苯并杂环化合物和含有其作为活性成分的用于预防或治疗癌症疾病的组合物
本发明涉及苯并杂环化合物和含有其作为活性成分的用于预防或治疗癌症疾病的组合物。
背景技术:
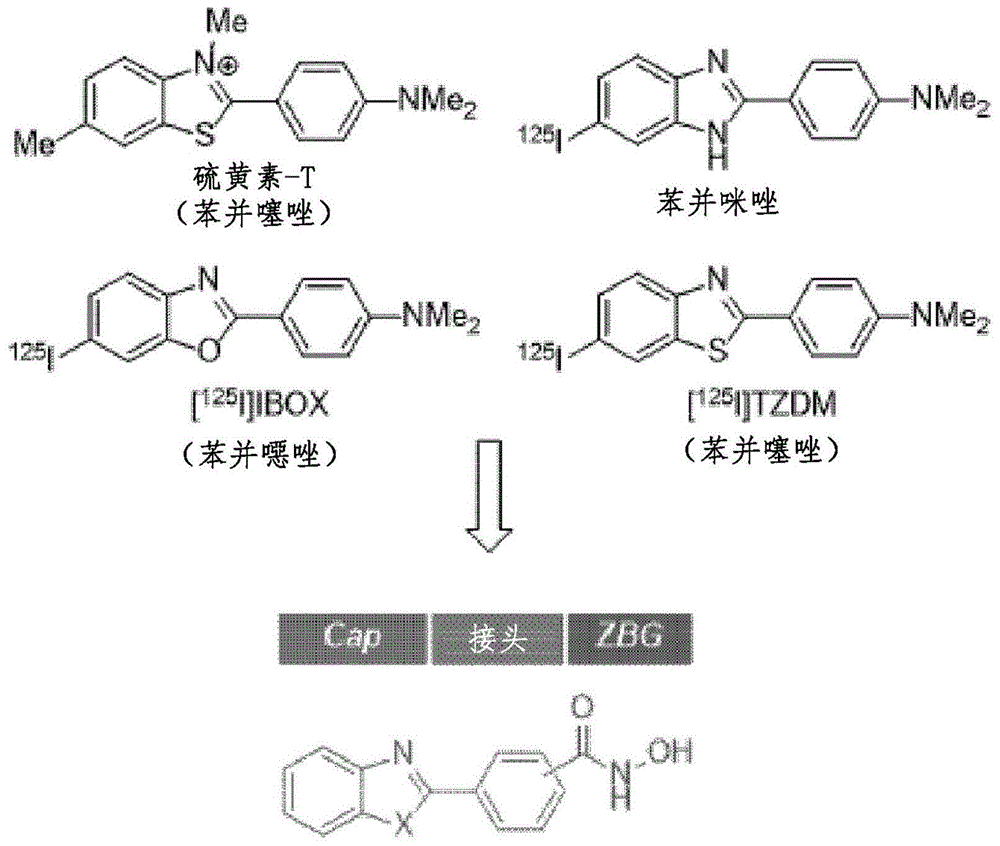
:脑瘤是指在颅骨中发生的任何肿瘤,并且包括在脑和脑周围结构中发生的所有肿瘤。根据脑的解剖学特性,在脑中形成没有发现癌细胞的肿瘤,也就是所谓的良性肿瘤,并且如果尺寸大,可能会因为脑压力升高而失去生命。这些中之一的脑癌是一种恶性脑瘤,是一种对脑造成巨大损害并具有极低的存活率的癌症。原发性脑癌不会转移到其它身体器官,但转移性脑癌由肺癌、乳腺癌和消化系统癌转移到脑引起。原发性脑癌包括神经瘤、星形细胞瘤、成神经细胞瘤、胶质瘤、脑膜瘤、少突神经胶质瘤、髓母细胞瘤、脊髓肿瘤和神经鞘瘤,但不限于此。脑癌的临床症状包括头痛、癫痫发作、呕吐、运动麻痹、重叠的对象(overlappingobjects)、视力下降、激素异常、听力丧失、头晕和言语障碍。作为脑癌的常规治疗方法,根据脑癌的类型或发生部位,通过手术切除最有效,但手术往往是不可能的,并且完全切除时的术后并发症的风险非常高。此外,由于存在抑制药物进入大脑中的血脑屏障(bbb),药物的吸收较低,因此与其它癌症相比,为了使用抗癌药物以化疗来治疗脑癌,需要给予高浓度的抗癌药物。结果,存在对身体的其它器官造成严重副作用的问题。技术实现要素:技术问题为了解决上述问题,本发明提供一种新的化合物或其药学上可接受的盐。本发明提供用于预防或治疗癌症疾病的药物组合物,所述药物组合物包含新的化合物或其药学上可接受的盐作为活性成分。此外,本发明提供用于预防或改善癌症疾病的健康功能食品组合物,所述健康功能食品组合物包含新的化合物或其药学上可接受的盐作为活性成分。技术方案本发明所述的新的化合物可以是由以下的化学式1表示的化合物或其药学上可接受的盐。[化学式1]在化学式1中,r选自o、s或nr1,且r1可选自氢、c1-c10烷基、c1-c4烷氧基、卤素或硝基。本发明所述的药物组合物可包含由化学式1表示的化合物或其药学上可接受的盐作为活性成分。本发明所述的健康功能食品组合物可包含由化学式1表示的化合物或其药学上可接受的盐作为活性成分。有益效果本发明所述的新的化合物或其药学上可接受的盐具有针对癌细胞系的高的抗增殖活性,并且可以用作有效的抗癌治疗剂。包含该化合物或其药学上可接受的盐作为活性成分的用于预防或治疗癌症疾病的药物组合物和健康功能食品组合物可以预防、治疗或改善各种癌症疾病,例如乳腺癌和肺癌以及脑癌。附图说明图1是衍生自淀粉样蛋白-β探针的结构的新的化合物的示意的设计图。图2示出了本发明的实验实施例所述的化合物9b的抑制效果。具体实施方式在下文中,将详细描述本发明。图1是衍生自淀粉样蛋白-β探针的结构的新的化合物的示意的设计图。已进行大量的研究以开发具有高的脑摄取的淀粉样蛋白-β探针,产生了用于体内淀粉样蛋白-β成像的多种放射性标记分子探针。在衍生自所述分子探针的支架(scaffold)中,硫黄素-t类似物如苯并噻唑、苯并噁唑和苯并咪唑不仅对淀粉样蛋白-β聚集体显示出出色的结合亲和力,而且显示出向大脑中的高摄取。由于在癌症治疗中的各种生理活性,这种苯并杂环在肿瘤学中受到关注,并且基于此,合成了用于开发具有高的中枢神经系统渗透性的抗癌剂的苯并杂环衍生物,从而完成本发明。本发明提供了由以下化学式1表示的化合物或其药学上可接受的盐:[化学式1]在化学式1中,r选自o、s或nr1,且r1可选自氢、c1-c10烷基、c1-c4烷氧基、卤素或硝基。优选地,化学式1中,r选自o、s或nr1,且r1可为氢或c1-c4烷基,并且更优选地,r可为o或s。更具体地,所述化合物可选自于由如下所组成的组:4-(1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺、n-羟基-4-(1-甲基-1h-苯并[d]咪唑-2-基)苯甲酰胺、4-(1-乙基-1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺、n-羟基-4-(1-丙基-1h-苯并[d]咪唑-2-基)苯甲酰胺、4-(1-丁基-1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺、4-(苯并[d]噁唑-2-基)-n-羟基苯甲酰胺、4-(苯并[d]噻唑-2-基)-n-羟基苯甲酰胺、3-(1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺、n-羟基-3-(1-甲基-1h-苯并[d]咪唑-2-基)苯甲酰胺、3-(1-乙基-1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺、n-羟基-3-(1-丙基-1h-苯并[d]咪唑-2-基)苯甲酰胺、3-(1-丁基-1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺、3-(苯并[d]噁唑-2-基)-n-羟基苯甲酰胺和3-(苯并[d]噻唑-2-基)-n-羟基苯甲酰胺。本发明所述的化合物可以以药学上可接受的盐的形式使用,并且该盐可以以药学上可接受的碱性盐或酸性盐的形式使用。所述碱性盐可以以有机碱盐或无机碱盐的形式使用,且其可以选自于由钠盐、钾盐、钙盐、锂盐、镁盐、铯盐、胺鎓盐(aminiumsalt)、铵盐(ammoniumsalt)、三乙基盐、铝盐和吡啶盐所组成的组,但不限于此。作为酸性盐,由游离酸形成的酸加成盐是有用的。无机酸和有机酸可用作游离酸,盐酸、溴酸、硫酸、亚硫酸、磷酸、双磷酸、硝酸等可用作无机酸;并且枸橼酸、醋酸、马来酸、苹果酸、富马酸、葡萄糖酸甲基磺酸、苯磺酸、樟脑磺酸、草酸、丙二酸、戊二酸、乙酸、乙醇酸、琥珀酸、酒石酸、4-甲苯磺酸、半乳糖醛酸、扑酸、谷氨酸、柠檬酸、天冬氨酸、硬脂酸等可用作有机酸。优选地,盐酸可用作无机酸,并且甲基磺酸可用作有机酸。此外,本发明所述的化合物可包括可通过常规方法制备的所有盐、水合物和溶剂化物,以及药学上可接受的盐。在本发明所述的化合物中,可通过常规方法制备加成盐,且可通过将化合物溶解于水混溶性有机溶剂(例如丙酮、甲醇、乙醇或乙腈)并加入过量的无机碱的水基溶液或有机碱,然后沉淀或结晶来制备加成盐。或者,可通过蒸发溶剂或过量碱并干燥以获得加成盐、或通过抽滤沉淀盐来制备混合物。或者,可通过从混合物中蒸发溶剂或过量碱并干燥以获得加成盐或抽滤沉淀盐来制备。如本文所用,“预防”是指通过给予本发明所述的药物组合物或健康功能食品组合物来抑制或延迟癌症疾病或其至少一种症状的发作的任何动作。此外,它包括用于预防性治疗和预防复发的对病情缓解的受试者的治疗。如本文所用,“治疗”是指通过给予本发明所述的药物组合物来改善或有益地改变病症的任何动作,例如缓和、减少或消除癌症疾病或其至少一种症状的发作。如本文所用,“改善”是指通过摄入本发明所述的健康功能食品组合物来改善或有益地改变病症的任何动作,例如缓和、减少或消除癌症疾病或其至少一种症状的发作。如本文所用,“药物组合物”是指为特定目的而给予的组合物,而对于本发明的目的,它是指为了预防或治疗癌症疾病或其至少一种症状而给予的组合物。如本文所用,“健康功能食品”具有与用于特定保健用途的食品(foshu)类似的含义,并且是指除了营养供应之外,还被加工以有效显示生物调节功能的具有高的医学和药学效果的食品。本发明提供用于预防或治疗癌症疾病的药物组合物,该组合物包含所述化合物或其药学上可接受的盐作为活性成分。在本发明所述的药物组合物中,由于该化合物或盐具有针对癌细胞的抗增殖活性,因此其可应用于各种癌症疾病。癌症疾病可以选自于由如下所组成的组:脑癌、乳腺癌、肺癌、皮肤癌、卵巢癌、子宫癌、前列腺癌、肾癌、结肠癌、胰腺癌、胃癌、肝癌、结直肠癌、头颈癌、喉癌、食管癌、血癌和白血病,但不限于此。根据上述配方,本发明所述的药物组合物可通过进一步包含适当的药学上可接受的载体、赋形剂或稀释剂来制备。“药学上可接受的”是指显示出对暴露于该药物组合物的细胞或人无毒的性质。本发明中可使用的载体、赋形剂或稀释剂包括乳糖、右旋糖、蔗糖、山梨醇、甘露醇、木糖醇、赤藓糖醇、麦芽糖醇、淀粉、阿拉伯树胶、海藻酸盐/酯、明胶、磷酸钙、硅酸钙、纤维素、甲基纤维素、微晶纤维素、聚乙烯吡咯烷酮、水、羟基苯甲酸甲酯、羟基苯甲酸丙酯、滑石粉、硬脂酸镁或矿物油,但不限于此,并可使用本领域已知的任何合适试剂。可以按照常规方法以口服制剂的形式来配制和使用本发明的药物组合物,例如粉剂、颗粒剂、片剂、胶囊、混悬剂、乳剂、糖浆、气雾剂、外用制剂、栓剂或无菌可注射溶液。当本发明所述的药物组合物以上述形式配制时,其可使用稀释剂或赋形剂(例如常用的填充剂、增重剂、粘合剂、润湿剂、崩解剂和表面活性剂)来制备。用于口服给予的固体制剂包括片剂、丸剂、粉剂、颗粒剂、胶囊等,并且这种固体制剂可以通过混合至少一种赋形剂(例如淀粉、碳酸钙、蔗糖、乳糖、明胶等)来制备。此外,除了简单的赋形剂外,还可以使用例如硬脂酸镁和滑石粉的润滑剂。除了口服使用的液体和液体石蜡外,还可以通过添加各种赋形剂(例如润湿剂、甜味剂、芳香剂、防腐剂等)来制备。用于胃肠外给予的制剂包括无菌水性溶液、非水性溶剂、混悬剂、乳剂、冻干制剂和栓剂。作为非水性溶剂和混悬剂,可以使用丙二醇、聚乙二醇、例如橄榄油的植物油、例如油酸乙酯的可注射酯等。作为栓剂的基质,可以使用witepsol、macrosol、吐温61、可可脂、月桂精、甘油明胶等。此外,可添加钙或维生素d3以增强治疗效果。在本发明所述的药物组合物中,可以以药学上有效的量施用药物组合物。“药学上有效的量”是指足以以适用于医学治疗的合理的收益/风险比治疗疾病而不产生副作用的量,且有效剂量水平可以根据包括如下的因素来确定:患者的健康状况、溃疡类型、严重程度、药物活性、对药物的敏感性、给予方法、给予时间、给予途径和排泄率、治疗持续时间、联合用药或同时用药以及医学领域众所周知的其它因素。作为本发明所述的药物组合物中的活性成分的化合物的量可以根据患者的年龄、性别、体重和疾病而变化,但可以以0.001mg/kg至100mg/kg、优选0.01mg/kg至10mg/kg每天给予一次到多次。此外,本发明所述的化合物的剂量可根据给予途径、疾病程度、性别、体重、年龄等而增加或减少。因此,上述剂量并不以任何方式限制本发明的范围。药物组合物可通过各种途径给予至哺乳动物,例如鼠、小鼠、家畜和人。所有给予的模式都可以预期,例如,可以通过口服、直肠或静脉内、肌肉内、皮下、支气管内吸入、子宫内中隔或脑室内注射而给予。此外,本发明提供用于预防或改善癌症疾病的健康功能食品组合物,所述健康功能食品组合物包含所述化合物或其药学上可接受的盐作为活性成分。在本发明所述的健康功能食品组合物中,由于该化合物或盐对癌细胞具有抗增殖活性,因此可以应用于各种癌症疾病。癌症疾病可以选自于由如下所组成的组:脑癌、乳腺癌、肺癌、皮肤癌、卵巢癌、子宫癌、前列腺癌、肾癌、结肠癌、胰腺癌、胃癌、肝癌、结直肠癌、头颈癌、喉癌、食管癌、血癌和白血病,但不限于此。本发明所述的健康功能食品可以以粉剂、颗粒剂、片剂、胶囊、糖浆或饮料的形式提供,并且除了本发明所述的化合物之外,健康功能食品与其它食品或食品添加剂一起使用,本发明所述的化合物是活性成分并可根据常规方法适当使用。活性成分的混合量可根据其用途(例如,预防、保健或治疗)的目的而适当地确定。本发明所述的健康功能食品中所含化合物的有效剂量可根据药物组合物的有效剂量来使用,但在出于保健和卫生目的或出于健康控制目的而长期摄入的情况下,其可为上述范围或更小,并且由于在安全性方面没有问题,活性成分可以以至少上述范围的量来使用。本发明所述的健康功能食品的类型不存在特别限制,并且实例包括肉、香肠、面包、巧克力、糖果、零食、糕点、披萨、拉面、其它面条、口香糖、包括冰淇淋的乳制品、各种汤、饮料、茶、饮品、酒精饮料、维生素复合物等。实施例在下文中,通过实施例更详细地描述本发明。这些实施例仅旨在更详细地说明本发明,并且对于本领域技术人员而言显而易见的是,根据本发明的要旨,本发明的范围不受这些实施例的限制。提供本发明的实施例以向本领域的普通技术人员更全面地解释本发明。[合成准备]所有试剂和溶剂均从商业供应商处购买,并使用而无需进一步纯化。处理湿敏化合物的所有实验均在氩气气氛下进行。使用旋转蒸发器在减压下进行浓缩或溶剂去除。在预涂硅胶f254tlc板(e,merck)上进行分析薄层色谱,在uv光下或通过使用碘气染色进行可视化。柱色谱在中压下在二氧化硅(mercksilicagel40-63m)上进行,或通过使用具有预填充的硅胶盒(biotage)的biotagesp1快速纯化系统进行。使用jeolresonance制造的jnm-ecz500r(500mhz)进行nmr分析。化学位移(δ)以百万分之报告。将样品溶剂的氘锁信号用作参考,并且耦合常数(j)以赫兹(hz)表示。裂分模式的缩写如下:s,单峰(singlet);d,二重峰(doublet);t,三重峰(triplet);q,四重峰(quartet);dd,双二重峰(doubletofdoublets);td,三二重峰(tripletofdoublet);m,多重峰(multiplet)。通过使用配备有vp-odsc18柱的双泵shimadzulc-6ad系统进行的分析hplc证实所有最终化合物的纯度高于95%。[实施例1]化合物合成(5,6a-6d)[反应方案1]上述反应方案1示出了化合物5和化合物6a-化合物6d的合成过程。参考方案1,简言之,邻苯二胺1与醛2在二甲基甲酰胺(以下为dmf)水溶液中的反应通过需氧氧化提供关键中间体化合物3,收率为88%。将化合物3在dmf中用氢化钠处理2小时,然后添加各种烷基碘化物以提供化合物4a-化合物4d,收率为45%-70%。最后,化合物3和化合物4a-化合物4d在甲醇中依次用nh2oh和koh处理以得到化合物5和化合物6a-化合物6d,收率为32%-35%。更详细地,该过程进行如下。(1-1)化合物3:4-(1h-苯并[d]咪唑-2-基)苯甲酸甲酯将邻苯二胺(5.84g,53.98mmol)和4-甲酰基苯甲酸甲酯(9.75ml,59.37mmol)的混合物在180mldmf和20ml水中在80℃下在开口烧瓶中搅拌36小时。将混合物在减压下浓缩并通过柱纯化,以提供化合物3,收率为88%。rf=0.23(3∶7=乙酸乙酯∶己烷)。1hnmr(500mhz,cd3od)δ8.19-8.14(m,4h),7.63(s,2h),7.30-7.27(m,2h),3.93(s,3h).13cnmr(125mhz,cd3od)δ166.5,150.6,133.9,131.2,129.9,126.4,123.1,51.5.(1-2)化合物4a:4-(1-甲基-1h-苯并[d]咪唑-2-基)苯甲酸甲酯在0℃下将在dmf(5ml)中的60%氢化钠(0.09g,3.75mmol)添加至处于dmf(5ml)中的化合物3(0.32g,1.25mmol)。在室温下搅拌混合物2小时。加入碘甲烷(0.09ml,1.38mmol)并搅拌8小时。用乙酸乙酯稀释反应物。有机层用水洗涤,用na2so4干燥,减压浓缩,并通过mplc纯化以提供化合物4a,收率为70%。rf=0.26(3∶7=乙酸乙酯∶己烷)。1hnmr(500mhz,cdcl3)δ8.17(d,j=8.5hz,2h),7.85-7.81(m,3h),7.38-7.36(m,1h),7.33-7.30(m,2h),3.94(s,3h),3.84(s,3h).13cnmr(125mhz,cdcl3)δ166.5,152.5,143.0,136.7,134.5,131.1,129.9,129.4,123.3,122.7,120.1,109.8,52.4,31.8.(1-3)化合物4b:4-(1-乙基-1h-苯并[d]咪唑-2-基)苯甲酸甲酯在0℃下将在dmf(5ml)中的60%氢化钠(0.09g,3.75mmol)添加至处于dmf(5ml)中的化合物3(0.32g,1.25mmol)。在室温下搅拌混合物2小时。加入碘乙烷(0.09ml,1.38mmol)并搅拌8小时。用乙酸乙酯稀释反应物。有机层用水洗涤,用na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物4b,收率为49%。rf=0.25(3∶7=乙酸乙酯∶己烷)。1hnmr(500mhz,cdcl3)δ8.19(d,j=8.5hz,2h),7.85-7.81(m,3h),7.45-7.43(m,1h),7.35-7.31(m,2h),4.32-4.27(m,2h),3.96(s,3h),1.47(t,j=7.0hz,3h).13cnmr(125mhz,cdcl3)δ166.6,152.3,143.3,135.6,134.9,131.2,130.0,129.3,123.2,122.7,120.3,110.1,52.4,39.8,15.4.esims(m/e)181.12[m+h]+.(1-4)化合物4c:4-(1-丙基-1h-苯并[d]咪唑-2-基)苯甲酸甲酯在0℃下将在dmf(5ml)中的60%氢化钠(0.08g,3.50mmol)添加至处于dmf(5ml)中的化合物3(0.32g,1.25mmol)。在室温下搅拌混合物2小时。加入碘丙烷(0.13ml,1.28mmol)并搅拌8小时。用乙酸乙酯稀释反应物。有机层用水洗涤,用na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物4c,收率为45%。rf=0.30(3∶7=乙酸乙酯∶己烷)。1hnmr(500mhz,cdcl3)δ8.19(d,j=8.5hz,2h),7.83-7.79(m,3h),7.42-7.40(m,1h),7.33-7.29(m,2h),4.19(t,j=7.5hz,2h),3.95(s,3h),1.85-1.80(m,2h),0.84(t,j=7.1hz,3h).13cnmr(125mhz,cdcl3)δ166.5,152.5,143.0,136.7,134.5,131.1,129.9,129.4,123.3,122.7,120.1,109.8,52.4,31.8,29.7,15.4.esims(m/e)195.14[m+h]+.(1-5)化合物4d:4-(1-丁基-1h-苯并[d]咪唑-2-基)苯甲酸甲酯在0℃下将在dmf(5ml)中的60%氢化钠(0.09g,3.60mmol)添加至处于dmf(5ml)中的化合物3(0.30g,1.20mmol)。在室温下搅拌混合物2小时。加入碘丁烷(0.15ml,1.32mmol)并搅拌8小时。用乙酸乙酯稀释反应物。有机层用水洗涤,用na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物4d,收率为46%。rf=0.35(3∶7=乙酸乙酯∶己烷)。1hnmr(500mhz,cdcl3)δ8.17(d,j=8.5hz,2h),7.85-7.81(m,3h),7.38-7.36(m,1h),7.33-7.30(m,2h),4.20(t,j=7.5hz,2h),3.94(s,3h),1.81-1.74(m,2h),1.24-1.22(m,2h),0.83(t,j=7.5hz,3h).13cnmr(125mhz,cdcl3)δ166.5,152.5,143.0,136.7,134.5,131.1,129.9,129.4,123.3,122.7,120.1,109.8,52.4,44.6,31.9,19.9,13.5.(1-6)化合物5:4-(1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(3.72g,53.51mmol)添加至处于甲醇(5ml)中的氢氧化钾(3.00g,53.51mmol)的溶液。将混合物在0℃下搅拌15分钟,并且去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物3(0.30g,1.19mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌2小时。用乙酸将混合物中和至ph7,并用乙酸乙酯萃取。将有机层用na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物5,收率为40%。rf=0.08(9∶1=乙酸乙酯∶己烷)。1hnmr(500mhz.dmso-d6)δ8.13(d,j=8.5hz,2h),7.89(d,j=8.0hz,2h),7.59-7.57(m,2h),7.20-7.18(m,2h).13cnmr(125mhz,dmso-d6)δ163.3,151.2,137.2,130.4,126.6,125.9.122.1.esims(m/e)254.09[m+h]+.(1-7)化合物6a:n-羟基-4-(1-甲基-1h-苯并[d]咪唑-2-基)苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(1.29g,18.6mmol)添加至处于甲醇(5ml)中的氢氧化钾(1.04g,18.6mmol)的溶液。将混合物在0℃下搅拌15分钟,并且去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物4a(0.11g,0.41mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌2小时。将混合物用3nhcl中和至ph7,并用乙酸乙酯萃取。将有机层经na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物6a,收率为36%。rf=0.17(9∶1=乙酸乙酯∶己烷)。1hnmr(500mhz,dmso-d6)δ8.11(d,j=8.0hz,2h),8.02(d,j=8.5hz,1h),7.91(dd,j=8.0hz,1.5hz,1h),7.86(d,j=8.5hz,1h),7.69(t,j=8.5hz,1h),7.62(d,j=8.0hz,1h),7.43(t,j=7.5hz,1h),3.71(s,3h).13cnmr(125mhz,dmso-d6)δ154.5,152.9,138.8,134.5,133.9,132.8,131.5,130.3,129.8,129.7,129.2,126.7,124.3,115.4,29.8.esims(m/e)167.11[m+h]+.(1-8)化合物6b:4-(1-乙基-1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(1.12g,16.05mmol)添加至处于甲醇(5ml)中的氢氧化钾(0.90g,16.05mmol)的溶液。将混合物在0℃下搅拌15分钟,并且去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物4b(0.10g,0.36mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌3小时。将混合物用乙酸中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物6b,收率为51%。rf=0.19(9∶1=乙酸乙酯∶己烷)。1hnmr(500mhz,dmso-d6)δ11.42(s,1h),9.22(s,1h),7.95(d,j=7.5hz,2h),7.87(d,j=8.5hz,2h),7.71(d,j=8.0hz,1h),7.67(d,j=8.0hz,1h),7.33-7.25(m,2h),4.34(q,j=7.5hz,2h),1.33(t,j=7.5hz,3h).13cnmr(125mhz,cd3od)δ167.1,153.6,143.4,136.4,135.2,134.2,130.7,128.7,124.6,124.1,119.9,111.9,40.8,15.4.esims(m/e)282.12[m+h]+.(1-9)化合物6c:n-羟基-4-(1-丙基-1h-苯并[d]咪唑-2-基)苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(1.06g,15.29mmol)添加至处于甲醇(5ml)中的氢氧化钾(0.86g,15.29mmol)的溶液。将混合物在0℃下搅拌15分钟,去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物4c(0.11g,0.34mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌2小时。将混合物用乙酸中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物6c,收率为49%。rf=0.19(9∶1=乙酸乙酯∶己烷)。1hnmr(500mhz,dmso-d6)δ11.41(s,1h),9.19(s,1h),7.93(d,j=8.5hz,2h),7.86(d,j=8.5hz,2h),7.70(d,j=8.5hz,2h),7.33-7.25(m,2h),4.29(t,j=7.5hz,2h),1.72-1.65(m,2h),0.72(t,j=7.5hz,3h).13cnmr(125mhz,dmso-d6)δ163.7,152.1,142.2,135.7,133.8,132.9,129.3,127.4,122.8,122.3,119.2,111.2,45.7,22.7,11.0.esims(m/e)296.14[m+h]+.(1-10)化合物6d:4-(1-丁基-1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(2.03g,29.19mmol)添加至处于甲醇(5ml)中的氢氧化钾(1.64g,29.19mmol)的溶液。将混合物在0℃下搅拌15分钟,并且去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物4d(0.20g,0.65mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌3小时。将混合物用乙酸中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物6d,收率为51%。rf=0.26(9∶1=乙酸乙酯∶己烷)。1hnmr(500mhz,dmso-d6)δ11.40(s,1h),9.18(s,1h),7.93(d,j=8.5hz,2h),7.86(d,j=8.0hz,2h),7.68(t,j=8.5hz,2h),7.32-7.24(m,2h),4.32(t,j=7.0hz.2h),1.67-1.61(m,2h),1.16-1.08(m,2h),0.74(t,j=7.5hz,3h).13cnmr(125mhz,dmso-d6)δ164.1,152.6,143.1,136.2,134.1,133.6,129.7,127.8.123.2,122.6,119.8,111.6,44.4,31.7.19.7,13.8.esims(m/e)310.16[m+h]+.[实施例2]化合物合成(9a-9b)[反应方案2]反应方案2示出了化合物9a-化合物9b的合成过程。参考反应方案2,通过2-氨基苯酚7a或2-氨基苯硫酚7b与醛2一起的碘介导的环化以32%-35%的收率合成化合物8a和化合物8b,其中2-氨基苯酚7a或2-氨基苯硫酚7b是相应的起始原料。通过在甲醇中依次用nh2oh和koh处理酯8a和8b,成功地获得了化合物9a和化合物9b,收率为42%-45%。更详细地,该过程如下进行。(2-1)化合物8a:4-(苯并[d]噁唑-2-基)苯甲酸甲酯将2-氨基苯酚(0.20g,1.83mmol)和4-甲酰基苯甲酸甲酯(0.30g,1.83mmol)溶解于二氯甲烷(20ml)中,并在室温下在氩气气氛下搅拌4小时。将反应混合物添加至碘(0.12g,0.92mmol)并在室温下在开口烧瓶中搅拌1小时。将混合物在减压下浓缩并通过柱纯化,以得到化合物8a,收率32%。rf=0.31(1∶9=乙酸乙酯∶己烷)。1hnmr(500mhz,cdcl3)δ8.32(d,j=8.5hz,2h),8.18(d,j=8.5hz,2h),7.80-7.79(m,1h),7.61-7.59(m,1h),7.40-7.37(m,2h),3.96(s,3h).13cnmr(125mhz,cdcl3)δ166.5,162.1,151.0,142.1,132.6,131.1,130.2,127.6,125.9,125.0,120.5,110.9,52.6.(2-2)化合物8b:4-(苯并[d]噻唑-2-基)苯甲酸甲酯将2-氨基苯硫酚(1.71ml,15.98mmol)和4-甲酰基苯甲酸甲酯(2.62g,15.98mmol)溶解于二氯甲烷(100ml)中,并在室温下在氩气气氛下搅拌4小时。将反应混合物添加至碘(1.01g,7.99mmol)并在室温下在开口烧瓶中搅拌45分钟。将混合物在减压下浓缩并通过柱纯化,以得到化合物8b,收率为35%。rf=0.31(1∶9=乙酸乙酯∶己烷)。1hnmr(500mhz,cdcl3)δ8.15-8.14(m,4h),8.09(d,j=7.5hz,1h),7.91(d,j=8.5hz,1h),7.51(t,j=8.0hz,1h),7.41(t,j=7.5hz,1h),3.95(s,3h).13cnmr(125mhz,cdcl3)δ166.5,154.2,137.6,135.4,132.2,130.4,129.7,127.6,126.7,125.8,123.7,121.9,52.5.(2-3)化合物9a:4-(苯并[d]噁唑-2-基)-n-羟基苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(2.47g,35.56mmol)添加至处于甲醇(5ml)中的氢氧化钾(1.99g,35.56mmol)的溶液。将混合物在0℃下搅拌15分钟,并且去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物8a(0.2g,0.79mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌2小时。将混合物用3nhcl中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物9a,收率为45%。rf=0.24(7∶3=乙酸乙酯∶己烷)。1hnmr(500mhz,dmso-d6)δ11.41(s,1h),9.17(s,1h),8.12(d,j=8.0hz,2h),8.01(d,j=8.5hz,2h),7.71(d,j=8.0hz,1h),7.66(d,j=8.0hz,1h),7.33(t,j=6.5hz.1h),7.29(t,j=7.5hz,1h).13cnmr(125mhz,dmso-d6)δ163.3,161.5,150.3,141.4,135.7,128.6,127.9,127.3,125.9,125.1,120.1,111.1.esims(m/e)255.08[m+h]+.(2-4)化合物9b:4-(苯并[d]噻唑-2-基)-n-羟基苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(43.44g,3.02mmol)添加至处于甲醇(5ml)中的氢氧化钾(43.44g,2.24mmol)的溶液。将混合物在0℃下搅拌15分钟,并且去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物8b(0.26g,0.79mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌2小时。将混合物用3nhcl中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物9b,收率42%。rf=0.30(7∶3=乙酸乙酯∶己烷)。1hnmr(500mhz,dmso-d6)δ11.41(s,1h),9.19(s,1h),8.19-8.17(m,3h),8.10(d,j=8.0hz,1h),7.94(d,j=9.0hz,2h),7.57(t,j=7.3hz,1h),7.49(t,j=7.3hz,1h).13cnmr(125mhz,dmso-d6)δ166.4,163.2,153.5,135.1,135.0,134.7,127.9,127.2,126.9,125.9,123.1,122.5.esims(m/e)271.05[m+h]+.[实施例3]化合物合成(13,14a-14d)[反应方案3]反应方案3示出了化合物13和化合物14a-化合物14e的合成过程,以探索间位区域异构体的生物活性。参考反应方案3,与反应方案1类似,由邻苯二胺1与醛10在dmf水溶液中得到化合物11,收率为86%。将化合物11在dmf中用氢化钠处理2h,然后与相应的烷基碘化物烷基化以得到化合物12a-化合物12d,收率为11%-91%。通过化合物11和化合物12a-化合物12d在甲醇中与nh2oh和koh的反应,最终得到化合物13和化合物14a-化合物14d,收率33%-55%。(3-1)化合物11:3-(1h-苯并[d]咪唑-2-基)苯甲酸甲酯将邻苯二胺(2.00g,18.50mmol)和3-甲酰基苯甲酸甲酯(3.03g,18.50mmol)的混合物在180mldmf和20ml水中在80℃下在开口烧瓶中搅拌48小时。将混合物在减压下浓缩并通过柱纯化,以得到化合物11,收率为86%。rf=0.25(3∶7=乙酸乙酯∶己烷)。1hnmr(500mhz,dmso-d6)δ8.80(s,1h),8.45(d,j=8hz,1h),8.05(d,j=7.5hz,1h),7.70(t,j=8.0hz,1h),7.63(s,2h),7.23-7.22(m,2h),3.92(s,3h).13cnmr(125mhz,dmso-d6)165.9,150.3,131.0,130.8,130.7,130.5,130.3,129.6,127.1,127.1,127.0,122.6,122.3,52.4.esims(m/e)253.10[m+h]+.(3-2)化合物12a:3-(1-甲基-1h-苯并[d]咪唑-2-基)苯甲酸甲酯在0℃下将在dmf(5ml)中的60%氢化钠(0.05g,1.24mmol)添加至处于dmf(5ml)中的化合物11(0.21g,0.82mmol)。在室温下搅拌混合物2小时。加入碘甲烷(0.06ml,0.91mmol)并搅拌2.5小时。用乙酸乙酯稀释反应物。将有机层用水洗涤,用na2so4干燥,减压浓缩,并用mplc纯化以得到化合物12a,收率为91%。rf=0.25(4∶6=乙酸乙酯∶己烷)。1hnmr(500mhz,cdcl3)δ8.44(s,1h),8.19(d,j=8.0hz,1h),8.02(d,j=7.5hz,1h),7.85-7.83(m,1h),7.63(t,j=8.0hz,1h),7,43-7.41(m,1h),7.37-7.33(m,2h),4.0(s,3h),3.90(s,3h).13cnmr(125mhz,dmso-d6)δ166.6,152.8,143.0,134.0,130.9,130.8,130.4,129.2,123.2,122.8,120.1,109.9,52.5,31.97.esims(m/e)267.11[m+h]+.(3-3)化合物12b:3-(1-乙基-1h-苯并[d]咪唑-2-基)苯甲酸甲酯在0℃下将在dmf(5ml)中的60%氢化钠(0.05g,2.04mmol)添加至处于dmf(5ml)中的化合物11(0.20g,0.81mmol)。在室温下搅拌混合物2小时。加入碘乙烷(0.07ml,0.89mmol)并搅拌2.5小时。用乙酸乙酯稀释反应物。将有机层用水洗涤,用na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物12b,收率为11%。rf=0.28(4∶6=乙酸乙酯∶己烷)。1hnmr(500mhz,cdcl3)δ8.41(s,1h),8.19(d,j=8.0hz,1h),7.97(d,j=8.0hz,1h),7.84(d,j=8.5hz,1h),7.63(t,j=8.0hz,1h),7.45(d,j=9.5hz,1h),7.36-7.26(m,2h),4.31(q,j=7.5hz,2h),3.96(s,3h),1.50(t,j=7.0hz,3h).13cnmr(125mhz,dmso-d6)δ166.6,152.4,143.3,135.5,133.9,131.1,131.9,130.8,130.3,129.2,123.1,122.7,120.3,110.2,52.5,39.9,15.4.esims(m/e)281.13[m+h]+.(3-4)化合物12c:3-(1-丙基-1h-苯并[d]咪唑-2-基)苯甲酸甲酯在0℃下将在dmf(5ml)中的60%氢化钠(0.07g,1.78mmol)添加至处于dmf(5ml)中的化合物11(0.30g,1.19mmol)。在室温下搅拌混合物2小时。加入碘丙烷(0.12ml,1.31mmol)并搅拌8小时。用乙酸乙酯稀释反应物。将有机层用水洗涤,用na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物12c,收率为28%。rf=0.32(3∶7=乙酸乙酯∶己烷)。1hnmr(500mhz,cdcl3)δ8.38(t,j=1.0hz,1h),8.17(td,j=8.0hz,1.0hz,1h),7.94(td,j=7.5hz,2.0hz,1h),7.83-7.81(m,1h),7.59(t,j=8.0hz,1h),7.42-7.40(m,1h),7.31-7.29(m,2h),4.19(t,j=7.5hz,2h),3.93(s,3h),1.87-1.82(m,2h),0.85(t,j=7.0hz,3h).13cnmr(125mhz,cdcl3)δ166.4,152.6,143.1,135.7,133.9,131.1,130.7,130.2,129.1,123.0.122.5,120.1,110.3,52.4,46.4,23.3,11.2.esims(m/e)295.14[m+h]+.(3-5)化合物12d:3-(1-丁基-1h-苯并[d]咪唑-2-基)苯甲酸甲酯在0℃下将在dmf(5ml)中的60%氢化钠(0.07g,1.78mmol)添加至处于dmf(5ml)中的化合物11(0.30g,1.19mmol)。在室温下搅拌混合物2小时。加入碘丁烷(0.15ml,1.31mmol)并搅拌8小时。用乙酸乙酯稀释反应物。将有机层用水洗涤,用na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物12d,收率为79%。rf=0.33(3∶7=乙酸乙酯∶己烷)。1hnmr(500mhz,cdcl3)δ8.39(t,j=1.5hz,1h),8.17(td,j=7.5hz,1.5hz,1h),7.95(td,j=8.0hz,1.5hz,1h),7.83-7.81(m,1h),7.60(t,j=8.0hz,1h),7.43-7.41(m,1h),7.32-7.28(m,2h),4.23(t,j=7.5hz,2h),3.93(s,3h),1.84-1.78(m,2h),1.31-1.24(m,2h),0.85(t,j=7.5hz,3h).13cnmr(125mhz,cdcl3)δ166.4,152.5,143.1,135.7,133.9.131.1,130.8,130.7,130.2,129.1,123.0,122.7,120.1,110.3,52.4,44.7,31.9,20.0,13.6.esims(m/e)309.16[m+h]+.(3-6)化合物13:3-(1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(3.72g,53.51mmol)添加至处于甲醇(5ml)中的氢氧化钾(3.00g,53.51mmol)的溶液。将混合物在0℃下搅拌15分钟,并且去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物11(0.3g,11.89mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌3小时。将混合物用3nhcl中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化,得到化合物13,收率为33%。1hnmr(500mhz,dmso-d6)δ10.12(brs,2h),8.91(s,1h),8.72(d,j=8.5hz,1h),8.22(d,j=8.0hz,1h),7.86-7.83(m,3h),7.56-7.54(m,2h).13cnmr(125mhz,cd3od)δ167.5,149.5,135.4,133.9,133.0,131.5,132.8,129.9,127.9,124.2,115.1.esims(m/e)254.09[m+h]+.(3-7)化合物14a:n-羟基-3-(1-甲基-1h-苯并[d]咪唑-2-基)苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(3.72g,53.51mmol)添加至处于甲醇(5ml)中的氢氧化钾(3.00g,53.51mmol)的溶液。将混合物在0℃下搅拌15分钟,并且去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物12a(0.30g,11.89mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌3小时。将混合物用乙酸中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化,得到化合物14a,收率为49%。1hnmr(500mhz,dmso-d6)δ11.4(s,1h),9.24(s,1h),8.23(s,1h),8.01(d,j=8hz,1h),7.94(d,j=8.5hz,1h),7.71-7.62(m,4h),7.33(m,2h),3.90(s,3h)13cnmr(125mhz,dmso-d6)δ163.6,152.4,142.5,136.5,133.3,131.9,130.4,129.0,128.1,127.7,122.6,122.2,119.1,110.8,31.8.esims(m/e)268.11[m+h]+.(3-8)化合物14b:3-(1-乙基-1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(3.72g,53.51mmol)添加至处于甲醇(5ml)中的氢氧化钾(3.00g,53.51mmol)的溶液。将混合物在0℃下搅拌15分钟,并且去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物12b(0.30g,11.89mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌3小时。将混合物用3nhcl中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化,得到化合物14b,收率为43%。1hnmr(500mhz,dmso-d6)δ8.33(s,1h).8.12(d,j=8hz,1h),8.03(d,j=8hz,1h),7.74-7.67(m,3h),7.33-7.25(m,2h),4.33(q,j=7hz,2h),1.36(t,j=7hz,3h)13cnmr(125mhz,cd3od)δ168.4,152,5,141.9,135.0,132.9,131.0,130.0,129.9,128.9,123.2,122.7,118.5,39.4,14.1.esims(m/e)282.12[m+h]+.(3-9)化合物14c:n-羟基-3-(1-丙基-1h-苯并[d]咪唑-2-基)苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(2.83g,40.71mmol)添加至处于甲醇(5ml)中的氢氧化钾(2.28g,40.71mmol)的溶液。将混合物在0℃下搅拌15分钟,并且去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物12c(0.28g,0.90mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌3小时。将混合物用乙酸中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化,得到化合物14c,收率为55%。rf=0.22(8∶2=乙酸乙酯∶己烷)。1hnmr(500mhz,dmso-d6)δ11.39(s,1h),9.14(s,1h),8.09(s,1h),7.89(d,j=6.5hz,2h),7.67-7.62(m,3h),7.28-7.21(m,2h),4.25(t,j=7.5hz,2h),1.66(q,j=6.5hz,2h),0.69(t,j=7.0hz,3h).13cnmr(125mhz,dmso-d6)δ163.5,152.3,142.6,135.8,133.2,131.7,131.2,129.1,128.2,127.6,122.6,122.1,119.3,111.1,45.6,22.7,11.0.esims(m/e)296.13[m+h]+.(3-10)化合物14d:3-(1-丁基-1h-苯并[d]咪唑-2-基)-n-羟基苯甲酰胺在0℃下将在甲醇(5ml)中的盐酸羟胺(2.83g,40.71mmol)添加至处于甲醇(5ml)中的氢氧化钾(2.28g,40.71mmol)溶液中。将混合物在0℃下搅拌15分钟,并且去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(10ml)中的化合物12d(0.28g,0.90mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌3小时。将混合物用乙酸中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化以得到化合物14d,收率为50%。rf=0.28(8∶2=乙酸乙酯∶己烷)。1hnmr(500mhz,dmso-d6)δ11.45(s,1h),9.21(s,1h),8.15(t,j=1.5hz,1h),7.95-7.92(m,2h),7.71-7.66(m,3h),7.33-7.25(m,2h),4.32(t,j=7.5hz,2h),1.69-1.63(m,2h),1.18-1.10(m,2h),0.74(t,j=7.5hz,3h).13cnmr(125mhz,dmso-d6)δ163.4,152.3,142.6,135.7,133.3,131.7,130.8,129.0,128.0,127.6,122.7,122.1,119.3,111.1,43.8,31.3,19.2,13.3.esims(m/e)310.15[m+h]+.[实施例4]化合物合成(17a-17b)[反应方案4]反应方案4示出了化合物17a-化合物17b的合成过程。在反应方案4中,与反应方案2不同的是,通过化合物15a-化合物15b与醛10的碘介导的环化来构建化合物16a-化合物16b的首次尝试并不好。因此,在dmf中进行氰化钠催化的化合物15a-化合物15b与醛10的环化,成功地以44%-67%的收率得到了化合物16a-化合物16b。最后,在甲醇中用nh2oh和koh将化合物16a-化合物16b转化为化合物17a-化合物17b。更详细地说,该过程进行如下。(4-1)化合物16a:3-(苯并[d]噁唑-2-基)苯甲酸甲酯向分子筛(分子过滤器)中添加在40mldmf中的2-氨基苯酚(0.50g,4.58mmol)和3-甲酰基苯甲酸甲酯(0.75g,4.58mmol)。然后,将反应混合物在60℃下在开口烧瓶中搅拌4小时。4小时后,将反应混合物在室温下冷却并添加至氰化钠(nacn,0.02g,0.46mmol)。将反应混合物在室温下搅拌24小时。将混合物在减压下浓缩并通过柱纯化,得到化合物16a,收率为67%。rf=0.25(1∶9=乙酸乙酯∶己烷)。1hnmr(500mhz,dmso-d6)δ8.70(s,1h),8.43(d,j=8.5hz,1h),8.17(d,j=8.0hz,1h),7.84-7.82(m,2h),7.77(t,j=7.5hz1h),7.48-7.41(m,2h),3.92(s,1h).13cnmr(125mhz,cdcl3)δ166.3,162.2,150.9,142.1,132.5,131.8,131.2,129.3,128.8,127.7,125.6,124.9,120.3,52.5.esims(m/e)254.08[m+h]+.(4-2)化合物16b:3-(苯并[d]噻唑-2-基)苯甲酸甲酯向分子筛(分子过滤器)添加在40mldmf中的2-氨基苯硫酚(0.50g,4.58mmol)和3-甲酰基苯甲酸甲酯(0.66g,4.58mmol)。然后,将反应混合物在60℃下在开口烧瓶中搅拌4小时。4小时后,将反应混合物在室温下冷却并添加至氰化钠(0.02g,0.46mmol)。将反应混合物在室温下搅拌24小时。将混合物在减压下浓缩并通过柱纯化,得到化合物16b,收率为44%。rf=0.26(1∶9=乙酸乙酯∶己烷)。1hnmr(500mhz,dmso-d6)δ8.57(s,1h),8.26(d,j=7.5hz,1h),8.13(d,j=7.5hz,1h),8.08(d,j=8.5hz,2h),7.68(t,j=7.5hz,1h),7.55(t,j=8.0hz,1h),7.46(t,j=8.0hz,1h),3.90(s,3h).13cnmr(125mhz,cdcl3)δ166.8,166.4,154.1,135.1,134.0,131.8,131.6,131.1,129.2,128.6,126.5,126.5,125.5,123.4,52.4.esims(m/e)270.06[m+h]+.(4-3)化合物17a:3-(苯并[d]噁唑-2-基)-n-羟基苯甲酰胺在0℃下将在甲醇(6ml)中的盐酸羟胺(3.70g,53.31mmol)添加至处于甲醇(6ml)中的氢氧化钾(2.99g,53.31mmol)的溶液。将混合物在0℃下搅拌15分钟,去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(12ml)中的化合物16a(0.30g,1.18mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌3小时。将混合物用3nhcl中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化,得到化合物17a,收率为50%。1hnmr(500mhz,dmso-d6)δ11.52(s,1h),9.24(s,1h),8.61(s,1h),8.33(d,j=7.5hz,1h),8.0(d,j=8.0hz,1h),7.83(t,j=8.0hz,2h),7.71(t,j=8.0hz,1h),7.46-7.43(m,2h).13cnmr(125mhz,dmso-d6)δ163.2,161.7,150.3,141.4,133.8,130.1,129.4,129.7,126.7,125.9,125.1,120.0,111.1.esims(m/e)255.08[m+h]+.(4-4)化合物17b:3-(苯并[d]噻唑-2-基)-n-羟基苯甲酰胺在0℃下将在甲醇(6ml)中的盐酸羟胺(2.32g,33.42mmol)添加至处于甲醇(6ml)中的氢氧化钾(1.87g,33.42mmol)的溶液。将混合物在0℃下搅拌15分钟,去除沉淀的氯化钾,并将滤液照此使用;在0℃下向在四氢呋喃(12ml)中的化合物16b(0.20g,0.74mmol)的溶液中加入新制备的羟胺,并在相同温度下搅拌3小时。将混合物用3nhcl中和至ph7,并用乙酸乙酯萃取。有机层经na2so4干燥,减压浓缩,并通过mplc纯化,得到化合物17b,收率为4%。1hnmr(500mhz,dmso-d6)δ11.51(s,1h),8.47(s,1h),8.24(d,j=8.0hz,1h),8.20(d,j=7.5hz,1h),8.11(d,j=8.0hz,1h),7.94(d,j=7.5hz,1h),7.67(t,j=7.5hz,1h),7.58(t,j=7.0hz,1h),7.50(t,j=7.0hz,1h).13cnmr(125mhz,dmso-d6)δ166.6,163.3,153.5,134.6,133.9,133.1,129.8,129.7,129.6,126.9,125.8,125.6,123.1,122.6.esims(m/e)271.05[m+h]+.[实验实施例1]在sh-sy5y细胞系中的抗增殖作用使用通过上述实施例1至实施例4合成的化合物研究针对人成神经细胞瘤细胞系sh-sy5y(人恶性转移性成神经细胞瘤的体外模型)的抗增殖活性。1.细胞培养将sh-sy5y细胞在具有l-谷氨酰胺并补充500mg/ml链霉素、100单位/ml青霉素和10%胎牛血清(fbs)的dmem(dulbecco改良的eagle培养基)中培养。细胞在潮湿气氛(37℃,5%co2)中生长汇合。2.实验结果下表1是根据实验实施例1的抗增殖活性实验的结果,并示出72小时后对细胞系sh-sy5y的细胞毒性值,即当该化合物使细胞增殖减少一半时的最大浓度值(gi50;半数最大生长抑制浓度)。[表1]参考表1,化合物9b对sh-sy5y细胞系发挥出最有效的抗增殖活性,gi50值为2.01μm。参比药物saha同样对sh-sy5y细胞系提供良好的抗增殖活性,gi50值为2.90μm。saha是用于治疗皮肤t细胞淋巴瘤(癌症的类型)的药物。与苯并噻唑类似物化合物9b相比,对位取代的苯并咪唑类似物5和6a-6d显示出相对较低的抗增殖活性,gi50值为38.1μm-60μm,但苯并咪唑基团(6d)的氮原子上较长的丁基取代基显示出增高的抗增殖活性,gi50值为26.8μm。苯并噁唑9a(其具有取代最有效的化合物9b的硫原子的氧原子)具有25.7μm的gi50值,提供比化合物9b低12倍的有效的抗增殖活性。这可能是因为硫原子的疏水性在与hdac酶的结合和在细胞渗透性中起关键作用。与甲基取代的化合物13和未取代的化合物14a相比,在间位取代的苯并咪唑部分的氮原子上的乙基、丙基和丁基取代基(包括化合物14b-化合物14d)也具有增高的针对sh-sy5y细胞系的细胞抗增殖活性。然而,与对位取代的苯并噻唑9b相比,所有的间位取代的化合物13、化合物14a-化合物14d和化合物17a-化合物17b示出相对差或中等的抗增殖活性。[实验实施例2]在乳腺癌细胞系mda-mb-231中的抗增殖作用使用通过上述实施例1至实施例4合成的化合物研究对乳腺癌细胞系mda-mb-231的抗增殖活性。下表2示出了根据对乳腺癌细胞系mda-mb-231的抗增殖活性试验的结果。参考表2,化合物9b对mda-mb-231细胞系显示出最强的抗增殖活性,gi50值为3.86μm,而参比药物saha对mda-mb-231细胞系也显示出优异的抗增殖活性,gi50值为3.16μm。苯并噁唑9a(其具有取代最有效的化合物9b的硫原子的氧原子)具有8.31μm的gi50值,其不如化合物9b有效,但提供了高的抗增殖活性。[表2][实验实施例3]化合物9b在h1975、a549和hela细胞系中的抗增殖作用使用化合物9b研究对非小细胞肺癌细胞系h1975、肺癌细胞系a549和宫颈癌细胞系hela的抗增殖活性,其在上述实验实施例1和实验实施例2中表现出高的抗增殖活性。[表3]化合物h1975(gi50;μm)a549(gi50;μm)hela(gi50;μm)9b3.7115.613.5表3显示了对非小细胞肺癌细胞系h1975、肺癌细胞系a549和宫颈癌细胞系hela的抗增殖活性试验的结果。参考表3,化合物9b通常对细胞系提供高的抗增殖活性,并且可以证实其在各种癌细胞系中显示出抗增殖作用。[实验实施例4]集落形成分析非贴壁依赖性细胞生长是细胞癌变的特征,并且表明转化细胞独立于固体表面生长的能力。软琼脂集落形成试验是广泛使用的用于测量非贴壁依赖性细胞体外生长能力的方法。因此,通过软琼脂集落形成试验,探索化合物9b对非贴壁依赖性细胞生长的抑制效果。1.实验方法将sh-sy5y细胞接种于具有1.2%琼脂(4×105细胞/孔)的6孔板中,并且加入浓度为1μm、3μm和5μm的化合物9b,并培养1~2周。将dmso(0.5%)作为阴性对照。孵育后,将集落用0.05%结晶紫染色10分钟。将集落用三蒸水清洗并捕获。2.实验结果图2示出了本发明实验实施例所述的化合物9b的抑制效果。参见图2,用化合物9b处理sh-sy5y细胞显示出对sh-sy5y成神经细胞瘤的集落形成的抑制效果。此外,随着化合物9b的浓度增加,sh-sy5y细胞形成集落的能力以剂量依赖性方式降低,并且在化合物9b的浓度为3μm时,sh-sy5y成神经细胞瘤的非贴壁依赖性细胞生长几乎被完全抑制。总之,实验数据示出化合物9b以浓度依赖性方式有效地抑制sh-sy5y细胞的非贴壁依赖性生长能力。以下,对本发明所述的含有化合物9b的组合物的制备实施例进行描述,但本发明并不限于此,而是旨在详细地进行描述。[配方实施例1]药物组合物的配方实施例[配方实施例1-1]粉剂的制备通过将20mg化合物9b、100mg乳糖和10mg滑石粉混合并填充在密封袋中制备粉剂。[处方实施例1-2]片剂的制备按照常规的片剂制备方法,通过将20mg化合物9b、100mg玉米淀粉、100mg乳糖和2mg硬脂酸镁混合并制片来制备片剂。[配方实施例1-3]胶囊的制备按照常规的胶囊制备方法,通过将10mg化合物9b、100mg玉米淀粉、100mg乳糖和2mg硬脂酸镁混合并将上述组分填充于明胶胶囊中来制备胶囊。[配方实施例1-4]注射剂的制备按照常规的注射剂制备方法,将10mg化合物9b、适量的注射用无菌蒸馏水和适量的ph调节剂混合并按各安瓿(2ml)加工上述组分来制备注射剂。[配方实施例1-5]软膏的制备按照常规的软膏制备方法,将10mg化合物9b、250mgpeg-4000、650mgpeg-400、10mg白凡士林、1.44mg对羟基苯甲酸甲酯、0.18mg对羟基苯甲酸丙酯和剩余量的纯净水混合并加工上述组分来制备软膏。[配方实施例2]健康功能食品[配方实施例2-1]健康食品的制造按照常规方法,将1mg化合物9b、适量的维生素混合物(70μg维生素a乙酸酯、1.0mg维生素e、0.13mg维生素b1、0.15mg维生素b2、0.5mg维生素b6、0.2μg维生素b12、10mg维生素c、10μg生物素、1.7mg烟酰胺、50μg叶酸、0.5mg泛酸钙)和适量的无机混合物(1.75mg硫酸亚铁、0.82mg氧化锌、25.3mg碳酸镁、15mg磷酸二氢钾、55mg磷酸二钙、90mg柠檬酸钾、100mg碳酸钙、24.8mg氯化镁)混合,然后制备颗粒剂以制备健康食品。[配方实施例2-2]健康饮品的制备将1mg化合物9b、1000mg柠檬酸、100g低聚糖、2g李子浓缩物、1g牛磺酸和纯化水混合,以使得总量为900ml,并根据常规的健康饮品制备方法混合上述成分,在85℃下加热搅拌约1小时,并将得到的溶液过滤并加入到灭菌的2l容器中,密封并灭菌,然后储存在冰箱中。至此,通过本发明的具体实施例来研究本发明。本发明所属领域的普通技术人员可以理解,在不脱离本发明的本质特征的情况下,可以以修改的形式实现本发明。因此,应当从解释性的观点而不是限制性的观点来考虑所公开的实施方式。本发明的范围在权利要求而不是在以上描述中示出,并且在与其等同的范围内的所有差异应当被解释为包括在本发明中。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1