控这些基因的tfbs的两两相关性揭示了几个tf簇在一起并且远离其他tfbs簇(图1b)。该观察结果与来自chip-seq实验的实验观察结果一致,从而表明我们的程序返回了与基因组调控的功能和结构相关原理一致的结果。此外,多个细胞系中的encodechip-seq数据也支持结合至单个顺式单元的实际tf(图9)。重要的是,不同的mgt#1和mgt#2slcr由高度独立的单个顺式单元组装,并且测量分别仅为827pb和1015bp的长度,各自可代表最高达60%的总体调控潜力。实施例2:使用包括mgt#1作为slcr的慢病毒载体在人胶质瘤起始细胞中间充质命运的遗传追踪携带slcr诸如mgt#1的典型的慢病毒载体,驱动荧光报道子mvenus或mcherry的亚型表达。为了促进体内遗传追踪,将mvenus驱动至质膜(通过igk前导和血小板源性生长因子受体(pdgfr)跨膜序列标签化;图1c)并且将mcherry通过nls穿梭至细胞核。为了使能够荧光可视化以及独立于报道子表达分选slcr,我们还包括了经由普遍存在的pgk启动子表达h2b-cfp融合的第二盒(图1c)。作为原型测试,我们在具有mgt#1-mvenusslcr的hek293t细胞中产生了慢病毒颗粒,并使用病毒颗粒感染具有mes基因型的人胶质瘤起始细胞(mes-hgic)。在瞬时转染以及在稳定转导和冷冻切片的肿瘤球体两者中均观察到膜mvenus表达(图1d)。接下来,用mgt#1慢病毒颗粒转导近等基因和特征化的mes-hgic和pn-hgic。pn-hgic具有idh1和tp53点突变的组合,其仅在pngbm中发现,而mes-hgic具有tp53、pten和nf1的三重敲减,以mesgbm背景为特征。有趣地,我们观察到在mes-hgic中基础荧光有微小但可测量的增加,表明mgt#1反映了这些细胞中基础更高的固有信号传导(图1e)。由于tnfα被认为是优势的mes-gbm信号传导通路,并且可以诱导pn至mes转变20,我们接下来通过使mes-hgics-mgt#1低和pn-hgics-mgt#1低暴露于tnfα来测试mgt#1是否忠实地再现mesgbm信号传导。在tnf的存在下,先前已示出mgt#1slcr的至少两个顺式单元直接接合tnf驱动的nfkbtf。令人放心的是,与每个亲本对照相比,tnfα在两种细胞类型中均诱导荧光增强。有趣的是,尽管facs分选步骤确保在两种细胞类型中均存在相等基础水平的mgt#1表达,但mes-hgics-mgt#1低转变为mes-hgics-mgt#1高,而pn-hgics-mgt#1低仅达到pn-hgics-mgt#1中水平(图1e-f),验证了mgt#1报告子用于mesgbm亚型特异性表达,并且利用该系统为hgics的适应性响应被刻入其肿瘤基因型中提供了证据。人gic和gsc在“nbe”条件下持续增殖,其代表补充有基础fgf和egf的无血清neurobasal培养基25。我们进一步用pdgf-aa补充我们的gic,因为这是在gbm中最常基因扩增的信号传导通路26。为了使用我们的遗传策略调查mes-gbm信号传导的基态,我们在mes-hgics-mgt#1低和pn-hgics-mgt#1低细胞中进行了中通量细胞因子筛选。gic在标准条件下增殖,并将它们重新接种在384孔格式中。接下来,用生物学和技术复制品中的个体细胞因子刺激gic,随后是在预定义的时程实验中连续荧光底部读数。在典型的实验中,我们从刺激后最多48小时纵向获取mgt#1荧光发射,并然后我们将荧光相对于初始gic归一化。与先前的报道和上述实验一致,在tnfα信号传导的存在下,mes-hgics-mgt#1低转变为mes-hgics-mgt#1高(图2a,8)。因此,mgt#1告知了对于具有不同基因型的肿瘤细胞之间的外源信号传导的差异响应。此外,mgt#1是筛选框架以鉴定支持mes-hgics-mgt#1低和pn-hgics-mgt#1低细胞的生长和亚型标识的相关信号传导的坚实基础。实施例3:使用mgt#1和mgt#2slcr作为读出用于研究gic中固有的和适应性响应在相同的实验条件下,第二独立报道子(mgt#2)示出一致的结果(图2a),这支持我们从基因表达谱开始生成功能性slcr的能力。有趣地,mgt#1和mgt#2报道子两者均指示fbs能够诱导间充质分化,与tnfα的情况不同,这伴随着由目视检查和流式细胞术所测量的gic分化(数据未显示)。这一发现可仅部分地由tgfb1的存在解释,tgfb1的确是fbs的已知组分。实际上,tgfb1是间充质诱导剂,但不会强烈诱导mgt#1,当在同一时间框内用作纯化的细胞因子时它不会促进分化(图2a)。也许更有趣地,在fbs上的这一观察结果与tcga报告高度一致,即mesgbm标签不能在任何小鼠脑细胞中找到,而只能在fbs培养的星形胶质细胞中找到16。胶质瘤的小鼠模型中tnfα的体内来源被认为是肿瘤微环境(tme),尤其是胶质母细胞瘤相关的小胶质细胞/单核细胞(gam)27。在hgam中也观察到了tnfα表达28。有趣地,通过gam的idh1野生型gbm浸润最近与nf1缺陷和meggbm亚型标识相关联14。为了给募集到gbm的gam会驱动nf缺陷型gbm细胞中的mes分化的假说提供实验支持,我们在体外进行了共培养idh1野生型和nf1缺失的mes-hgics-mgt#1微弱细胞与从具有gbm的患者中纯化的macs-纯化的cd11b细胞。令人惊讶地,在存在il-6刺激下,共培养的hgics-mgt#1微弱细胞与cd11b+hgam诱导mgt#1表达(图2b)。先前已示出il-6可以刺激gams29,并且可以由gscs30或来自tme的间充质干细胞31产生。值得注意的是,无论是未刺激的还是当暴露于tlr4内源性配体生腱蛋白-c(tnc32),其是另一种gsc衍生的促炎症因子33,hgam都不足以驱动在mes-hgic中的mgt#1表达。此外,无论是否存在hgam,tnfα均在mes-hgic中驱动mgt#1诱导(图2b)。因此,我们的数据揭示了涉及围绕il6信号传导并导致mesgbm特化(specification)的在gbmtme中的潜在细胞串扰。这些数据也突显了slcr在离体机制剥离非细胞自主相互作用的潜力。我们的数据支持slcr作为用于研究gic中固有的和适应性响应的有效读出,但不排除这种读出很大程度上受限于报道子的唯一规则的可能性。为了理解报道子调控是否伴随细胞标识的差异,我们在mes-hgics-mgt#1低和pn-hgics-mgt#1低细胞中进行了免疫印迹、球形基因表达谱分析和靶向mrna验证。尽管在相同的实验条件下增殖,但通过测试的所有实验方式,mes-hgics-mgt#1低和pn-hgics-mgt#1低细胞在信号传导通路激活和基因表达上持续示出有限但可测量的基础差异(图2c-d-e-f)。值得注意的是,尽管tnfα刺激在两种细胞类型中均诱导nfkb-p65、stat3和p38-mapk的磷酸化,但这导致了明显不同的基因表达输出(图2c-d-e-f)。因此,mgt#1告知了活性信号传导(例如tnfα)的影响,并且即使当先前存在的依赖于上下文的差异已到位(例如间充质信号传导扩增或转变),它也确实反映了相似的细胞命运转变。有趣地,总体的和靶向基因表达谱分析两者均表明,tnfα驱动pn-hgic至更接近其初始状态的mes-hgic的状态(图2c-d-e-f)。实施例4:使用mgt#1从功能上测试环境损害(例如电离辐射)是否可能以gbm细胞自主方式诱导间充质转分化gbm中的间充质分化最初被描述为放射治疗后复发的显性事件19,并且之后经由tnf驱动的nfkb活化与获得的辐射抗性相联系。反复地,相关证据支持炎症信号传导、emt和辐射抗性之间的联系。为了从功能上测试辐照是否可能以细胞自主方式诱导间充质转分化,将mes-hgics-mgt#1低和pn-hgics-mgt#1低细胞单独或与tnfα组合暴露于电离辐射(ir)。在此实验中,我们围绕着递送10gy的单次辐射剂量,有两个原因:(i)我们实验性地确定这是亚致死的(单独或与其他治疗组合,包括tnfα或替莫唑胺;数据未显示),并且(ii)10gy接近于实验性地证明引发作为固有辐射抗性的方式以及在多个人gsc中增强的修复能力34,35的二次响应的剂量。辐照二十四小时后残留的dna损伤标志物h2a磷酸化证实了双链断裂和修复两者的发生。然而,只有一小部分gic从任一遗传背景转变为mgt#1高状态(图2g-h)。相反,mes-hgics-mgt#1低和pn-hgics-mgt#1低细胞两者与tnfα组合均示出增强的间充质分化,表明tnf信号传导和ir协同诱导了这种细胞命运特化。综上所述,这些数据支持以下结论:亚致死ir与其他机制协同驱动gbm中的间充质转化。这些数据也支持以下推测:nfkb激活是由于由基因毒性应激引起的非经典信号传导而增强的36。实施例5:使用slcr的gbm亚型化和重编程。原神经gbm被认为代表了常见的gbm祖先亚型,并且也反映了少突胶质细胞的起源26,37。先前的研究表明,fbs中的长期增殖会影响单个细胞系的表型标识25,16。为了测试pnslcr是否会反映原神经状态,我们决定使用在pn标识下的主tf来诱导fbs驱动的常规细胞系重编程为pn-gic。为此,我们将mgt#1或pngt#2转导至t98g细胞系中,其以更可能与pn表型相关的tp53突变(https://portals.broadinstitute.org/ccle)为特征16。与基因型驱动的预测一致,当从fbs替换为nbe增殖条件时,t98细胞示出pngt#2的基础表达,而不是mgt#1的基础表达(图3a-b)。重要地,sall3、sox2和pou3f2的瞬时过表达进一步增强了pngt#2的激活,但对mgt#1表达是中性的(图3b)。值得注意的是,这些实验组是用带有核定位信号传导的mcherry荧光蛋白进行的,因此排除了荧光蛋白强度(mcherry比mvenus更亮)、定位和稳定性(mvenus是跨膜且稳定的)在观察的表型转变中起主要作用。总体而言,这些实验表明,使用本文所描述的系统和合成生物学方法,通过gbm细胞中的单个slcr可以拦截已知在gbm生物学中起关键作用的多个固有和外部触发因子。实施例6:使用mgt#1剖析乳腺癌和肺癌细胞中的上皮-至-间充质转化间充质转分化是被上皮起源的多种肿瘤劫持的生理过程39。为了研究我们的遗传追踪策略是否延伸超出了gbm内稳态,我们接下来将mgt#1转导至特征明确的上皮和间充质乳腺癌细胞中。肿瘤亚型被遗传刻印在乳腺癌细胞中40。一致地,在第一轮慢病毒转导后,上皮mcf7细胞与被认为已经历emt的mda-231细胞相比,示出更低的mgt#1表达(图10a-b)。为了确认mgt#1表达反映了实际的乳腺癌亚型标识,我们facs分选并亚克隆了最高mcf7-mgt#1和中间mda-231-mgt#1表达的细胞。然而,facs分选的群体的进一步增殖重新建立了预分选的内稳态,mcf7表达的mgt#1水平比mda-231更低。这样的水平似乎是稳定的,因为用emt诱导剂tgfb2的短期处理在mcf7和mda-231中均未强烈修饰基础mgt#1荧光(图4a)。ezh2抑制可以在几种小鼠和人肺癌细胞中支持kras驱动的emt41。在这种情况下,我们测试了slcr在反映对生物和化学刺激的细胞和分子响应中的用途。与先前的发现一致,在上皮a549细胞中的纵向测量揭示了高mgt#1荧光由ezh2抑制剂gsk126和tfgb信号传导协同诱导(图4b)。暴露于tgfb信号传导的上皮肺癌细胞很容易改变其形态,以及开始表达高水平的mgt#1,如通过流式细胞术测量的(图11a-b)。有趣地,在早期时间点,流式细胞术揭示了tfgb信号传导和通过gsk126的ezh2抑制诱导了相似程度的分子转变,但gsk126并未诱导细胞形态改变。在组合设置中,tfgb信号传导和gsk126协同地诱导mgt#1激活,并且还观察到中间形态变化(图11a-b),这引起了有趣的可能性,即gsk126除了作为比tgfb信号传导的放大器外还通过额外机制对emt起作用。实施例7:使用ezh2抑制和mgt#1用于研究nsclc细胞中上皮-至-间充质转化的信号传导和遗传基础为了利用ezh2抑制和mgt#1作为框架来阐明nsclc细胞中emt的信号传导基础,我们接下来在gsk126-和媒介物处理的a549-mgt#1低细胞中进行了细胞因子筛选。与上述数据和我们最近发表的观察结果(serresietal.,j.exp.med,2018,doi:10.1084/jem.20180801)一致,tnfα被证明也是上皮肺癌细胞中对mgt#1表达的主要信号传导,对在纵向中通量微板读数器筛选中测量的总体高荧光输出具有gsk126的适度的累加效应。同时,我们证实,当gsk126存在时,a549细胞经由细菌lps不同地响应于tlr刺激,并且也在这些实验条件下,我们示出了tgfb1当与gsk126组合时更显著地诱导mgt#1。用几种细胞因子及其组合筛选的系统分析表明,ezh2抑制增强了对emt的外部信号传导的转录响应(图12)。总体而言,mgt#1响应表明在emt期间可能聚集多个信号传导通路,并且意味着转录抑制控制细胞亚稳定性。接下来,我们希望利用ezh2抑制和mgt#1作为高通量筛选的框架,以阐明nsclc细胞中emt的遗传基础。首先,我们用mgt#1报告子转导a549和h1944kras驱动的nsclc细胞两者。随后,我们在两种细胞系中引入了tet可诱导的krab-dcas9和靶向一整套人激酶组的sgrna文库(543个基因,总计5,901个grna;~5个grna/基因)。此外,我们还包括了靶向grna的必需和非必需基因,以用作筛选过程的对照。该系统允许系统地敲减单个细胞中的单个基因(图4c)。通过如先前所述采用gsk126处理,我们facs纯化了nsclc细胞,这些细胞在支持荧光报告子的表达的能力中有所改进或受损,并且示出上皮或间充质表型(图4d-e)。基因集合富集分析支持筛选的整体质量,如通过必需基因但不是非必需基因所测量的,与输入群体相比,在体外在两种细胞系中都显著缺失(数据未显示)。通过比较a549-mgt#1低和h1944-mgt#1低与其mgt#1高对应物,我们仅检索到小部分的grna在两种细胞系中在两种状态的任一状态下统计学差异地富集或缺失(14/5912,0.24%),表明大多数人激酶对于gsk126驱动的emt是不必要的。然而,两种grna均具有统计学意义,并示出与a549-mgt#1低和h1944-mgt#1低细胞关联的高倍变化,表明它们的表达可导致激酶相关基因的转录抑制,从而使得在ezh2抑制后肺癌细胞emt(图4e)。有趣地,一种grna靶向先前被报道可增强nf-kb驱动的emt的acvr1受体42,并且一种grna靶向cnksr2,cnksr2是参与ras依赖的信号传导的支架蛋白,其是用于控制肺癌中的emt的非显而易见的候选。我们使用常规crispr/cas9技术验证了筛选的结果,并且两个独立的克隆cnksr2ko克隆示出了与亲本对照相比增强的上皮特征,并且与预期为ezh2功能丧失表型所需的arid1ako相似(图4f)。先前已示出ras驱动的emt通过hippo通路发生43。我们通过使用潜在的slcr生成的数据揭示了可能通过ras/mapk依赖的信号传导直接促进emt的额外的机制。综上所述,用三种不同癌症类型中的上皮-间充质转化获得的结果强调了我们的slcr揭示肿瘤这种内稳态的组织非依赖性能力。实施例6:mgt#1作为体内肿瘤内稳态的遗传追踪报道子在证明了slcr在剖析离体细胞和分子状态中的实用性之后,我们接下来希望测试mgt#1作为体内肿瘤内稳态的遗传追踪报告子的作用。我们将mes-hgics-mgt#1微弱细胞颅内移植到nsg小鼠中并且纵向监测肿瘤形成。在高级别疾病阶段的神经体征发作时,我们处死动物并进行组织化学和免疫组织化学以及内源性和表面标志物分析。从组织结构上,所有肿瘤均显示为ⅳ级gbm,小鼠脑的大部分被恶性细胞浸润,表明广泛的增殖和侵袭(图5a)。对于每只动物(n=10),我们使用成像引导的肿瘤切除术来生成单细胞制备(prep),同时保留浸润的脑组织。免疫组织化学染色揭示,表达mgt#1的细胞非随机分布在肿瘤块中,而很好地局限在侵袭前端(图5a-b)。鉴于对病毒的响应,染色质修饰和基因沉默都可能潜在地影响slcr表达,为了证实mgt#1反映功能性肿瘤内异质性并排除表达mgt#1的细胞仅仅是逃脱者,我们使用了两种方法。首先,我们检查了其中对于其他标志物以及mgt#1独立的h2b-cfp的表达不存在mesgbm信号传导的所有密集区域。我们证实了借助于微管蛋白染色在免疫染色中绝大多数被染色的肿瘤组织对于抗原是可接近的,并且我们证实了其中可以通过染色质浓缩推断出活性增殖的几个mgt#1“深色”细胞确实是h2b-cfp阳性的(图5c-d)。其次,我们通过流式细胞术进行了平行的体外/体外表面标志物和内源性分析。与免疫组织化学染色一致,内源性mvenus荧光表达在体内示出显著的异质性水平。与体外增殖的mes-hgics-mgt#1微弱细胞相比,异种移植物衍生的肿瘤细胞示出明亮mes-hgics-mgt#1细胞的少量群体,而绝大多数肿瘤细胞转变为mgt#1低或暗状态(图5e)。细胞表面受体cd133,其通常用于标记患者来源的异种移植物中的肿瘤增殖细胞,示出了从体外总体cd133高群体至低或阴性状态的相似的转变。值得注意的是,表达cd133的细胞包括相当部分的表达mgt#1和未表达的细胞,从而支持mgt#1描述功能异质性的能力(图5e)。总体而言,我们的实验强调了slcr阐明肿瘤内异质性的能力(图5f)。进一步的实验以证明本发明的可行性和实现:实施例7:合成基因座控制区(slcr)的进一步表征slcr被设计为模拟内源性cre,诸如α-珠蛋白lcr,其示出位置无关的细胞类型和发育阶段特异性表达并接合转录因子。这些元件通常被定义为超级增强子,并浓缩成辅激活物点(puncta)。为了测试slcr是否与内源性lcr共享特征,我们通过rna-fish测量了mgt#1转导的细胞中新生的rna,并使用if搜索了brd4或med1浓缩物。双重if和rna-fish在固定的表达mgt#1的肿瘤细胞中鉴定了brd4或med1与mgt#1的新生rna之间的共定位(图1g)。此外,可诱导的mgt#1驱动的mvenus和“管家”pgk驱动的h2b-cfpmrna两者均存在于肿瘤细胞细胞质中,但在细胞核中仅可检测到mvenus(图16),表明了两种cre的差异强度。接下来,我们将原神经(pngt#1-2)和间充质(mgt#1-2)slcr慢病毒颗粒转导至自发永生化人神经祖细胞中,其获得了pdgfra、c-myc和cdk4的高拷贝数。为了重现常见的pn和mesgbm遗传背景,我们进一步工程化hgic,以使其缺失pten并且带有idh1r132和tp53r273h点突变或者进一步缺失tp53和nf1,从而分别生成pn-hgic和mes-hgic。这些细胞示出与gbm患者相似的dna甲基化谱,并在体内获得亚型特异性基因表达,并且因此代表了两种不同的gbm亚型。在体外生长因子定义的条件下,pngt#1-2在两种细胞类型中均示出强表达,而mgt#1-2在两种基因型中均显示整体低表达,从而强调了针对不同调控网络的设计特异性。值得注意的是,与pn-hgic相比,mgt#1在mes-hgic中具有更高的基础表达,表明了基因型特异性响应(图1h)。因此,我们设计了在保留内源性cre的关键特征的同时系统地生成反映给定细胞标识的合成lcr的方法。实施例8:支持通过slcr的功能性报道子活性的额外证据为了调查对mes-hgics-mgt#1低和pn-hgics-mgt#1低细胞中外部信号传导的适应性响应,我们接下来进行了表型筛选。用选定的因子(细胞因子、生长因子、化合物)刺激nbe增殖的hgic,并在刺激后48小时分析facs(图13b)。相对于初始hgic归一化,slcr揭示了在mes-和pn-hgics-mgt#1低中的共享的和专有的响应,并突出了tnfα信号传导以及人血清或fbs和激活素a作为mes-gbm调控子。该结果在两个独立的mes-gbmslcr(mgt#1-2)之间可重现并跟进验证。相反,pn表型似乎对由外部信号传导诱导的变化响应更低。(图13b-c和17)。mesgbm特化似乎是对先前存在的内源性表型的补充,如通过cd133和pngt#2的表面表达所测量的。确实,先前曾报道过tnfα是重要的mes-gbm信号传导通路,并且是pn-至-mes转变的诱导剂。此外,发现nfkb(已知的tnf诱导的tf)在tnfα刺激下接合mgt#1slcr中包括的至少两种cre(图9b)。facs分选的pn-hgics-mgt#1低具有与mes-hgics-mgt#1低可比较的mgt#1表达水平,但仍未达到与tnfα相似的响应(图2g和8和13a)。一致地,尽管在相同的信号传导条件下增殖,但mes-hgics-mgt#1低和pn-hgics-mgt#1低细胞在所选信号传导通路的激活和内源性表达上显示出差异(图2)。tnfα刺激在两种细胞类型中均诱导了nfkb-p65、stat3和p38-mapk的磷酸化,但这导致了明显不同的基因表达输出(图2d)。这些分析表明,尽管tnfα驱动mes-hgic中的mesgbm标签,但pn-hgic致力于类似初始mes-hgic的状态的状态(图2e-f)。总体而言,我们的结果表明slcrmgt#1-2反映了内源性间充质gbm基因表达程序,同时捕获了信号传导通路(例如tnfα)的激活状态和任何先前存在的背景相关差异(例如mes与pn背景)。促分化信号传导(即人血清或fbs)驱动报道子激活的观察结果与示出mes-gbm标签可能归因于fbs培养的星形胶质细胞而不是任何小鼠脑细胞的先前发现一致。值得注意的是,洗出(washout)实验表明mes-gbm状态在几天的时间框内是可逆的(图18),表明mesgbm状态可以被获取并反向。gbm中的间充质转分化被发现为标准护理后复发的显性事件,并且经由tnf驱动的nfkb活化与获得的辐射抗性相联系。大量相关证据支持炎症信号传导、emt、先天免疫细胞浸润和辐射抗性之间的联系。为了实验测试辐照是否可以以细胞自主方式诱导间充质转分化,将mes-hgics-mgt#1低和pn-hgics-mgt#1低细胞单独或与tnfα组合暴露于电离辐射(ir)。无论是单剂量还是分次剂量,mgt#1激活均示出对ir升高的剂量响应(图2g和19)。mes-hgics-mgt#1低和pn-hgics-mgt#1低细胞两者与tnfα组合均示出增强的间充质转分化。单次10gy辐射剂量在多种人gsc中是亚致死的。同样,无论单独还是与其他处理(例如tnfα或替莫唑胺)组合,我们的gic都保持健康,并在辐照后二十四小时显示残留的dna损伤标志物γh2ax磷酸化,从而证实已经发生了双链断裂并且正在修复中(图2h)。经典nfkb激活可以发生在下游tnfα信号传导以及通过非经典基因毒性应激。为了对nfkb在固有的和获得的mes-gbm状态中的重要性提供实验支持,我们在mes-hgic中使用crispr/cas9删除了p65/rela,这导致固有的mgt#1表达显著下调(图13c)。值得注意的是,虽然tnfα在多克隆和单克隆relako细胞中诱导mes-gbm信号传导的能力显著受损,但ikb激酶(ikk)抑制剂-16进一步限制了对tnfα的适应性响应。在单克隆relakogic中,我们排除了由于relako逃脱者而发生的补偿,这表明relako细胞中的其他nfkb转录因子可以转导tnf信号传导(图19b)。在患者中,gbm干细胞状态在维持肿瘤内稳态中是遗传库的主导。接下来,我们希望通过进行全基因组汇集的crispr/cas9筛选测试slcr是否可用于发现调控mesgbm状态的基因。在mes-hgics-mgt#1低中的遗传筛选以其初始状态进行或者当mes-gbm状态被外部信号传导或基因毒性应激诱导时进行(即分别是fbs+tnfα或tnz+ir;图13d)。在73,179个grna中,表型筛选分别返回了与mgt#1高和低部分有关的333和1,164个grna(图13e)。文库和处理对mgt#1表达的影响,与适合度相关但非对照的基因的平均统计缺失,以及靶向rela的两个sgrna在初始状态下的缺失(图20a-d),都表明该筛选可以发现功能基因。有趣地,一些临床相关的药物靶标诸如parp1和eed,似乎在所有条件下是mgt#1激活的关键调控子,但对于增殖不是必需的。据报道,parp1活性是ir诱导的nf-kb激活所必需的,并且在其他情况下,polycomb阻遏因子复合物2支架eed抑制促进emt。为了测试这种方法是否可用于优先考虑导致细胞命运改变的药物治疗,我们搜索了命中的上游调控子。其中,一些grna与靶标下游rar/rxr激动剂和mek1抑制剂先前相关,分别具有mgt#1-低和-高部分富集的统计趋势(图13e和20)。为了验证两种药物都可能对细胞命运决定有影响的预测,我们将mes-hgics-mgt#1暴露于mek1选择抑制剂tak-733或全反式维甲酸(atra)。在两种情况下,与单独的tnfα相比,mes-hgics-mgt#1对短期tnfα刺激(4小时)的响应具有mgt#1和mes-gbm内源性标志物两者的更高的上调(图13f),表明预处理使这些细胞对mesgbm程序激活敏感。atra和tak-733使mgt#1敏感高于eed/ezh2抑制剂gsk126的效果,从而支持治疗的特异性。因此,相比于仅基于适合度的先前大量研究,slcr提供了药物基因组学信息的表型层。总体而言,这些结果为间充质gbm是短暂且可逆的细胞状态提供了实验证据,并且支持设计的slcr在表型筛选应用中的稳健性和有效性。实施例9:slcr能够区分分子上不同的实体原发性癌症类型可以基于其分子谱分组。染色质可及性是癌症类型相似性的最强预测因子,并且可用于鉴定个体癌症类型的常见维度空间内的亚型标识。为了研究由slcr描述的所获得的异质性是否伴随着全基因组染色质可及性的变化,我们在体外和体内对mes-hgics-mgt#1高细胞进行了atac-seq。染色质可及性的差异分析揭示了许多正在经历重构的基因,尤其是在pn-至-mes转变wwtr1(taz)的驱动子处和几个tnf受体基因位点处,表明重构事件的遗传追踪仅发生在生理相关的肿瘤微环境中(图14a-b)。来自tcga和胶质瘤干细胞的atac-seq数据的集成进一步揭示了mes-hgics-mgt#1高细胞代表了普通胶质瘤空间内的特异性实体(图14c)。重要地,gic的无监督染色质谱分析,由mgt#1高和低表达划分,将那些样本分组为定义的簇(图14d),表明mgt#1表达强调了染色质可及性中独特模式的获得。这些结果突显了slcr在揭示肿瘤内异质性中的效力,并能够与主要的癌症数据一起对肿瘤模型进行深入的细胞和分子表征。实施例10:slcr有助于发现肿瘤和免疫细胞之间的非细胞自主性串扰的治疗意义通过胶质母细胞瘤相关的小胶质细胞/单核细胞(gam)的idh1野生型gbm浸润最近已与nf1缺陷和mes-gbm亚型标识相关联,但gam和mes-gbm之间是否存在因果关系仍未解决。为了实验性地测试先天免疫细胞是原因而不是由nf1缺陷型gbm细胞中的mes转分化募集的这一假设,我们进行了idh1野生型和nf1缺失的mes-hgics-mgt#1低细胞与永生化人小胶质细胞系(hmg;cl.c20)的体外共培养。首先,我们比较了在gbm肿瘤球体和多细胞类器官培养条件下由单细胞的pn-和mes-slcr表达两者的表达。球状体培养支持具有受限的自发分化和细胞死亡的干细胞和祖细胞的扩增50,51,而胶质瘤类器官使表型多样化的细胞群体增加。类似于体内表达模式(图14a),我们发现mes-hgic在类器官条件下和在人小胶质细胞的存在下显示出异质性pn-和mes-slcr表达模式,与它们在纯球状体培养物中的同质性表达相反(图15a)。接下来,我们使用穿透小室插入物在同质gbm肿瘤球体和hmg细胞之间建立共培养。令人惊讶地,hmg细胞在mes-hgic中驱动mgt#1诱导的程度与tnfα相当(图15b-c和21)。与先前的实验一致,hmg也在pn-hgic中激活mgt#1至更低程度。相比之下,在体外,由人cd34+衍生的髓系来源的抑制细胞(mdsc)仅在两种细胞系中轻度刺激mgt#1表达(图21)。来自两种条件下的mes-hgics-mgt#1高细胞的全转录组分析揭示了共同和专有的nfκb相关基因激活,并提供了证据表明适应性免疫细胞驱动特异性mes-gbm状态,其在很大程度上与患者的标签共享靶标(图15d)。有趣地,我们没有发现由任一细胞类型表达tnfα的证据。而是,以胆固醇生物合成途径中的基因为特征的代谢转录组重构似乎构成了针对与hmg细胞共培养特异性的mes-hgic标签(图15e-g)。这些数据表明肿瘤细胞中nfkb的激活主要归因于先天免疫细胞。实际上,源自适应性免疫系统的炎症介质ifnγ和il-2,以及基质衍生的il-6没有在相当程度上触发直接的mgt#1激活(图17),共同提供了对导致体内mes-gbm状态的级联事件的实验见解。emt与化疗的抗性有关,但也提供了治疗机会。dna损伤应激是gbm中护理标准的主要治疗组分,也称为stupp方案。gbm中的tnf-nfkb标签先前与大量患者和pdx模型中的间充质状态和辐射抗性有关。因此,我们接下来利用slcr鉴定mes内稳态的能力,以探索小胶质细胞驱动的gbm状态的治疗意义。为此,我们在hmg驱动的转化后,facs分选mgt#1-2高和mgt#1-2低mes-和pn-hgic细胞,并将这些细胞暴露于一组选定的标准和靶向化疗剂。令人惊讶地,与它们的slcr低对应物相反,mes-hgics-mgt#1高或-mgt#2高细胞两者均证明对基于dna损伤的治疗剂(奥拉帕利、atr抑制剂ve-821、拓泊替康、丝裂霉素c)和lxr623(调控胆固醇外排的lxr激动剂)具有更高的抗性。(图15h和21)。重要地,mes-hgics-mgt#1高细胞保留了与靶向的剂诸如bay11-7085(iκb)、wp1066(stat3;图15h和21)的相似的敏感性谱。mes-hgics-mgt#1高的改变的化学敏感性谱与由hmg细胞驱动的基因表达变化一致,包括在mes-hgics-mgt#1高细胞中的dna损伤基因标签表达受损,细胞周期谱与患者来源的mes-gbm的过表达和胆固醇生物合成标签一起变换(图21)。用原神经基因型获得了相似的结果,表明hmg细胞可以将hgics转变为两种功能和治疗上不同的状态,并且支持slcr在靶发现平台中的使用,以整合与肿瘤异质性相关的复杂响应。总体而言,我们的结果偶然地将先天免疫细胞与mes-gbm状态相关联,并突出了slcr在体内和离体中机械剖析相关非细胞自主相互作用的潜力。本发明的进一步优点和实现:目前,我们对生物水平上复杂的细胞和分子机制的理解主要依赖于体内实验,并且受到可用的遗传追踪技术的限制。我们已经建立了允许生成能够拦截细胞固有和非细胞自主信号传导的合成报道子的系统生物学框架。这些slcr可用于说明体外和体内基因型-至-分子和细胞表型转变。在实验上,将生物、化学和环境刺激关联至细胞命运转变,包括通过化学和正向遗传筛选,slcr可用于表征分子机制。我们已应用这种方法来研究gbm亚型表达谱的细胞和分子特征。在表达平台(微阵列、rna-seq)、读出(基因表达、dna甲基化)和患者群体(西方和中国)中,原神经和间充质gbm亚型的鉴定是一致的。尽管如此广泛的努力,但当涉及到其起源、位置或时空演变时,gbm亚型的意义仍然是难以捉摸的。通过组合近等基因模型和messlcr,我们示出了mes-gbm特化的最重要组件本质上是适应性的。尽管以mes-hgic为例的基因型指导的固有mes信号传导当与pn-hgic相比时示出了在messlcr的表达中可测量的但中等的差异,但tnf信号传导以及促分化刺激(例如fbs)是mes信号传导的主要触发因子。有趣地,tnfα和fbs两者都通过差异影响细胞形态来触发mes转分化。如通过mgt#1表达的异质性程度以及未分化和自我更新的肿瘤细胞的标志物推断的,这两种响应似乎都刻印在体内。我们的实验将gbm细胞中的mgt#1读出关联至与迁移相关的标志物诸如cd44的表达,对促炎性微环境的反应以及对亚致死剂量的遗传毒性应激的抗性,所有这些都代表了肿瘤进展的标志,包括在单细胞水平上的gbm中18。这些发现说明了mgt#1阐明gbm中细胞和分子机制的能力。这项技术能够将细胞和分子谱分析转化成表型图,其可以满足与健康和疾病中的细胞和分子特征(包括单细胞水平)的连续映射相关的实验需求。实际上,slcr改进了体内表型测定,其仍然代表全面理解生物水平上的复杂细胞和分子机制所必不可少的步骤。这样,它提供了重要的离体机会。我们示出,反映体内调控网络的slcr准确地拦截了细胞固有的和非细胞自主的信号传导,并且成功地应用于剖析体内和体外基因型-至-分子和细胞表型转变。我们通过研究gbm亚型表达谱的细胞和分子基础证明了该系统的实用性。在表达平台(微阵列、rna-seq和单细胞rna-seq)、读出(基因表达、dna甲基化)和患者种族(西方和中国)中,原神经和间充质gbm亚型的鉴定是一致的。尽管这样广泛的努力,但当涉及其起源、位置或时空演变以及更重要地其治疗意义时,gbm亚型的意义仍是难以捉摸的。原神经和间充质gbm程序依赖于特异性转录因子的活性。在这里,我们整合了近等基因模型和细胞系与slcr,并且结果与是默认gbm实体的pn-gbm一致,其强烈依赖于rtk信号传导,并因此受到神经干细胞培养条件的促进。取而代之,我们示出了mes-gbm特化的最重要组件本质上是适应性的。在不存在肿瘤微环境下,即使在具有meg-gbm基因型(例如nf1缺失)的细胞中,pn状态似乎是硬线连接的,但mes标识通过急性炎症和促分化刺激(例如tnf信号传导以及牛或人血清)而迅速扩增。有趣地,在不同的细胞类型中,通过slcrs测量的mes转分化可能与差异地影响细胞形态一起发生。我们的实验将gbm细胞中的mes-slcr读出、对促炎症微环境的前馈响应、对亚致死剂量的基因毒性应激的抗性以及迁移相关的标志物诸如cd44的表达相关联,所有这些都代表了人癌症进展的标志,包括在单细胞水平的gbm中。这些特征似乎刻印在组织内稳态中,如通过体内和离体肿瘤模型中的聚类细胞表达模式(“内稳态”)和异质性推断的。三种不同癌症类型中mes-gbm主要组件的遗传追踪强调了我们slcr揭示肿瘤内稳态的独立于组织的能力,并提供了emt代表了劫持发育细胞过程的进一步证据。这些发现说明了slcr在阐明多因素疾病中的细胞和分子机制中的多功能性。进一步的,在药物基因组学中使用slcr可以通过揭示表型特异性依赖性和抗性来显著促进转化医学。最终,slcr能够机械剖析先天免疫细胞和肿瘤细胞之间的病理生理相关的非细胞自主相互作用。gam被认为构成胶质瘤小鼠模型和人肿瘤两者中tnfα的来源。我们的结果为mes-gbm亚型与特异性免疫状况之间的临床关联提供了实验支持,并且揭示了对mes-gbm的tnfα非依赖性路径。重要地,本文鉴定的gam驱动的mes-gbm状态示出与患者标签有一定程度的重叠,其与个体患者标签本身的重叠相当。总之,通过将生物、化学和环境刺激关联至细胞命运转变,包括通过化学和遗传筛选,已证明slcr能够用于表征分子机制。先前使用大规模平行测序或混合模型生成合成报道子的尝试揭示了这种方法的潜在用途和与对设计的有限控制相关的局限性。我们的方法实质上解决了这个问题,并且代表了未来发展的基础,从对基本设计组件的线性改进(例如,使用tfbs和顺式元件的策划资源)至系统生成和验证大量slcr,随后是机器学习成功的特征。。同时,可以通过组合slcr与dna条形码来延伸稳健的细胞类型或状态特异性和粒度。可以通过将slcr转录输入与能够进行布尔(boolean)逻辑输出的合成效应蛋白偶联来实现可调操作。因此,通过slcr的遗传追踪是可扩展的,并且可以延伸到几乎任何给定系统,无论是离体还是体内,以剖析控制正常和疾病内稳态的细胞固有和非细胞自主机制。参考文献1.kretzschmar,k.&watt,f.m.lineagetracing.cell148,33–45(2012).2.barker,n.etal.identificationofstemcellsinsmallintestineandcolonbymarkergenelgr5.nature449,1003–1007(2007).3.barker,n.,tan,s.&clevers,h.lgrproteinsinepithelialstemcellbiology.development140,2484–2494(2013).4.livet,j.etal.transgenicstrategiesforcombinatorialexpressionoffluorescentproteinsinthenervoussystem.nature450,56–62(2007).5.liu,c.etal.mosaicanalysiswithdoublemarkersrevealstumorcelloforigininglioma.cell146,209–221(2011).6.schwitalla,s.etal.intestinaltumorigenesisinitiatedbydedifferentiationandacquisitionofstem-cell-likeproperties.cell(2012).doi:10.1016/j.cell.2012.12.0127.schepers,a.g.etal.lineagetracingrevealslgr5+stemcellactivityinmouseintestinaladenomas.337,730–735(2012).8.driessens,g.,beck,b.,caauwe,a.,simons,b.d.&blanpain,c.definingthemodeoftumourgrowthbyclonalanalysis.nature(2012).doi:10.1038/nature113449.oshimori,n.&fuchs,e.paracrinetgf-βsignalingcounterbalancesbmp-mediatedrepressioninhairfolliclestemcellactivation.cellstemcell10,63–75(2012).10.chen,j.etal.arestrictedcellpopulationpropagatesglioblastomagrowthafterchemotherapy.nature(2012).doi:10.1038/nature1128711.zhu,l.etal.multi-organmappingofcancerrisk.cell166,1132–1146.e7(2016).12.church,g.m.,elowitz,m.b.,smolke,c.d.,voigt,c.a.&weiss,r.realizingthepotentialofsyntheticbiology.natrevmolcellbiol15,289–294(2014).13.stupp,r.etal.effectsofradiotherapywithconcomitantandadjuvanttemozolomideversusradiotherapyaloneonsurvivalinglioblastomainarandomisedphaseiiistudy:5-yearanalysisoftheeortc-ncictrial.lancetoncol.10,459–466(2009).14.wang,q.etal.tumorevolutionofglioma-intrinsicgeneexpressionsubtypesassociateswithimmunologicalchangesinthemicroenvironment.cancercell32,42–56.e6(2017).15.noushmehr,h.etal.identificationofacpgislandmethylatorphenotypethatdefinesadistinctsubgroupofglioma.cancercell17,510–522(2010).16.verhaak,r.g.w.etal.integratedgenomicanalysisidentifiesclinicallyrelevantsubtypesofglioblastomacharacterizedbyabnormalitiesinpdgfra,idh1,egfr,andnf1.cancercell17,98–110(2010).17.sottoriva,a.etal.intratumorheterogeneityinhumanglioblastomareflectscancerevolutionarydynamics.procnatlacadsciusa110,4009–4014(2013).18.lee,j.-k.etal.spatiotemporalgenomicarchitectureinformsprecisiononcologyinglioblastoma.naturegenetics49,594–599(2017).19.phillips,h.s.etal.molecularsubclassesofhigh-gradegliomapredictprognosis,delineateapatternofdiseaseprogression,andresemblestagesinneurogenesis.cancercell9,157–173(2006).20.bhat,k.p.etal.mesenchymaldifferentiationmediatedbynf-κbpromotesradiationresistanceinglioblastoma.cancercell24,331–346(2013).21.encodeprojectconsortiumetal.identificationandanalysisoffunctionalelementsin1%ofthehumangenomebytheencodepilotproject.nature447,799–816(2007).22.thurman,r.e.,day,n.,noble,w.s.&stamatoyannopoulos,j.a.identificationofhigher-orderfunctionaldomainsinthehumanencoderegions.genomeres17,917–927(2007).23.kim,t.h.etal.analysisofthevertebrateinsulatorproteinctcf-bindingsitesinthehumangenome.cell128,1231–1245(2007).24.ong,c.-t.&corces,v.g.ctcf:anarchitecturalproteinbridginggenometopologyandfunction.natrevgenet15,234–246(2014).25.lee,j.etal.tumorstemcellsderivedfromglioblastomasculturedinbfgfandegfmorecloselymirrorthephenotypeandgenotypeofprimarytumorsthandoserum-culturedcelllines.cancercell9,391–403(2006).26.ozawa,t.etal.mosthumannon-gcimpglioblastomasubtypesevolvefromacommonproneural-likeprecursorglioma.cancercell26,288–300(2014).27.quail,d.f.etal.thetumormicroenvironmentunderliesacquiredresistancetocsf-1rinhibitioningliomas.science352,aad3018(2016).28.szulzewsky,f.etal.humanglioblastoma-associatedmicroglia/monocytesexpressadistinctrnaprofilecomparedtohumancontrolandmurinesamples.glia64,1416–1436(2016).29.adzaye,o.d.etal.gliomastemcellsbutnotbulkgliomacellsupregulateil-6secretioninmicroglia/brainmacrophagesviatoll-likereceptor4signaling.j.neuropathol.exp.neurol.75,429–440(2016).30.inda,m.-d.-m.etal.tumorheterogeneityisanactiveprocessmaintainedbyamutantegfr-inducedcytokinecircuitinglioblastoma.genesdev24,1731–1745(2010).31.hossain,a.etal.mesenchymalstemcellsisolatedfromhumangliomasincreaseproliferationandmaintainstemnessofgliomastemcellsthroughtheil-6/gp130/stat3pathway.stemcells33,2400–2415(2015).32.midwood,k.etal.tenascin-cisanendogenousactivatoroftoll-likereceptor4thatisessentialformaintaininginflammationinarthriticjointdisease.natmed15,774–780(2009).33.jachetti,e.etal.tenascin-cprotectscancerstem-likecellsfromimmunesurveillancebyarrestingt-cellactivation.cancerres75,2095–2108(2015).34.stanzani,e.etal.radioresistanceofmesenchymalglioblastomainitiatingcellscorrelateswithpatientoutcomeandisassociatedwithactivationofinflammatoryprogram.oncotarget8,73640–73653(2017).35.bao,s.etal.gliomastemcellspromoteradioresistancebypreferentialactivationofthednadamageresponse.nature444,756–760(2006).36.hinz,m.etal.acytoplasmicatm-traf6-ciap1modulelinksnucleardnadamagesignalingtoubiquitin-mediatednf-κbactivation.molcell40,63–74(2010).37.lei,l.etal.glioblastomamodelsrevealtheconnectionbetweenadultglialprogenitorsandtheproneuralphenotype.plosone6,e20041(2011).38.rheinbay,e.etal.anaberranttranscriptionfactornetworkessentialforwntsignalingandstemcellmaintenanceinglioblastoma.cellrep(2013).doi:10.1016/j.celrep.2013.04.02139.kalluri,r.&weinberg,r.a.thebasicsofepithelial-mesenchymaltransition.journalofclinicalinvestigation119,1420–1428(2009).40.baird,r.d.&caldas,c.geneticheterogeneityinbreastcancer:theroadtopersonalizedmedicine?bmcmed11,151(2013).41.serresi,m.etal.polycombrepressivecomplex2isabarriertokras-driveninflammationandepithelial-mesenchymaltransitioninnon-small-celllungcancer.cancercell29,17–31(2016).42.wamsley,j.j.etal.activinupregulationbynf-κbisrequiredtomaintainmesenchymalfeaturesofcancerstem-likecellsinnon-smallcelllungcancer.cancerres75,426–435(2015).43.shao,d.d.etal.krasandyap1convergetoregulateemtandtumorsurvival.cell158,171–184(2014).44.ohinata,y.,sano,m.,shigeta,m.,yamanaka,k.&saitou,m.acomprehensive,non-invasivevisualizationofprimordialgermcelldevelopmentinmicebytheprdm1-mvenusanddppa3-ecfpdoubletransgenicreporter.reproduction136,503–514(2008).45.gargiulo,g.etal.invivornaiscreenforbmi1targetsidentifiestgf-β/bmp-erstresspathwaysaskeyregulatorsofneural-andmalignantglioma-stemcellhomeostasis.cancercell23,660–676(2013).46.gargiulo,g.,serresi,m.,cesaroni,m.,hulsman,d.&vanlohuizen,m.invivoshrnascreensinsolidtumors.natprotoc9,2880–2902(2014).47.li,p.,markson,j.s.,wang,s.,chen,s.,vachharajani,v.,andelowitz,m.b.(2018).morphogengradientreconstitutionrevealshedgehogpathwaydesignprinciples.science360,543–548.48.blankvoort,s.,witter,m.p.,noonan,j.,cotney,j.,andkentros,c.(2018).markeddiversityofuniquecorticalenhancersenablesneuron-specifictoolsbyenhancer-drivengeneexpression.currbiol28,2103–2114.e2105.49.takahashi,k.,andyamanaka,s.(2006).inductionofpluripotentstemcellsfrommouseembryonicandadultfibroblastculturesbydefinedfactors.cell126,663–676.50.suvà,m.-l.,rheinbay,e.,gillespie,s.m.,patel,a.p.,wakimoto,h.,rabkin,s.d.,riggi,n.,chi,a.s.,cahill,d.p.,nahed,b.v.,etal.(2014).reconstructingandreprogrammingthetumor-propagatingpotentialofglioblastomastem-likecells.cell51.frith,m.c.,fu,y.,yu,l.,chen,j.-f.,hansen,u.,andweng,z.(2004).detectionoffunctionaldnamotifsviastatisticalover-representation.nucleicacidsres32,1372–1381.52.phillips,h.s.,kharbanda,s.,chen,r.,forrest,w.f.,soriano,r.h.,wu,t.d.,misra,a.,nigro,j.m.,colman,h.,soroceanu,l.,etal.(2006).molecularsubclassesofhigh-gradegliomapredictprognosis,delineateapatternofdiseaseprogression,andresemblestagesinneurogenesis.cancercell9,157–173.53.verhaak,r.g.w.,hoadley,k.a.,purdom,e.,wang,v.,qi,y.,wilkerson,m.d.,miller,c.r.,ding,l.,golub,t.r.,mesirov,j.p.,etal.(2010).integratedgenomicanalysisidentifiesclinicallyrelevantsubtypesofglioblastomacharacterizedbyabnormalitiesinpdgfra,idh1,egfr,andnf1.cancercell17,98–110.54.sturm,d.,witt,h.,hovestadt,v.,khuong-quang,d.-a.,jones,d.t.w.,konermann,c.,pfaff,e.,m.,sill,m.,bender,s.,etal.(2012).hotspotmutationsinh3f3aandidh1definedistinctepigeneticandbiologicalsubgroupsofglioblastoma.cancercell22,425–437。序列表<110>马克思-德布鲁克-分子医学中心亥姆霍兹联合会(max-delbrück-centrumfürmolekularemedizininderhelmholtz-gemeinschaft)<120>用于工程合成顺式调控dna的方法<130>ppi21170396de<150>ep18192715.3<151>2018-09-05<160>6<170>patentinversion3.5<210>1<211>827<212>dna<213>人工序列<220><223>slcr<400>1atatttatttttaggaccagaaagttaaagtgaattggatttgatccattttctgaaagg60ctggcaagaattcttgacattgcacaggaatttccatgtcagcatgttctcacatgtatg120atctaatttagagattattttggggggcgggggttgaggaaatggcatgactcagagttt180aaaagccccaaatcttagctgtgcctgtgtagctttaccacataacccattgataactta240gttgtgcaaccatcaccaccatctgttttcagaactcttttcattttgcgaaactgaaac300ccgttaagcactgatttcccactctccctcctcccagcccatagcaaaccaccatcccac360cagcactttcatttcgcaaatggcaaaactgaagccgatattgtggttgtgacttatccc420aaagtaatatacacataaacctctatggatgaggaaaaagacagagggaaactaaaaatt480caaaagaacaaatttgactcacagatttgctgactcatagttgtgacacttcctggctca540ggaagttgaatttcattaagcctttgtggtttggggctctgctgtgctttgacagctctg600atctcctcccttccggctgggctgtctggggcgctctaaaatgagtgttgatttaatgca660ctgccttcgcacccgtgctggtgcgtcccggggacaggggtggctgtgcggtgccgcggc720ggccggcggggctccttccccagcaggggtggggacgctgagtcacggatctgtcaccgc780tttgcacctctccgagccctcgggggccaaagcaaaagcgaaagcga827<210>2<211>1015<212>dna<213>人工序列<220><223>slcr<400>2ctagaacagcagggccacctccttctctcccccgcgggcatgggcccccacccccactgc60cggcagagtgctgaggactcgtgcaccatgagaacttctgaccatgagaactttgacttc120cggatttgggggatctgcccaggtgaacacaatgcaaggggctgcatgacctaccaggac180agaactttccccaattacagggtgactcacagccgcattggtgactcacttcaatgtgtc240atttccggctgctgtgtgtgagcagtggacacgtgaggggggggtgggtgagagagacag300gccacattgtgcaacagatctctagagctttttcatcttgcaaaactgaaactgtatacc360catggaacaacagctccctgctcccctccccctcagctcctgggtagtgacatttcttga420ttctcagtaaactatcacaagaacaaaaaaccaaacaccgcatattctcactcataggtg480ggaattgaacaatgagatcacatggacacaggaaggggaatatcacactctggggactgt540ggtggggtggggggagtggggagggatagcactgggatgtcccaagagaaggggaagagg600gggaggtgttagagaacttgtgtgttcaaccgaaacatgatgaaaacagggaaagccccc660aagatacctgtcattcccgatgatgtcagattcagcaaattcaatgataacaaaacatta720tgaaaaaattagtaattaaaataatacagcaatgtgtatgaacaaaataatcaatgaaag780tgaaacctaatagtaattccacaaacttattacaaagctattaatttaaagagtagtggc840aattgaaaaccacaaccaacaccagtgcttacagcagcaatacttttactcagacttcct900gtttctggaacttgccttcttttttgctgtgtttatacttcccttgtctgtggttagata960agtataaagccctagatctaagcttctctgtcttcctccctccctcccttcctct1015<210>3<211>1050<212>dna<213>人工序列<220><223>slcr<400>3agagctcctggccaaggtctttgtgttcagaccagaagaggaaggagggctccctccccc60tggggctgtggaggctgaggctcctggggggttgtccacatctggaccgtgggagctgtt120ggggggaatgggggcaggtggagaagaggataagcagctgattgggcccagactactctg180ggctggctccatcttacatgactgccacaaacagctgcaggagtgtgacagatcacaaca240ctagcattgtacctcaaaatatgcttgtaccctaaggcacaagaactggtttgacttaca300accgcagcccccgtccgggcaccccgaggcccgcgggagccaccctcgaaccccggccgc360gcacgggcggggcggcgcgcacctgccgggagcccgtgtttgtaaacaaaccgcgcgcct420aattagcctggcgggagcgcgcgcgcggggcggggggcggggcgtcggtgcgcgcgggca480ggtcggccccgcccggggaggagccgcgctctgccgcgccctccgtgtcaccatctcccc540cacccgacttggcggggcgcgggcttgctggagcctgcgggacccagagcccgctccgga600gccagccctgggagtggccagcttgaacccgagggccccgcagaccgttactccggcccc660cgcccggggcggggcgcgcgggggcgcggcgcagcccaacccgcacagccgcgtccccaa720acaccaccgaggagggaaaacagacggagaggggtggggctgcgggcggggccggcgcct780aattgggccgcgggcgcctcgaggtgggcggggcataagggggcggggccgcggagaccc840cgggcgggagcagggagaggaaagaagagactgagtacgcggagaccgagattcggaaat900atttctgccttaattgttcttccattgtctttctcctgtgggtcccctctcacctttctg960tatggtcctggatcaccccccgaggctttgtctcccccatccacgggcttattctctcgg1020cacccccttcctctcccgtcatcggttgat1050<210>4<211>1164<212>dna<213>人工序列<220><223>slcr<400>4ttaattaatccctcctctaatccctccagcgggatcagggaggaggtgcgggacctgctg60ccccgggcttgcccccatcccggcctcacgcatgggcgcctgtctcagccctctcccagg120acgctgcaggtgtggctgggccagcgctaattagtgggccgcgcgggggccccgctgagc180ctttgacagaaaaggcggtagggaggtgggggcagggaggcgctccaccagccagaagtc240cggagcgcaacccaaagtactccatctcaaaagaaaaaaggcgggggcggtggggggggg300gggtgatttcagtacaaagcctacagacattataaaaatattaagatttttgttcgtttg360ttttttgtttttgagacagagtctcactgtcacccccaggctggagtctgtgccggcgcc420cgctgcttcgcatctgcgcgcccgcccggtgccgggccccgccctccgcctcagccccaa480gctcggcccgcgggcccggccacaggtgccccggcggccccgcctggcccgagggaagag540ggcagctgggaggggcccatgagagaaccaaaactgtgcccccaggcttggaaagaaatc600acatgtatggccagcaggaaggttccggaaggttccggaggacacctgcaggtgggactg660agaacaggggtctcggctgggagtggctgaggccatatgaggacctcgactgccacaaac720agctgcaggagtgtgacagatcacaacactagcattgtacctcaaaatatgcttgtaccc780taaggcacaagaactggtttgacttacaaaactgatctcagagttgggatcaaagttttt840ctaccactctactatgagccctcggccgggccccgccccgccagctccgcgcggctctgg900gctctctaggggtggggctgcgggcggggccggcgcctaattgggccgcgggcgcctcga960ggtgggcggggcataagggggcggggccgcggagaccccgggcgggagtttttttctgca1020agcgagagggggggtgttgttggtatcgccccctccttctcctccccccaggggtgaaag1080tgcaagaggaagtgcagccgctgccatctttcctccgctccgaacacacggagcccgggg1140ccgcacagccgccgctcctgtaca1164<210>5<211>1110<212>dna<213>人工序列<220><223>slcr<400>5atggtctcaatctcctgaccttgtgatccgcccacctcggcctcccaaagtgctgggatt60acaggtgtgagccaccacgcccagccgacagtcccttatctggttcatcttcgtacctct120aaaagtcagcatggatgctctattaatgatatatttatacatattagcaacaaacaattg180gaaactaaaactttaaaaagacattctcacacctgtaatcccagcatgttgggaggtcga240ggcaggcgaatcacgaggtcaggagttcgagaccagcctggccaacatggtgaaactctg300gaagaccgaaactattcagcaagaactaagaaccacaatgttaagggggtccattgttta360tttttttttctttagaggatgaaaaccaaaggtcaggtgatttaatttaaaattaacact420cttattttttgcccgcccgcctgcctgcctctttacaatttacagaatgtcttaaggtag480ttaagtttcaagtttttctttctcagtatcctaccttcatgcatcaaagtgggtggcctt540tatcccattaacggcaattacgtaagacagatgtccctagatgaaatcttacagttcttt600tagtcagaccccccaccccgccaccgccaccagacaccaccatcgctgtgtagtgtgggt660ttttattcgtgttcgtgtgtgtgtgtggacacattttccttttcggttgctctgtccttt720ggttcgtgctcgcctcgctttttccacactcctgctctctggctctctgtgtctctcgct780ctttcgaaaattttcctaagtccgggcgcgcgctccctccccttccgcccaccccagccc840ctcggcggcgcccgcgggagggggaggaggcctcgggggcgccgggcgacgcggtccggg900gggtggagcgttggcgtcgtgcgaggggtcgtcactggcgcggagacgccccctctcccc960cctcggctcagccgggctgctgcccgagcccggggggtggggggcgtctccccggcccgt1020cccgtccccggccgggcgcgggcggagggaccccctccccgggctcccggggggccgcct1080ccctccgccggctcccgccctcccagccgc1110<210>6<211>1112<212>dna<213>人工序列<220><223>slcr<400>6ttaattaagaatatctggctggccacgtgtttgtaaagaaaaaccaagacggccaggcga60ggtggctcacacctgtaatcccagcactttgggaggccgaggcgggcgctgcccttcggc120cttcaaggaggaattcctactgtttatgaagatcgggtttgggtttttggtttttttttt180ctttttcttttttccgtggtggtggtgggtgggcttttgttctttttgttttttctgtgg240tggtggtgggtgggctttatgaatataccatattttgcctattgtttttctatttatcag300gtggtgtcatttgagttgttttcaccctcttgtgactatgaataatgatactataaacaa360tcttatacagcatcagtgtcaaaaatcactaacattcctatacacagacgtgactaaact420tccagcttggggtcccgtggacctgcagccaggtgcagcaggtcacagggcaaggacacg480tgtcattggtgaccttcactattcagtgcccagatgctcagtgctctgtgcaggccacct540ggctggtctcaggtaccgctgctctgtctcgctcaccggccgggctatgttgattgtccc600ctcgcggcgcccggaagcgaccctcagtaaacaaagccgtgtgtgggcgcagccccagaa660gcctggggcgcgcagtccagcccaagagaggcgggggaggaatgttgtgaatgaaccccg720ggcccgccccgaaactccgcataaggcctgggccgcgggggtcctcccactctgattggc780ctctggcgccccgtgattgacagcgcccctcgctgtgcgctctggttgggtaaacaagaa840aagactggcatcgcagtcatcgagtgagcagcgaggcttggacacgggtctggcggcgca900gccaatggcgggggagggccgaggaggccgagggggggccaatagggacaggcggtgggg960gcgggacgacggcggagctaaagcggcggctgaagcagcttcattgttgtgaagagtctt1020aaaggggccgcatcaccctgccggcccggcgcgggtcgggggtgggtgcggtaggggtcc1080cggggcggccgagcgcagaggacggatgtaca1112当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1