增强慢病毒载体产生的方法
本发明涉及一种慢病毒载体的高效产生方法。
背景技术:
来源于人免疫缺陷病毒1型(hiv-1)的慢病毒载体是在体外和体内两者中用于将外源基因导入分裂细胞和非分裂细胞的重要工具。慢病毒载体的安全性和方便性通过各种方法得到改善(非专利文献1)。水疱性口炎病毒糖蛋白(vsv-g)假型的慢病毒载体在人巨细胞病毒(cmv)最早期启动子的调控下在hek293t细胞内产生,可以介导向大范围的细胞中的非常高效的转导(非专利文献2)。以造血干细胞和t细胞为靶标的慢病毒载体在最近的临床应用中非常成功(非专利文献3)。在体内实验和非分裂细胞的转导中,多数情况下需要高效价的慢病毒载体,因此必须通过大型培养来制造,利用超速离心进行浓缩,接着通过色谱和超滤进行纯化(非专利文献4)。该操作步骤由于使用大量的质粒和细胞培养材料以及用于浓缩和纯化的装置来制备载体导致高成本,因此目前成为了很大的障碍。为了改善用于从培养上清的慢病毒载体的浓缩和纯化的操作步骤而付出很多努力,但关于用于增强载体的实际产生的简单方法的报告比较少。显示有通过阻止细胞内自然免疫应答,增加病毒载体产生(非专利文献5),以及通过免疫应答蛋白siglec-9的稳定的表达使转染效率增加,从而增加慢病毒载体产生(非专利文献6)。其他研究者对在培养基中添加咖啡因(非专利文献7)或丁酸钠(非专利文献8)而使慢病毒载体效价增加的情况进行了研究。但是,为了制备高效价的慢病毒载体,仍需要进一步改善。
现有技术文献
非专利文献
非专利文献1:matraietal.,moleculartherapy:thejournaloftheamericansocietyofgenetherapy18,477-490,doi:10.1038/mt.2009.319(2010).
非专利文献2:burns,j.c.etal.,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica90,8033-8037(1993).
非专利文献3:dunbar,c.e.etal.,science359,doi:10.1126/science.aan4672(2018).
非专利文献4:mccarron,a.etal.,journalofbiotechnology240,23-30,doi:10.1016/j.jbiotec.2016.10.016(2016).
非专利文献5:devriesetal.,genetherapy15,545-552,doi:10.1038/gt.2008.12(2008).
非专利文献6:shoji,t.etal.,cytotechnology67,593-600,doi:10.1007/s10616-013-9679-7(2015).
非专利文献7:ellis,b.l.etal.,humangenetherapy22,93-100,doi:10.1089/hum.2010.068(2011).
非专利文献8:ansorge.s.etal.,thejournalofgenemedicine11,868-876,doi:10.1002/jgm.1370(2009).
技术实现要素:
发明所要解决的问题
慢病毒载体是用于传递外源基因以在细胞内稳定表达的重要工具。从含慢病毒载体的培养基中纯化慢病毒载体的技术开发已取得重大进展。但是,对于从产生细胞增加慢病毒载体产生的方法,尚未进行充分研究。
本发明的目的是提供一种在293t细胞中增强慢病毒载体产生的方法和用于该增强的试剂。
解决问题的技术方案
人t细胞白血病病毒1型(htlv-1)的tax蛋白(下面,记为“htlv-1tax”或“tax”)促进htlv-1的ltr驱动转录,是激活的细胞转录因子的反式作用蛋白,所述细胞转录因子包括camp应答元件结合蛋白(creb)/激活转录因子(atf)、核因子b(nf-κb)和激活蛋白-1(ap-1)1。本发明人发现htlv-1tax能够增强hek293t细胞中的cmv启动子驱动基因表达,并将该发现用于慢病毒载体的产生。
本发明人发现如果同时表达少量tax,则在产生细胞中gag的表达和慢病毒载体粒子的释放这两者显著增加,转导效率提高了超过10倍,通过产生细胞中的启动子激活,制备高效价的慢病毒载体,从而完成了本发明。
即,本发明如下所述:
[1]一种产生慢病毒载体的方法,所述方法在以包装混合物共转染293t细胞时,同时表达激活启动子的因子,从而增强慢病毒载体产生量,其中,所述包装混合物包含:含有编码形成慢病毒粒子所必需的蛋白的基因的多于一个质粒,以及含有转录为整合到慢病毒粒子中的rna的基因和要表达的目的导入基因的质粒。
[2]根据[1]的产生慢病毒载体的方法,其中,激活启动子的因子是激活cmv启动子的因子。
[3]根据[1]或[2]的方法,其中,激活启动子的因子是选自htlv-1tax、hiv-1tat、nf-κbrela、ap-1和creb/atf中的一种或更多种。
[4]根据[1]~[3]中任一项的产生慢病毒载体的方法,其中,所述方法在以包装混合物共转染293t细胞时,同时在293t细胞中表达htlvtax或nf-κbrela,从而增强慢病毒载体产生量,其中,所述包装混合物包含:含有编码形成慢病毒粒子所必需的蛋白的基因的多于一个质粒,以及含有转录为整合到慢病毒粒子中的rna的基因和要表达的目的导入基因的质粒。
[5]根据[1]~[3]中任一项的产生慢病毒载体的方法,其中,所述方法在以包装混合物共转染293t细胞时,同时在293t细胞中表达选自htlv-1tax、hiv-1tat和nf-κbrela中的2种,或者表达htlv-1tax、hiv-1tat和nf-κbrela这3种,从而增强慢病毒载体产生量,其中,所述包装混合物包含:含有编码形成慢病毒粒子所必需的蛋白的基因的多于一个质粒,以及含有转录为整合到慢病毒粒子中的rna的基因和要表达的目的导入基因的质粒。
[6]根据[4]或[5]的产生慢病毒载体的方法,其中,构成包装混合物的多于一个质粒由至少含有ltr即5’ltr和3’ltr、包装信号ψ和目的导入基因即转基因的慢病毒载体质粒和彼此独立地含有gag、pol、rev和env的多于一个质粒组成。
[7]根据[6]的产生慢病毒载体的方法,其中,构成包装混合物的多于一个质粒由如下3个质粒组成:(1)至少含有ltr即5’ltr和3’ltr、包装信号ψ和目的导入基因即转基因的慢病毒载体质粒、(2)含有包装所必需的gag和pol且任选含有调控基因rev和tat的包装质粒,以及(3)含有编码vsv-g的基因的包膜质粒。
[8]根据[6]的产生慢病毒载体的方法,其中,构成包装混合物的多于一个质粒由如下4个质粒组成:(a)至少含有ltr即5’ltr和3’ltr、包装信号ψ和目的导入基因即转基因的慢病毒载体质粒、(b)含有包装所必需的gag和pol的包装质粒、(c)含有编码vsv-g的基因的包膜质粒,以及(d)含有rev的rev表达质粒。
[9]根据[6]的产生慢病毒载体的方法,其中,构成包装混合物的多于一个质粒由如下3个质粒组成:(a)至少含有ltr即5’ltr和3’ltr、包装信号ψ和目的导入基因即转基因的慢病毒载体质粒、(b)含有包装所必需的gag和pol的包装质粒,以及(c)含有包膜表达单元和rev表达单元的质粒,所述包膜表达单元含有编码vsv-g的基因,所述rev表达单元含有rev。
[10]根据[8]或[9]的产生慢病毒载体的方法,其中,慢病毒载体质粒中的5’ltr和3’ltr被修饰。
[11]根据[3]~[10]中任一项的产生慢病毒载体的方法,其中,tax未被整合到慢病毒载体中。
[12]一种用于在293t细胞中产生慢病毒载体的试剂盒,所述试剂盒包含包装混合物和表达载体,其中,所述包装混合物包含:含有编码形成慢病毒粒子所必需的蛋白的基因的多于一个质粒,以及含有转录为整合到慢病毒粒子中的rna的基因和要表达的目的导入基因的质粒;所述表达载体含有编码激活启动子的因子的基因。
[13]根据[12]的用于产生慢病毒载体的试剂盒,其中,激活启动子的因子是激活cmv启动子的因子。
[14]根据[12]或[13]的试剂盒,其中,激活启动子的因子是选自htlv-1tax、hiv-1tat、nf-κbrela、ap-1和creb/atf中的一种或更多种。
[15]根据[12]~[14]中任一项的用于在293t细胞中产生慢病毒载体的试剂盒,所述试剂盒包含包装混合物和表达载体,其中,所述包装混合物包含:含有编码形成慢病毒粒子所必需的蛋白的基因的多于一个质粒,以及含有转录为整合到慢病毒粒子中的rna的基因和要表达的目的导入基因的质粒;所述表达载体含有编码htlv-1tax或nf-κbrela的基因。
[16]根据[12]~[14]中任一项所述的用于在293t细胞中产生慢病毒载体的试剂盒,所述试剂盒包含包装混合物和表达载体,其中,所述包装混合物包含:含有编码形成慢病毒粒子所必需的蛋白的基因的多于一个质粒,以及含有转录为整合到慢病毒粒子中的rna的基因和要表达的目的导入基因的质粒;所述表达载体含有编码选自htlv-1tax、hiv-1tat和nf-κbrela中的2种或含有编码htlv-1tax、hiv-1tat和nf-κbrela这3种的基因。
[17]根据[16]的用于产生慢病毒载体的试剂盒,其中,含有编码选自htlv-1tax、hiv-1tat和nf-κbrela中的2种或者含有编码htlv-1tax、hiv-1tat和nf-κbrela这3种的基因的表达载体是分别不同的载体。
[18]根据[15]~[17]中任一项的用于产生慢病毒载体的试剂盒,其中,构成包装混合物的多于一个质粒由至少含有ltr即5’ltr和3’ltr、包装信号ψ和目的导入基因即转基因的慢病毒载体质粒和彼此独立地含有gag、pol、rev和env的多于一个质粒组成。
[19]根据[18]的用于产生慢病毒载体的试剂盒,其中,构成包装混合物的多于一个质粒由如下3个质粒组成:(1)至少含有ltr即5’ltr和3’ltr、包装信号ψ和目的导入基因即转基因的慢病毒载体质粒、(2)含有包装所必需的gag和pol且任选含有调控基因rev和tat的包装质粒,以及(3)含有编码vsv-g的基因的包膜质粒。
[20]根据[18]的用于产生慢病毒载体的试剂盒,其中,构成包装混合物的多于一个质粒由如下4个质粒组成:(a)至少含有ltr即5’ltr和3’ltr、包装信号ψ和目的导入基因即转基因的慢病毒载体质粒、(b)含有包装所必需的gag和pol的包装质粒、(c)含有编码vsv-g的基因的包膜质粒,以及(d)含有rev的rev表达质粒。
[21]根据[18]的用于产生慢病毒载体的试剂盒,其中,构成包装混合物的多于一个质粒由如下3个质粒组成:(a)至少含有ltr即5’ltr和3’ltr、包装信号ψ和目的导入基因即转基因的慢病毒载体质粒、(b)含有包装所必需的gag和pol的包装质粒,以及(c)含有包膜表达单元和rev表达单元的质粒,所述包膜表达单元含有编码vsv-g的基因,所述rev表达单元含有rev。
[22]根据[20]或[21]的用于产生慢病毒载体的试剂盒,其中,慢病毒载体质粒中的5’ltr和3’ltr被修饰。
[23]一种293t细胞,其含有[15]~[22]中任一项的试剂盒中所含的质粒和载体。
本说明书包含作为本申请优先权的基础的日本专利申请号2018-176230号的公开内容。
发明效果
在293t细胞中共转染慢病毒包装所必需的多于一个质粒时,通过以单独同时表达htlv-1tax或者nf-κbrela,或者同时表达htlv-1tax与nf-κbrela、htlv-1tax与hiv-1tat或nf-κbrela与hiv-1tat的两者或者htlv-1tax与nf-κbrela与hiv-1tat的三者,能够增强慢病毒载体产生。通过本发明的方法,能够改善慢病毒载体产生,容易且安全地产生慢病毒载体。该结果有助于降低细胞培养和转染操作步骤成本,通过组合目前的浓缩和纯化技术,能够用于多种多样的体外和体内的应用。
而且,在本发明的方法中,因为tax蛋白未被整合到慢病毒载体粒子中,因此确保了制备物的安全性。
附图说明
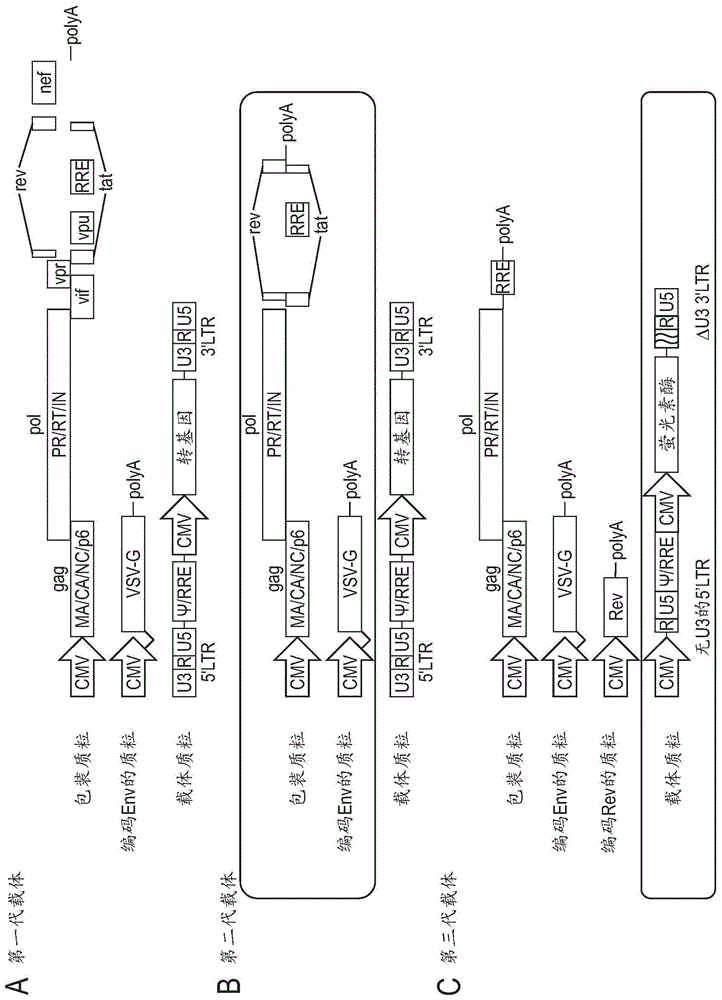
图1-1是表示代表性的第一代、第二代和第三代慢病毒载体系统的结构图。
图1-2是表示vsv-g和rev包含在一个质粒中的第三代慢病毒载体系统的结构图。
图2是表示tax在hek293t细胞中强力激活cmv启动子的图。
图3是表示慢病毒载体产生细胞中的tax共表达所引起的慢病毒载体的产生增强图。
图4是表示慢病毒载体产生细胞中的tax表达和gag表达的水平图。
图5是表示tax引起的病毒从产生细胞释放的增强图。
图6是表示tax被整合到慢病毒载体粒子中的可能性低的图。
图7-1是表示单独地同时表达htlv-1tax(a)、hiv-1tat(b)或nf-κbrela(c)时的慢病毒载体的产生增强效果图。
图7-2是表示在同时表达htlv-1tax与hiv-1tat(a)、htlv-1tax与nf-κbrela(b)或nf-κbrela与hiv-1tat(c)的两者或者htlv-1tax、nf-κbrela、hiv-1tat的三者(d)时的慢病毒载体的产生增强效果图。
图8是表示psv质粒的结构图。
图9是表示在实施例2中使用的转移质粒的结构图。
图10是表示单独或同时表达hiv-1tat或nf-κbrela时的慢病毒载体的产生增强的结果图。
图11是表示实施例3中使用的转移质粒的结构图。
图12是表示使用venus作为报告基因时的效价测定方法的概要图。
图13是表示使用venus作为报告基因时的效价测定的结果(效价的绝对值)图。
图14是表示使用venus作为报告基因时的效价测定的结果(效价的增加程度)图。
具体实施方式
下面,详细说明本发明。
本发明是一种产生慢病毒载体方法,并且是一种增强慢病毒载体产生量的方法。
在本发明的方法中,作为具有产生慢病毒载体能力的包装细胞,使用293t(hek293t)细胞,在293t细胞中产生慢病毒时,共表达作用于启动子并激活启动子的因子。启动子并没有限定,例如,可举出:cmv启动子。作为激活启动子的因子,可举出:tax、nf-κbrela、hiv-1tat(下面,记为“hiv-1tat”或“tat”)、ap-1、creb/atf等,也可以共表达其中之一,还可以组合共表达多于一个,如2个、3个、4个或5个。在本发明中,作为所述激活启动子的因子,优选共表达选自tax、nf-κbrela和tat中的一个以上的因子。
另外,在本发明的方法中,作为具有产生慢病毒载体能力的包装细胞,使用293t(hek293t)细胞,在293t细胞中产生慢病毒时,共表达tax、nf-κbrela等转录因子、tat或它们的组合。
慢病毒载体是基于人免疫缺陷病毒1型(hiv-1)而开发的。
为了制造慢病毒载体,将hiv-1前病毒基因组分开并含有在多于一个质粒中,并将各质粒共转染(同时(共)转染)入细胞即可。这时,无需将病毒基因组的全部分开并含有在多于一个质粒中,含有除去辅助基因等基因之外的病毒基因组一部分即可。即,将含有编码形成慢病毒粒子所必需的蛋白的基因的多于一个质粒和含有转录为整合到慢病毒粒子中的rna的基因和要表达的目的导入基因的质粒(慢病毒载体质粒)共转染入包装细胞即可。作为形成慢病毒粒子所必需的蛋白,可举出:包装所必需的gag、pol、tat、rev等蛋白、vsv-g(gglycoproteinofvesicularstomatitisvirus:水疱性口炎病毒糖蛋白)等包膜(env)蛋白。vsv-g在具有广泛的宿主范围,物理强度高这方面为优选,但也可以使用其他的包膜。gag基因编码内部结构(细胞间质、衣壳和核衣壳)蛋白,pol基因编码rna依赖性dna聚合酶(逆转录酶)、蛋白酶和整合酶,env基因编码病毒包膜糖蛋白。此外,慢病毒载体质粒是含有包装信号(ψ)且插入目的基因的质粒。
编码包装所必需的gag、pol、tat、rev等蛋白的核酸、编码vsv-g等包膜(env)蛋白的核酸被分开并含有在多于一个质粒、例如3~5个质粒,优选为3或4个质粒中,这些多于一个质粒与慢病毒载体质粒共转染即可。
共转染的结果,由慢病毒载体质粒转录的重组病毒rna基因组在包装信号(ψ)的作用下整合到包装蛋白中,形成病毒核。
这样,编码病毒形成所必需的蛋白的核酸分散并含有在不同的质粒中,从而防止通过同源重组生成野生型hiv-1。感染细胞的慢病毒载体是非感染性且安全的,因为它们无法在细胞中自我复制。
例如,可以使用慢病毒载体质粒、含有编码gag、pol、tat、rev等蛋白的核酸的质粒和含有编码vsv-g等包膜(env)蛋白的核酸的质粒。此外,调控基因rev和tat也可以单独地整合到其他质粒中。
具体地,可以使用如下3个质粒:(1)至少含有ltr(5’ltr和3’ltr)、包装信号(ψ)和目的导入基因(转基因)的慢病毒载体质粒、(2)含有包装所必需的gag和pol且任选含有调控基因rev和tat的包装质粒,以及(3)含有env基因的env表达质粒,所述env基因编码vsv-g等包膜(env)(包膜质粒)。而且,也可以使用如下4个质粒:(a)至少含有ltr(5’ltr和3’ltr)、包装信号(ψ)和目的导入基因(转基因)的慢病毒载体质粒、(b)含有包装所必需的gag和pol的包装质粒、(c)含有env基因的env表达质粒,所述env基因编码vsv-g等包膜(env)(包膜质粒),以及(d)含有rev的rev表达质粒。此外,还可以使用如下3个质粒:(a)至少含有ltr(5’ltr和3’ltr)、包装信号(ψ)和目的导入基因(转基因)的慢病毒载体质粒、(b)含有包装所必需的gag和pol的包装质粒,以及(c)含有env表达单元和rev表达单元质粒,所述env表达单元含有env基因,所述env基因编码vsv-g等包膜(env),所述rev表达单元质粒含有rev。
5’ltr和3’ltr可以被修饰,例如,5’ltr和3’ltr的u3可以被缺失或突变,这时,5’ltr的u3可以被cmv启动子等启动子置换。
在细胞内产生慢病毒载体中使用的上述多于一个质粒称为用于产生慢病毒的包装混合物。
优选在除了慢病毒载体质粒之外的质粒的3’末端侧连接polya加尾序列(多腺苷酸化序列,polya)。polya加尾序列(多腺苷酸化序列,polya)的来源没有限定,可举出:来源于生长激素基因的polya加尾序列例如来源于牛生长激素基因的polya加尾序列或来源于人生长激素基因的polya加尾序列,来源于sv40病毒的polya加尾序列,来源于人或兔的β球蛋白基因的polya加尾序列等。通过将polya加尾序列包含在表达盒中,使rna的稳定性和翻译效率提高。
上述慢病毒载体质粒在含有包装信号(ψ)的两端hiv-ltr之间整合有内部启动子和作为目的的基因。
慢病毒载体质粒和包装质粒中也可以含有rre(revresponseelement,rev应答元件)。
另外,在各质粒的5’末端侧也可以连接启动子。而且,如上所述,在慢病毒载体质粒中的目的基因的上流含有启动子。使用的启动子没有限定,可以使用cmv启动子、cmv-i启动子(hcmv+内含子启动子)、sv40启动子、ubc启动子、ef1a启动子、rsv启动子、β-肌动蛋白启动子、cag启动子、组织特异性启动子、tet表达调控启动子等,其中优选cmv启动子。
优选vif、vpr、vpu、vpx、nef等hiv的辅助基因中的至少一个被除去或失活。
而且,慢病毒载体质粒可以含有cppt(centralpolypurinetract,中央多聚嘌呤区)和wpre(woodchuckhepatitisvirusposttranscriptionalregulatoryelement,土拨鼠肝炎病毒转录后调节元件)。cppt使逆转录效率提高,wpre使rna的表达效率提高。
质粒中的各元件需要功能性连接。在此,功能性连接是指各个元件发挥其功能,以增强要表达的基因的表达地进行连接。
用于产生慢病毒载体的慢病毒载体系统为了防止同源重组引起的野生型病毒的发生,而改良成第一代慢病毒载体系统、第二代慢病毒载体系统、第三代慢病毒载体系统。
使用上述(1)~(3)这3个质粒的慢病毒载体系统是第二代慢病毒载体系统,使用(a)~(d)这4个质粒的慢病毒载体系统是第三代慢病毒载体系统。
第三代慢病毒载体系统中,可以将慢病毒载体质粒的5’ltr的u3置换为cmv启动子,在3’ltr的u3中引入突变使失去其启动子活性。将这样的慢病毒载体质粒称为sin(selfinactivating,自我失活)质粒。已报告了在将5’ltr的u3置换为cmv启动子时,不需要tat,可以从包装质粒除去tat(miyoshietal.,j.virol.,70,8150,1998;dulletal.,j.virol.,72,8463,1998.)。第三代慢病毒载体是不含tat的tat非依赖性慢病毒载体系统。
图1-1中表示代表性的第一代慢病毒载体系统、第二代慢病毒载体系统、第三代慢病毒载体系统的构成(根据biochemj.(2012)443,603-618进行修改)。此外,图1-2中表示vsv-g和rev包含在一个质粒中的第三代慢病毒载体系统的构成。
在本发明中,优选使用第二代慢病毒载体系统或第三代慢病毒载体系统。第二代慢病毒载体系统具有慢病毒载体产生量高的优点,第三代慢病毒载体系统具有安全性高的优点。
在本发明的方法中,在用293t细胞共转染构成上述慢病毒载体系统的质粒时,在细胞中同时共表达tax、nf-κbrela等转录因子、tat等。
tax是人t细胞白血病病毒1型(htlv-1)病毒基因的转录激活因子,通过激活ap-1、nf-κb和creb/atf等转录因子,激活的转录因子结合cmv启动子等启动子中的结合序列,从而作用于启动子而共转染入293t细胞中,促进构成该慢病毒载体系统的质粒的表达,从而增强产生慢病毒载体。
在共表达tax中,除了构成慢病毒载体系统的质粒之外,还将含有编码tax蛋白的核酸的表达载体共转染入293t细胞并共表达即可。作为表达载体,还可以使用除了慢病毒载体之外的病毒载体,例如,腺病毒、腺相关病毒、γ-逆转录等。tax表达载体中的启动子也没有限定,可以使用cmv启动子、cmv-i启动子(hcmv+内含子启动子)、sv40启动子、ef1α启动子、rsv启动子、β-肌动蛋白启动子、cag启动子、组织特异性启动子等。表达可以是暂时性表达,也可以是稳定地表达。稳定地表达例如可以将编码tax蛋白的核酸整合到293t细胞的基因组中。
htlv-1tax是强力的病毒反式作用蛋白,因此如果其反式作用功能充分,则被整合到慢病毒载体粒子中可能在靶细胞中引起不良作用。在本发明的方法中,不允许tax整合入慢病毒粒子内。
而且,也可以共表达作用于构成慢病毒载体系统的质粒中的cmv启动子等启动子的转录因子。
由于在cmv启动子等启动子中存在ap-1、nf-κb和creb/atf识别位点,因此可以将tax用于慢病毒载体产生。这是因为tax激活全部这些转录因子。如果同时表达少量的tax,则在产生细胞中gag的表达和慢病毒载体粒子的释放这两者显著增加,转导效率提高了超过10倍。作为被激活的转录因子的候选,可举出:ap-1、nf-κb和creb/atf。如上所述,tax激活ap-1、nf-κb和creb/atf等转录因子,被激活的转录因子作用于cmv启动子等启动子。可以共表达这些转录因子,在共表达这些转录因子时,293t细胞中的转录因子量增加,容易被tax激活,结果促进了共转染入293t细胞的构成慢病毒载体系统的质粒的表达,可以期待增强慢病毒载体产生。这种情况下,nf-κb表达含有转录激活区域的nf-κbrela即可。
另外,可以与tax一起,或者与tax转录因子一起共表达tat。通过共表达tax与tat,累加性增强慢病毒产生。在第三代慢病毒载体系统中是tat非依赖性慢病毒载体系统,但在第三代慢病毒载体系统中,也是通过共表达tax和tat来增强慢病毒载体产生。
转录因子和tat的表达可以通过与tax的表达相同的方法来进行。即,将含有编码各蛋白的核酸的表达载体共转染入293t细胞,进行共表达即可。这时,也可以使用含有各自的不同的多于一个载体,也可以使用含有tax、转录因子和tat中2个以上或全部的载体进行共表达。各质粒的给予量只要适当设定即可,只要将编码tax、tat和nf-κbrela的核酸以载体量换算成为相同量,或者以2至3倍左右的量比进行转染即可。例如,在使用12孔板进行共转染时,含有编码tax或tat的核酸的载体以约0.1μg/12孔,含有编码nf-κbrela的核酸的载体以约0.1~0.2μg/12孔进行转染即可。
另外,同时表达tax、转录因子和tat中的至少一个时,由于慢病毒的产生量增大,因此可以减少含有编码形成慢病毒粒子所必需的蛋白的基因的质粒的量。例如,与现有方法(慢病毒载体,三好浩之,http://cfm.brc.riken.jp/wp-content/uploads/subteam_for_manipulation_of_cell_fate_j/protocols_j_files/lentiviral%20vector%20prep.pdf)相比,即使使用1/2~1/3量也能够达到与现有方法同等以上的慢病毒产生量。作为原因,认为是通过减少用于表达慢病毒载体组成元件的质粒量,改善了慢病毒载体产生细胞的状态,以及仅通过激活cmv增强子/启动子就足以获得最大效果。
如上所述,将构成慢病毒载体系统的质粒共转染入包装细胞293t细胞,进一步共表达tax或者tax与tat和/或转录因子即可。具体地,共表达tax单独、rela单独、tax与tat的2种、tax与rela的2种、tat与rela的2种、tax与tat与rela的3种即可。
293t(hek293t)细胞是来源于人胎儿肾脏细胞系的293细胞(hek293细胞)的衍生株,是表达sv40larget抗原的细胞系。293t细胞可商购,可以从atcc(americantypeculturecollection,美国典型培养物保藏中心)等获得。在本发明中,也可以使用来源于293t细胞的衍生细胞,作为这样的细胞,可举出:lenti-x(商标)293t细胞(宝生物株式会社)。在本发明中,这样的293t细胞的衍生株也称为293t细胞。
利用质粒的293t细胞转染可以通过已知的方法来进行。例如,可举出:使用基因导入试剂的方法、磷酸钙法、脂质体法、deae葡聚糖法、电穿孔法、微注射法等。作为基因导入试剂,可举出:聚乙烯亚胺(pei,直链型或者支链型)和市售的基因导入试剂。作为市售的基因导入试剂,可举出:lipofectamine(注册商标)、lipofectamineplus(注册商标)、jetpei(注册商标)、transit(注册商标)、xfect(商标)、transfectin-lipid(注册商标)、oligofectamine(注册商标)、silentfect(注册商标)、dmrie-c(注册商标)、effectene(注册商标)、fugene(注册商标)等。在聚乙烯亚胺(pei)中,直链pei和含有伯胺、仲胺、叔胺的支链pei均可以使用。此外,pei的分子量也没有限定。进一步还可以使用经脱酰基化等化学修饰的pei。
共转染入293t细胞后,在培养数天后回收培养上清,从培养上清中回收慢病毒粒子。293t细胞的培养中使用的培养基、培养条件是通常的动物细胞的培养中使用的培养基、条件。用于产生慢病毒的培养期间例如为12~72小时,优选为24~48小时。这时,为了回收足够滴度的病毒载体,优选测定培养上清的滴度(效价)。在此,足够的滴度是指1×105ifu/ml以上,优选为5×105ifu/ml以上。滴度可以使用实时rt-pcr、p24的测定elisa、流式细胞仪等进行测定。此外,得到的慢病毒载体的量可以通过向细胞的转导效率进行确认。
根据本发明的方法,慢病毒的产生量在以向细胞,例如向利用萤光素酶报告基因测定方法的mt-4细胞的转导效率为指标时,增强至少3倍,优选为5倍,进一步优选8倍,进一步优选12倍,进一步优选14倍,进一步优选16倍,进一步优选18倍,进一步优选20倍,进一步优选22倍,进一步优选24倍。共表达tax、tat、转录因子的2种或3种时的增强效果明显,共表达tax和tat时增强至少5倍,共表达tax和rela时增强至少12倍,共表达tat和rela时增强至少16倍,共表达tax和tat和rela时增强至少22倍。
回收的慢病毒载体可以通过超速离心等进行浓缩、纯化后使用。
得到的慢病毒载体冷冻保存即可。
这样得到的慢病毒载体不论增殖中的细胞、停止增殖的细胞,都能够用于将基因导入哺乳动物细胞中,导入的基因可以稳定地整合到宿主细胞的基因组中,从而长时间的基因表达。因此,可以适合用作基因治疗用载体。
本发明包括用于增强使用293t细胞的慢病毒载体产生的试剂或试剂盒。
该试剂或试剂盒包含包装混合物和表达载体,其中,所述包装混合物包含:含有上述编码形成慢病毒粒子所必需的蛋白的基因的多于一个质粒,以及含有转录为整合到慢病毒粒子中的rna的基因和要表达的目的导入基因的质粒(慢病毒载体质粒);所述表达载体表达htlv-1tax。该试剂或试剂盒还可以含有表达选自ap-1、nf-κb和creb/atf中的转录因子的表达载体和/或表达hiv-1tat的表达载体。
实施例
通过下面的实施例具体地说明本发明,但本发明不限于这些实施例。
[实施例1]htlv-1tax引起的慢病毒载体产生的增强
材料和方法
细胞
在补充了10%胎牛血清(fbs)、100u/ml青霉素和100μg/ml链霉素的dulbeco改良eagle培养基(nacalaitesque)中培养细胞系hek293t(invitrogen公司,carlsbad,ca)。mt-4细胞保持在补充了10%fbs、100u/ml青霉素和100μg/ml链霉素的完全rpmi1640培养基(nacalaitesque)中。
质粒
pcsii-cmv-mcs-ires2-bsd、pcag-hivgp、pcmv-vsv-g-rsv-rev通过文部科学省国立生物资源项目由理研brc提供。为了生成pcsii-cmv-luc-ires2-bsd,用xhoi和noti消化pcsii-cmv-mcs-ires2-bsd和pcmv-luc。接着,将保持有来源于pcmv-luc的萤火虫萤光素酶基因的xhoi-noti片段插入pcsii-cmv-mcs-ires2-bsd的xhoi-noti位点中。将得到的质粒命名为pcsii-cmv-luc-ires2-bsd。phcmv-vsv-g由i.s.y.chen博士提供。pcmvdeltar8.2、pcmv-neo-bam-tax、pcmv-neo-bam按照yamaoka,s.etal.,cell93,1231-1240(1998);naldini,l.etal.proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica93,11382-11388(1996);hironaka,n.etal.,neoplasia6,266-278,doi:10.1593/neo.03388(2004);baker,s.j.etal.,science249,912-915(1990)中所述的方法制备。
另外,ph2rneotax、psv2tat和prc/cmvrela分别按照yamaokaetal.,emboj.15,873-87,1996;subramanisetal.,mol.cell.biol.,1,854,1981;schmitzandbaeuerle,emboj.,10,3805-3817,1991中所述的方法制备。
蛋白质印迹
使用溶解缓冲液(25mmtris,ph7.8,8mmmgcl2,1mmdtt、1%triton-x100,15%甘油)制备细胞溶解物。蛋白浓度由考马斯亮蓝(bradford)测定方法来确定。通过sds-page分离蛋白,转印至聚偏二氟乙烯(pvdf)膜上,与如下抗体反应:针对tax的小鼠单克隆抗体(mi73)(mori,k.etal.,thejournalofgeneralvirology68(pt2),499-506,doi:10.1099/0022-1317-68-2-499(1987).)、针对hiv-1p24的小鼠单克隆抗体(39/5.4a,abcam公司)、针对亲环素a的兔多克隆抗体(bml-sa296,enzolifesciences公司)或针对α-微管蛋白的小鼠单克隆抗体(dm1a,sigma-aldrich公司)。然后,将辣根过氧化物酶标记的兔抗小鼠igg(a206ps,americanqualexinternational公司)或山羊抗大鼠igg(sc-2006,santacruzbiotechnology)与膜一起孵育,利用westernlightningplus-ecl(perkinelmer)使蛋白可视化。
病毒液的准备
为了在tax的存在下的产生慢病毒载体,使用pei(聚乙烯亚胺)以每6孔板1.4μg的pcsii-cmv-luc-ires2-bsd、0.9μg的pcmvdeltar8.2和0.4μg的phcmv-vsv-g与pcmv-neo-bam-tax和/或pcmv-neo-bam进行共转染。在转染48小时后收获上清中的病毒。上清中的hiv-1衣壳(ca)的量利用hiv-1ca(p24)elisa(enzyme-linkedimmunosorbentassay,酶联免疫吸附测定)(zeptmetrix公司)进行定量。
另外,tax、tat或rela以单独、组合2种或者3种全部进行共表达。在tax、tat或rela单独共表达,tax、tat和rela中两者共表达,tax、tat和rela三者共表达中的第三代慢病毒载体产生量的效果使用pei以每24孔板0.14μg的pcag-hivgp、0.14μg的pcmv-vsv-g-rsv-rev、0.24μg的pcsii-cmv-luc-ires2-bsd与各自的表达载体或其对照空载体(ev1、ev2和ev3)0.05μg进行共转染。
萤光素酶报告基因测定方法
感染慢病毒载体的24孔板中的约2×105个mt4细胞在感染24小时后回收,用溶解缓冲液(25mmtrisph7.8,8mmmgcl2,1mmdtt,1%triton-x100,15%甘油)溶解,然后测定萤光素酶活性。萤火虫萤光素酶活性按照制造商的操作手册和glomax-multidetection系统(promega公司)进行测定。
萤光素酶的活性相对于通过考马斯亮蓝(bradford)测定方法确定的蛋白浓度进行了标准化。
慢病毒载体的纯化
将2ml的20%蔗糖溶液加入到型号sw55型超速离心管底部,用2ml的含慢病毒载体的培养上清进行分层。接着,在4℃、35,000rpm下对试样离心分离60分钟。将可沉淀的组分重悬于pbs(-)中,进行蛋白质印迹。
结果
htlv-1tax极大地增强了慢病毒载体产生。
利用hek293t细胞产生慢病毒载体通过将能够表达质粒(phcmv-vsv-g)的慢病毒转移载体与cv(对照载体:空载体)共转染入hek293t细胞来进行,所述质粒(phcmv-vsv-g)表达萤火虫萤光素酶(pcsii-cmv-luc-ires2-bsd)、包装质粒(pcmvdeltar8.2)和水疱性口炎病毒糖蛋白。
而且,假设同时激活ap-1、nf-κb和creb/atf的蛋白高效地增加慢病毒产生。这样的候选蛋白是htlv-1tax,熟知的是激活包含ap-1、nf-κb和creb/atf19在内的数个细胞转录因子(curreretal.,frontiersinmicrobiology,3,article406,2012)。既往的研究报告了tax在hek293细胞中激活cmv启动子,但在hek293t细胞中未激活(lwa,t.r.etal.,biotechnologyprogress27,751-756,doi:10.1002/btpr.571(2011).)。但是,本发明人发现tax在hek293t细胞中强力激活cmv启动子。具体地,在约2×105个hek293t细胞中,利用0.1μg的pcmv-luc和0.2μg的pcmv-neo-bam-tax(tax)作为报告载体或利用pcmv-neo-bam作为对照载体(cv)进行共转染。将30μg的细胞溶解物供于使用抗htlv-1tax或抗-α-微管蛋白抗体的蛋白质印迹(图2下方的图)。图2上方的图表示萤光素酶测定的结果与cv相比的倍数。小横线表示由3个独立实验计算出的标准误差。
在约1.5×106个hek293t细胞中,将0.4μg的phcmv-vsv-g、0.9μg的pcmvδr8.2和1.4μg的pcsii-cmv-luc-ires2-bsd与cv或依次增量的pcmv-neo-bam-tax中任一进行共转染。效应质粒的总量调整至0.4μg。转染48小时后,收集含慢病毒载体的培养上清。暴露于培养上清中的mt-4细胞中的转导效率在暴露24小时后进行评价。萤光素酶活性使用蛋白浓度进行校正,结果表示为与对照(0μg的pcmv-neo-bam-tax)相比较的倍数。示出了由3次独立实验计算出的平均值和标准误差。之后,报告测定的结果使用蛋白浓度进行校正。慢病毒载体产生细胞中的tax共表达如图3的感染细胞中的萤光素酶活性所示,观察到慢病毒载体产生最大增加12倍。
转染48小时后回收如图3所示的转染细胞。将同组的30μg溶解产物供于使用抗htlv-1tax和抗-α-微管蛋白抗体或抗hiv-1p24和抗亲环素a抗体的蛋白质印迹。如图4所示,根据蛋白质印迹,确认到产生细胞中的tax表达的剂量依赖性增加。产生细胞中gag的表达水平与慢病毒载体转导的结果高度相关。
为了评价tax是否增加了病毒从产生细胞释放,利用特异性elisa测定了hiv-1gag的浓度。转染的细胞上清中的gag蛋白量利用hiv-1ca(p24)elisa进行定量,其结果表示为与对照(0μg的pcmv-neo-bam-tax)相比较的倍数。示出了由3个独立实验计算出的平均值和标准误差。如图5所示,上清中的gag的量在tax的存在下同样地增加,与产生细胞中的转导效率和gag表达水平高度相关(图3和4)。
最后,从安全性与去除污染的角度考虑,对tax是否被整合入慢病毒载体粒子中进行了研究。
由于htlv-1tax是强力的病毒反式作用蛋白,因此如果其反式作用功能充分,则混入到慢病毒载体粒子中,有可能在靶细胞中引起不期望的效果。
培养上清中的病毒粒子通过蔗糖垫层并利用超速离心以沉淀物进行回收。即,利用20%蔗糖的超速离心,回收慢病毒粒子。接着,将细胞溶解物和沉淀的病毒粒子两者供于蛋白质印迹。将同组的30μg溶解产物供于使用抗htlv-1tax和抗-α-微管蛋白抗体或者抗hiv-1p24和抗亲环素a抗体的蛋白质印迹。如图6所示,tax蛋白在通过超速离心得到的沉淀物中的蛋白质印迹未被检测出。该结果表明tax被整合到慢病毒载体粒子的可能性非常低。
本实施例显示出htlv-1tax等转录激活因子的共表达,通过增强产生细胞中的病毒蛋白的表达而大幅提高了慢病毒载体产生(图2~图6)。tax增强了由cmv启动子驱动的报告基因表达(图2),而且增强了产生细胞中的gag表达。这与慢病毒载体转导的结果高度相关(图3和图4)。在本实施例的实验环境中,cmv启动子驱动慢病毒转移载体(pcsii-cmv-luc-ires2-bsd)、包装质粒(pcmvdeltar8.2)和表达包膜的质粒(phcmv-vsv-g)。因此,这些结果强烈表明来自这些质粒的基因表达的增强使慢病毒载体高效产生。但是,认可的是还可能tax激活某种宿主hiv-1依赖性因子的表达,接着有助于增强的慢病毒表达。值得注意的是,tax表达载体0.4μg在进行共转染时,慢病毒载体产生从峰值下降(图3)。该结果表示过量的质粒对慢病毒载体产生带来不良影响。实际上在这样的条件下,产生细胞中gag的表达减少(图4)。
[实施例2]htlv-1tax、tat或nf-κbrela单独或同时表达时的慢病毒载体的产生增强
接着,使用第三代慢病毒载体系统pcag-hivgp、pcmv-vsv-g-rsv-rev、pcsii-cmv-luc-ires2-bsd,以mt4细胞中的萤光素酶活性为指标,对下面组合的共表达所引起的慢病毒载体产生增大效果进行了研究(图7)。
图7-1中表示tax单独共表达(图7-1a)、tat单独共表达(图7-1b)和rela单独共表达(图7-1c)对第三代慢病毒载体产生量的效果。
图7-2中表示在tax、tat、rela中两者共表达(图7-2a:tax和tat;图7-2b:tax和rela;图7-2c:tat和rela)对第三代慢病毒载体产生量的效果。
图7-2d中表示tax、tat和rela三者共表达对第三代慢病毒载体产生量的效果。
如图7-1所示,在单独共表达时,得到tax中为4.8倍、tat中为5.1倍、rela中为15.6倍的慢病毒载体产生增大效果,此外,如图7-2所示,在两者共表达时,得到tax+tat中为5.8倍、tax+rela中为14.0倍、tat+rela中为17.1倍的慢病毒载体产生增大效果,在三者共表达时,得到tax+tat+rela中为24.9倍的慢病毒载体产生增大效果。倍率表示相对于在使用空载体(ev1、ev2和ev3)时值的倍数诱导(foldinduction)。
[实施例3]tat或nf-κbrela单独或同时表达时慢病毒载体的产生增强
(1)在被胶原蛋白包被的24孔板中,用0.5ml的含10%牛胎儿血清的dmem将人胚肾(humanembryonickidney)(hek)293t细胞,以3.0×105细胞/孔进行播种并使细胞粘着。
(2)2小时后,将下面的质粒与opti-mem(gibco)900μl混合,加入1μg/μl的聚乙烯亚胺(polyethylenimine,pei)30μl并在室温下孵育30分钟,然后滴加到细胞中。
(i)0.25μgpcag-hivgp
(ii)0.125μgpcmv-vsv-g-rsv-rev
(iii)0.375μgpcsii-cmv-luc-ires2-bsd
(iv)0.05μgpsv2tat或psv2
(v)0.025μgprc/cmvrela或prc/cmv(invitrogen公司制造)
psv质粒的结构如图8所示。
(3)24小时后,更换培养液。
(4)48小时后,回收含慢病毒载体的培养上清,用0.22μm滤器(millex)过滤,然后立刻使用其中100μl感染2×105mt4细胞(人t细胞系)。
(5)24小时后,离心回收mt4细胞,用含tritonx1%的缓冲液(25mmtrisph7.8,8mmmgcl2,1mmdtt,1%triton-x100,15%甘油)50μl溶解。离心后,使用其中20μl与实施例2同样地进行萤光素酶测定,按照制造商的操作手册和glomax-multidetection系统进行测定。
图9中表示(iii)的转移质粒(表达含有导入基因的病毒载体基因组)的结构。图10中表示萤光素酶测定的结果。如图10所示,感染靶细胞中的基因(萤光素酶)表达量在tat单独表达中增加4倍左右,在rela单独表达中增加5倍左右,在两者共表达中增加11倍左右。
[实施例4]使用venus作为报告基因时,共表达tat、rela、tat+rela时的测定效价
(1)在被胶原蛋白包被的24孔板中,用0.5ml的含10%胎牛血清的dmem将人胚肾(humanembryonickidney)(hek)293t细胞,以3.0×105细胞/孔进行播种并使细胞粘着。
(2)2小时后,将下面的质粒与opti-mem900μl混合,加入1μg/μl的聚乙烯亚胺(polyethylenimine,pei)30μl并在室温下孵育30分钟,然后滴加到细胞中。
(i)0.25μgpcag-hivgp
(ii)0.125μgpcmv-vsv-g-rsv-rev
(iii)0.375μgpcsii-cmv-hsvtk-ires2-venus
(iv)0.05μgpsv2tat或psv2
(v)0.025μgprc/cmvhrela或prc/cmv
pcsii-cmv-hsvtk-ires2-venus按照以下的方法制作:
在csii-cmv-rfa-ires2-venus(由三好浩之博士提供,riken)的nhei识别位点与bamhi识别位点之间,插入以pnl4-3tk(horietal.,jvi2013)为模板通过pcr扩增的来源于hsv-1的tk(thymidinekinase,胸苷激酶)dna片段。
pcr中使用的引物如下所述:
csiinhei中的5’-tk:gctctagagctagcatggcttcgtacccctgccatcaacac(seqidno:3)
csiibamhi中的3’-tk:cgcggatcctcagttagcctcccccatctcc(seqidno:4)
(3)24小时后,更换培养液。
(4)48小时后回收含慢病毒载体的培养上清,用0.22μm滤器过滤,然后立刻将培养上清二倍逐级稀释至1/8~1/64,使用其中10μl感染1×105mt4细胞(人t细胞系)。
(5)48小时后,使用facs分析装置(bectondickinson公司的facscalibur)测定venus荧光阳性细胞的比例。
图11中表示(iii)的转移质粒(表达含有导入基因的病毒载体基因组)的结构。
图12表示使用venus作为报告基因时的效价测定方法的概要。
图13和图14中表示效价测定的结果。图13表示效价的绝对值(慢病毒载体的效价),图14表示将使用对照载体时的效价作为100%时的效价增加程度。如图13和图14所示,观察到效价的增加在tat单独中为3倍左右,在rela单独表达中为4倍左右,在两者共表达中为6倍左右,达到2×107transductionunit(tu,转导单位)/ml的高效价。
工业上的可利用性
根据本发明的方法,能够安全高效率地产生慢病毒载体,其可用于基因治疗等的基因导入用。
序列表自由文本
seqidno:1引物
seqidno:2引物
seqidno:3引物
seqidno:4引物
本说明书所引用的所有出版物,专利和专利申请通过引用整体并入本文。
序列表
<110>国立大学法人东京医科齿科大学
<120>增强慢病毒载体产生的方法
<130>ph-8070-pct
<150>jp2018-176230
<151>2018-09-20
<160>4
<170>patentinversion3.5
<210>1
<211>26
<212>dna
<213>人工的(artificial)
<220>
<223>引物
<400>1
ccggatccgccaccatggcttccaag26
<210>2
<211>27
<212>dna
<213>人工的(artificial)
<220>
<223>引物
<400>2
aactcgagttagacgttgatcctggcg27
<210>3
<211>41
<212>dna
<213>人工的(artificial)
<220>
<223>引物
<400>3
gctctagagctagcatggcttcgtacccctgccatcaacac41
<210>4
<211>31
<212>dna
<213>人工的(artificial)
<220>
<223>引物
<400>4
cgcggatcctcagttagcctcccccatctcc31
- 还没有人留言评论。精彩留言会获得点赞!