新的偶联核酸分子及其用途的制作方法
本发明涉及医学领域,特别是肿瘤学领域。
背景技术:
dna损伤应答(ddr)检测dna病变并促进它们的修复。dna病变类型的广泛多样性需要多种很大程度上不同的dna修复机制例如错配修复(mmr)、碱基切除修复(ber)、核苷酸切除修复(ner)、单链断裂修复(ssb)和双链断裂修复(dsb)。例如,聚腺苷酸核糖聚合酶(parp)基本上参与修复ssb,而修复dna中的dsb使用两种主要机制:非同源末端连接(nhej)和同源重组(hr)。在nhej中,dsb被ku蛋白识别,然后结合并活化蛋白激酶dna-pkc,导致末端加工酶的召集和活化。已证明癌细胞修复治疗诱导的dna损伤的能力影响治疗功效。
这导致了靶向dna修复途径和蛋白质,以开发出提高对传统基因毒性疗法(化学治疗剂、放射疗法)的敏感性的抗癌药剂。用于癌症疗法的合成致死方法提供了新的机制以特异性靶向癌细胞并同时使非癌细胞不受损伤,从而降低与治疗相关的毒性。
在这些合成致死方法中,dbait分子是模拟双链dna病变的核酸分子。它们充当dna损伤信号传导酶parp和dna-pk的诱饵,诱导“假的”dna损伤信号,并最终抑制参与dsb和ssb途径的许多蛋白质在损伤部位处的召集。
dbait分子已在pct专利申请wo2005/040378、wo2008/034866、wo2008/084087和wo2017/013237中广泛描述。dbait分子可以由它们的治疗活性所必需的许多特征例如它们的最小长度来定义,所述最小长度可以变化,只要足以允许包含ku和dna-pkc蛋白的ku蛋白复合体的适当结合即可。因此已显示,dbait分子的长度必须大于20bp,优选约为32bp,以确保与这种ku复合体的结合并且能够使dna-pkc活化。
使用此类dbait分子的治疗的潜在预测性生物标志物已被表征。正如在pct专利申请wo2018/162439中所述,对dbait分子的敏感性事实上与具有微核(mn)的细胞的高自发频率相关。已提出将高的mn基础水平作为使用dbait分子的治疗的预测性生物标志物,随后在来自于各种不同组织的43种实体肿瘤细胞系以及16种细胞和患者来源的异种移植物模型中进行了验证。
此外最近已提出,微核(mn)会提供一个关键平台,作为dna损伤诱导的免疫应答的一部分(gekarajcellbiol.2017oct2;216(10):2999-3001)。最近的研究证实了mn的形成在dna损伤诱导的免疫活化中的作用。有趣的是,事实上,已发现胞质dna感应途径是dna损伤与先天免疫之间的主要联系。dna通常存在于细胞核和线粒体中,因此,它在细胞质中的存在充当触发免疫应答的危险相关分子模式(damp)。环鸟苷单磷酸(gmp)-腺苷单磷酸(amp)合酶(cgas)是检测作为damp的dna并诱导i型ifn和其他细胞因子的传感器。dna以序列不依赖性方式与cgas结合;这种结合诱导cgas的催化中心的构象变化,使得这种酶可以将鸟苷三磷酸(gtp)和atp转变成第二信使环gmp-amp(cgamp)。这种cgamp分子是衔接物蛋白ifn基因刺激物sting的内源高亲和力配体。因此,sting途径的活化可以包括例如炎性细胞因子ip-10(也被称为cxcl10)和ccl5或受体ngk2和pd-l1的刺激。
最近的证据表明sting(干扰素基因的刺激物)途径参与抗肿瘤免疫应答的诱导。因此,现在广泛地将sting激动剂开发为一类新的癌症治疗剂。已显示,癌细胞中sting依赖性途径的活化可以导致肿瘤被免疫细胞的浸润和抗癌免疫应答的调节。
sting是一种内质网衔接物,其促进先天免疫信号传导(一种对抗环境侵害、包括但不限于病原体例如细菌或病毒的快速非特异性免疫应答)。据报道,sting能够活化nf-kb、stat6和irf3转录途径,以诱导i型干扰素(例如ifn-α和ifn-β)的表达并在表达后发挥有效的抗病毒状态。然而,迄今为止开发的sting激动剂能够在所有细胞类型中活化sting途径,并可能触发与其在树突状细胞中活化有关的严重副作用。因此,sting激动剂被局部给药。
因此,对于找到在肿瘤细胞中特异性活化sting途径的方法,存在着真正的需求。
因此,对用于癌症治疗的疗法,尤其是依赖于几种机制的药物特别是dna修复途径和sting途径活化剂,以及对可能有助于检查点抑制剂在更多患者和范围更广的癌症中起作用的药物,仍存在需求。
癌细胞具有用于维持快速增殖的独特能量代谢。在常氧条件下对厌氧糖酵解的偏好是癌症代谢的独特特征,并被称为warburg效应。增强的糖酵解还支持作为癌细胞分裂的构造元件的核苷酸、氨基酸、脂质和叶酸的生成。烟酰胺腺嘌呤二核苷酸(nad)是一种在包括糖酵解在内的许多代谢途径中介导氧化还原反应的辅酶。提高的nad水平增强糖酵解并为癌细胞供能。在这种情况下,nad水平枯竭随后通过抑制能量产生途径例如糖酵解、三羧酸(tca)循环和氧化磷酸化来抑制癌细胞增殖。nad还充当几种酶的底物,从而通过这些酶调控dna修复、基因表达和应激反应。因此,nad代谢除了能量代谢之外还涉及癌症发病机理,并被认为是癌症治疗的有希望的治疗靶点,特别是针对由于dna修复基因缺陷(例如ercc1和atm缺陷)或idh(异柠檬酸脱氢酶)突变而表现出nad缺陷的癌细胞。
对于成功针对癌细胞群体而不出现对疗法有抗性的癌细胞的新的治疗方法,仍存在着需求。
技术实现要素:
本发明提供了在癌细胞中特异性靶向dna修复途径并刺激sting途径的新的偶联核酸分子。更具体来说,所述核酸分子能够活化parp而对dna-pk没有任何活化。
本发明涉及一种偶联核酸分子,其包含:双链核酸组成部分,其中第一链的5’末端与互补链的3’末端通过环连接在一起;和任选的连接到所述环的促进胞吞的分子,
其中
-所述双链核酸组成部分的长度为10至20个碱基对;
-所述双链核酸组成部分的序列与人类基因组中的任何基因具有低于80%的序列同一性;
-所述双链核酸组成部分包含脱氧核糖核苷酸和相对于所述核酸分子的核苷酸总数至多30%的核糖核苷酸或修饰的脱氧核糖核苷酸;并且
-所述环具有选自下述结构式之一的结构:
-o-p(x)oh-o-{[(ch2)2-o]g-p(x)oh-o}r-k-o-p(x)oh-o-{[(ch2)2-o]h-p(x)oh-o-}s(i)
其中r和s独立地是整数0或1;g和h独立地是1至7的整数,并且g+h之和为4至7;
其中k是
其中i、j、k和l独立地是0至6、优选地1至3的整数;
或
-o-p(x)oh-o-[(ch2)d-c(o)-nh]b-chr-[c(o)-nh-(ch2)e]c-o-p(x)oh-o-(ii)
其中b和c独立地是0至4的整数,并且b+c之和为3至7;
d和e独立地是1至3、优选地1至2的整数;并且
其中r是-lf-j,
x是o或s,l是连接物,并且f是0或1的整数,j是促进胞吞的分子或者是h。
所述核酸分子可以包含下述序列之一:
5’cccagcaaacaagcct-∫(seqidno1)
3’gggtcgtttgttcgga-∫
和
5’cagcaacaag-∫(seqidno2)
3’gtcgttgttc-∫
或其中1至3个核苷酸被核糖核苷酸或修饰的脱氧核糖核苷酸或核糖核苷酸替换的序列。
所述促进胞吞的分子可以选自胆甾醇、单链或双链脂肪酸、靶向能够进行受体介导的胞吞的细胞受体的配体或转铁蛋白。
更具体来说,所述促进胞吞的分子是胆甾醇。
或者,所述促进胞吞的分子是σ-2受体(σ2r)的配体。例如,所述σ-2受体(σ2r)的配体包含下述结构式:
其中n是1至20的整数。
在一种情况下,位于所述核酸分子的双链组成部分的游离末端处的核苷酸的1、2或3个核苷酸间键可以具有修饰的磷酸二酯骨架例如硫代磷酸酯键,优选地在两条链上都具有。例如,1至3个胸苷可以被2’-脱氧-2’-氟阿拉伯糖尿苷代替,或者1至3个鸟苷可以被2’-脱氧-2’-氟阿拉伯糖鸟苷代替,或者1至3个胞苷可以被2’-脱氧-2’-氟阿拉伯糖胞苷代替。
所述环可以具有结构式(i),并且k是
任选地,f是1,并且l-j选自-c(o)-(ch2)m-nh-[(ch2)2-o]n-(ch2)p-c(o)-j、-c(o)-(ch2)m-nh-c(o)-[(ch2)2-o]n-(ch2)p-j、c(o)-(ch2)m-nh-c(o)-ch2-o-[(ch2)2-o]n-(ch2)p-j、-c(o)-(ch2)m-nh-c(o)-[(ch2)2-o]n-(ch2)p-c(o)-j和-c(o)-(ch2)m-nh-c(o)-ch2-o-[(ch2)2-o]n-(ch2)p-c(o)-j,其中m是0至10的整数;n是0至15的整数;并且p是0至3的整数。
任选地,所述环具有结构式(i)
-o-p(x)oh-o-{[(ch2)2-o]g-p(x)oh-o}r-k-o-p(x)oh-o-{[(ch2)2-o]h-p(x)oh-o-}s(i)
其中x是s,r是1,g是6,s是0,并且k是
其中f是1,并且l是c(o)-(ch2)5-nh-[(ch2)2-o]3-(ch2)2-c(o)-j、-c(o)-(ch2)5-nh-c(o)-[(ch2)2-o]3-(ch2)3-j、-c(o)-(ch2)5-nh-c(o)-ch2-o-[(ch2)2-o]5-ch2-c(o)-j、-c(o)-(ch2)5-nh-c(o)-ch2-o-[(ch2)2-o]9-ch2-c(o)-j、-c(o)-(ch2)5-nh-c(o)-ch2-o-[(ch2)2-o]13-ch2-c(o)-j或-c(o)-(ch2)5-nh-c(o)-j。
任选地,f是1,并且l-j是-c(o)-(ch2)m-nh-[c(o)]t-[(ch2)2-o]n-(ch2)p-[c(o)]v-j或-c(o)-(ch2)m-nh-[c(o)-ch2-o]t-[(ch2)2-o]n-(ch2)p-[c(o)]v-j,其中m是0至10的整数,n是0至15的整数;p是0至4的整数;t和v是整数0或1,其中t和v中的至少一者是1。
在一种特定情况下,l可以选自-c(o)-(ch2)m-nh-[(ch2)2-o]n-(ch2)p-c(o)-j、-c(o)-(ch2)m-nh-c(o)-[(ch2)2-o]n-(ch2)p-j、c(o)-(ch2)m-nh-c(o)-ch2-o-[(ch2)2-o]n-(ch2)p-j、-c(o)-(ch2)m-nh-c(o)-[(ch2)2-o]n-(ch2)p-c(o)-j和-c(o)-(ch2)m-nh-c(o)-ch2-o-[(ch2)2-o]n-(ch2)p-c(o)-j,其中m是0至10的整数;n是0至15的整数;并且p是0至3的整数。
在非常特定情况下,l可以选自-c(o)-(ch2)5-nh-[(ch2)2-o]3-(ch2)2-c(o)-j、-c(o)-(ch2)5-nh-c(o)-[(ch2)2-o]3-(ch2)3-j、-c(o)-(ch2)5-nh-c(o)-ch2-o-[(ch2)2-o]5-ch2-c(o)-j、-c(o)-(ch2)5-nh-c(o)-ch2-o-[(ch2)2-o]9-ch2-c(o)-j、-c(o)-(ch2)5-nh-c(o)-ch2-o-[(ch2)2-o]13-ch2-c(o)-j或-c(o)-(ch2)5-nh-c(o)-j。
在特定情况下,所述偶联核酸分子选自以下结构式或其可药用盐:
其中核苷酸间键“s”是指硫代磷酸酯核苷酸间键;斜体的u是2’-脱氧-2’-氟阿拉伯糖尿苷;斜体的g是2’-脱氧-2’-氟阿拉伯糖鸟苷;斜体的c是2’-脱氧-2’-氟阿拉伯糖胞苷。
本发明还涉及一种药物组合物,其包含根据本公开所述的偶联核酸分子。任选地,所述药物组合物还包含另外的治疗剂,其优选地选自免疫调节剂例如免疫检查点抑制剂(ici)、基于t-细胞的癌症免疫疗法例如过继细胞转移(act)、遗传修饰的t-细胞或工程化改造的t-细胞例如嵌合抗原受体细胞(car-t细胞),或常规的化学治疗、放射治疗或抗血管生成药剂、hdac抑制剂(例如贝利司他)或靶向免疫毒素。
本发明还涉及根据本公开所述的偶联核酸分子或药物组合物,其用作药物,特别是用于治疗癌症。它还涉及一种在需要的对象中治疗癌症的方法,所述方法包括重复或长期给药治疗有效量的根据本发明所述的偶联核酸分子或药物组合物。任选地,所述方法包括给药重复的治疗周期,优选为至少两个给药周期,甚至更优选为至少三个或四个给药周期。
根据本发明所述的偶联核酸分子的重复或长期给药不导致癌细胞发展出对所述疗法的抗性。它可以与免疫调节剂例如免疫检查点抑制剂(ici)、基于t-细胞的癌症免疫疗法包括过继细胞转移(act)、遗传修饰的t-细胞或工程化改造的t-细胞例如嵌合抗原受体细胞(car-t细胞)相组合使用。
因此,所述偶联核酸分子或药物组合物与另外的治疗剂相组合用于治疗癌症,所述另外的治疗剂优选地选自免疫调节剂例如免疫检查点抑制剂(ici)、基于t-细胞的癌症免疫疗法例如过继细胞转移(act)、遗传修饰的t-细胞或工程化改造的t-细胞例如嵌合抗原受体细胞(car-t细胞),或常规的化学治疗、放射治疗或抗血管生成药剂、hdac抑制剂(例如贝利司他)或靶向免疫毒素。
在特定情况下,本发明还涉及一种用于可能的选择策略或临床分层策略的方式,以获得具有在nad+合成中带有缺陷的肿瘤的患者。这些患者可能是根据本发明所述的药物治疗的更好的响应者,特别是具有带有两种dna修复途径缺陷(例如ercc1和atm缺陷)或idh突变的肿瘤的患者。
在特定情况下,所述偶联核酸分子或药物组合物在癌症治疗中用于获得针对在nad+合成中带有缺陷的肿瘤细胞的靶向效应。更具体来说,所述肿瘤细胞还带有选自ercc1或atm缺陷的dna修复途径缺陷或idh突变。
附图说明
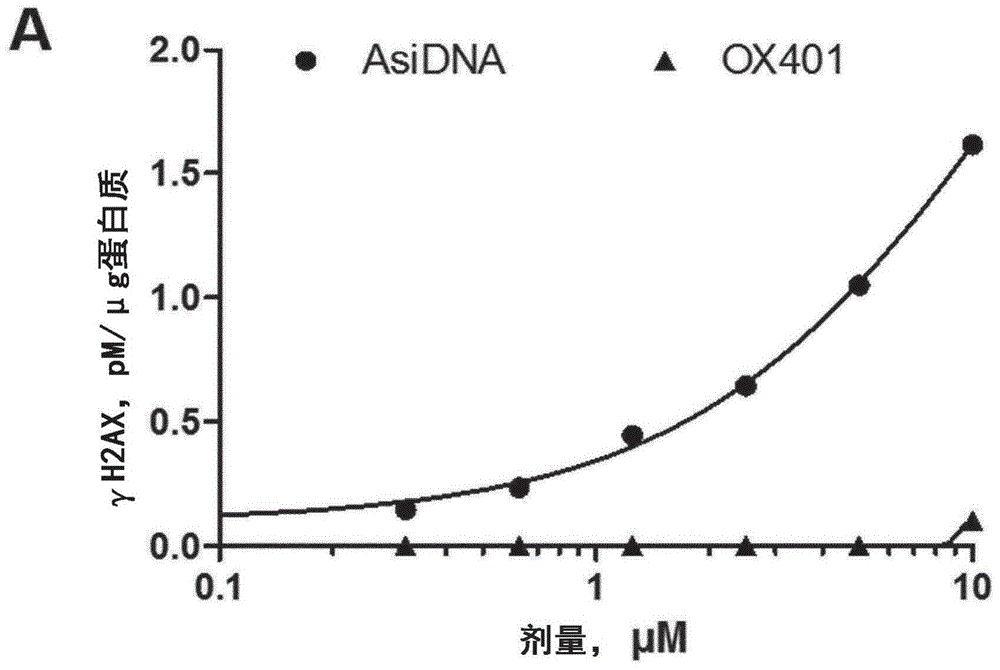
图1:ox401诱导的靶点作用。将细胞用剂量逐渐提高的ox401或asidnatm处理24小时,(a)通过h2ax磷酸化(γh2ax)评估dna-pk活化并(b)通过测量细胞par化(通过检测聚(adp-核糖)(par)聚合物)评估parp超活化。***,p<0.001。
图2:ox401表现出肿瘤特异性细胞毒性。将(a)肿瘤细胞和(b)非肿瘤细胞用ox401或asidnatm处理,并使用xtt测定法评估细胞存活率。细胞存活率被计算为活的处理过的细胞与活的未处理细胞的比率。ic50使用graphpadprism软件根据剂量响应曲线来计算。
图3:ox401触发肿瘤免疫应答。评估用ox401或asidnatm长期处理的mda-mb-231细胞的(a)微核阳性细胞的%,(b)分泌的ccl5和cxcl10趋化因子的量(使用elisa测定法),以及(c)总pd-l1(通过western印迹)和(d)表面结合的pd-l1的水平(通过流式细胞术分析)。cgamp,sting激动剂;**,p<0.01。
图4:ox402诱导parp活化。将细胞用剂量逐渐提高的ox402处理24小时,并通过测量细胞par化(通过检测聚(adp-核糖)(par)聚合物)来评估parp超活化。
图5:ox401在肿瘤细胞中诱导par化和高效nad+耗竭。将细胞用ox401(5μm)处理48小时、7天或13天,并评估parp超活化(通过par化蛋白的western印迹分析)(a、d)、nad+细胞内水平(b、e)和细胞存活率(c、f)。nad+的%和存活率被表示为处理过的细胞与未处理细胞(nt)的比率。(a、b、c)mda-mb-231肿瘤细胞,(d、e、f)mrc5肺成纤维细胞。
图6:ox401废止同源重组修复途径。将细胞用ox401(5μm)处理48小时,并使用(a)流式细胞术以检测磷酸化形式的h2ax(γh2ax)或(b)免疫荧光以检测γh2ax灶点来评估dsb水平。(c-d)在伴有或不伴有ox401(5μm)的奥拉帕尼(5μm)处理48小时后,通过(c)rad51蛋白向dsb部位的召集的检测和(d)rad51灶点的定量来分析同源重组途径的效能。***,p<0.001。
图7:用ox401处理的肿瘤细胞不发展出耐药性。(a)将细胞用他拉唑帕尼(2μm)或ox401(1.5μm)处理,并在每个处理和扩增循环后进行计数。(b)细胞存活率通过用处理过的细胞的数目除以未处理细胞的平均数来估算,并在每个处理时段后确定。(c)在用剂量逐渐增加的他拉唑帕尼处理4天后,使用xtt测定法确认了与u937亲代细胞相比在三个分离的群体(tal1、tal2和tal3)中存在对他拉唑帕尼的耐药性。存活率百分数使用未处理的条件进行归一化。
图8:ox401增强抗肿瘤免疫应答。将mda-mb-231细胞与t淋巴细胞(效应细胞与靶肿瘤细胞的比例为4:1)在存在或不存在ox401(5μm)的情况下共培养48小时,并评估(a)肿瘤细胞增殖,(b)分泌的颗粒酶b的量(使用elisa测定法)和(c、d)sting途径的活化(通过western印迹(c)或elisa测定法以定量分泌的ccl5趋化因子(d))。lta,活化的t淋巴细胞;mda,mda-mb-231肿瘤细胞。
图9:ox401、ox402、ox406、ox407、ox408、ox410和ox411与parp-1的结合动力学(kon)和相互作用强度(kd)。
发明详述
本发明涉及新的与促进胞吞的分子偶联的核酸分子例如胆甾醇-核酸偶联物,其特异性靶向并活化parp,诱导细胞nad的极大下调,并因此特别专用于癌症治疗,尤其是针对由于dna修复基因缺陷(例如ercc1和atm缺陷)或idh(异柠檬酸脱氢酶)突变而表现出nad缺陷的癌细胞。
本发明涉及新的与促进胞吞的分子偶联的核酸分子例如胆甾醇-核酸偶联物,其靶向ddr机制并且也是sting激动剂,允许它们与免疫检查点疗法(ict)相组合用于癌症的最佳治疗。
因此,本发明人令人吃惊地发现:
1)本发明的偶联核酸分子活化parp而不活化dna-pk,与dbait分子相比,单独使用即可导致具有微核、胞质染色质片段(ccf)和细胞毒性的癌细胞的增加。
2)癌细胞中微核(mn)和胞质染色质片段(ccf)的特异性增加导致sting途径活化的早期增加,正如通过炎性细胞因子(cxcl10和ccl5)的释放和癌细胞上pd-l1和nkg2表达的增加所显示的。这些效应特异性针对癌细胞。这种癌细胞特异性避免了普遍且广泛的炎症以及随后可能的有害副作用。
3)通过dna修复途径的抑制以及微核和ccf的产生来活化sting途径,代表了特别是通过先天免疫的活化来特异性活化肿瘤细胞中的sting途径的一种非常有吸引力的方式。
基于这些观察,本发明涉及:
-一种如下文所述的偶联核酸分子;
-一种药物组合物,其包含如下文所述的偶联核酸分子和可药用载体,特别是用于治疗癌症;
-一种如下文所述的偶联核酸分子,其用作药物,特别是用于治疗癌症;
-如下文所述的偶联核酸分子的一种用途,其用于制造特别是用于治疗癌症的药物;
-一种在需要的患者中治疗癌症的方法,所述方法包括给药有效量的本文中所公开的偶联核酸分子;
-一种药物组合物,其包含如下文所述的偶联核酸分子、另外的治疗剂和可药用载体,特别是用于治疗癌症;
-一种产品或药剂盒,其含有作为组合制剂的(a)如下文所公开的偶联核酸分子和任选的(b)另外的治疗剂,用于同时、分开或顺序使用,特别是用于治疗癌症;
-一种组合制剂,其包含(a)如下文所公开的发夹核酸分子,(b)如下文所描述的另外的治疗剂,用于同时、分开或顺序使用,特别是用于治疗癌症;
-一种药物组合物,其包含如下文所公开的偶联核酸分子,所述药物组合物与另外的治疗剂相组合用于治疗癌症;
-包含如下文所公开的偶联核酸分子的药物组合物的用途,其用于制造药物,所述药物与另外的治疗剂相组合用于治疗癌症;
-一种在需要的患者中治疗癌症的方法,所述方法包括给药有效量的a)如下文所公开的偶联核酸分子和b)有效量的另外的治疗剂;
-一种在需要的患者中治疗癌症的方法,所述方法包括给药有效量的包含本文中所公开的偶联核酸分子的药物组合物和有效量的另外的治疗剂;
-一种用于在需要的患者中提高使用治疗性抗肿瘤剂治疗癌症的效率或增强肿瘤对使用治疗性抗肿瘤剂的治疗的敏感性的方法,所述方法包括给药有效量的如下文所公开的偶联核酸分子;
-一种治疗癌症的方法,所述方法包括通过重复的治疗周期,优选地至少两个给药周期、甚至更优选地至少三个或四个给药周期,重复或长期给药本文中所公开的偶联核酸分子;
-一种在具有肿瘤细胞的患者中治疗癌症的方法,所述肿瘤细胞带有nad+合成中的缺陷并任选地带有选自ercc1或atm缺陷的dna修复途径缺陷或idh突变。
定义
每当在整个本说明书中关于本发明的药物组合物、药剂盒、产品和组合制剂提到“治疗癌症”等时,意味着:a)治疗癌症的方法,所述方法包括将本发明的药物组合物、药剂盒、产品和组合制剂给药到需要这种治疗的患者;b)用于治疗癌症的本发明的药物组合物、药剂盒、产品和组合制剂;c)本发明的药物组合物、药剂盒、产品和组合制剂在制备用于治疗癌症的药物中的用途;和/或d)用于治疗癌症的本发明的药物组合物、药剂盒、产品和组合制剂。
在本发明的上下文中,术语“治疗”表示治愈性、对症性和预防性治疗。本发明的药物组合物、药剂盒、产品和组合制剂可用于已患有癌症或肿瘤,包括处于癌症进展的早期或晚期阶段的人类。本发明的药物组合物、药剂盒、产品和组合制剂不必定治愈所述已患有癌症的患者,但会延迟或减缓所述疾病的进展或阻止其进一步进展,从而改善所述患者的状况。具体来说,本发明的药物组合物、药剂盒、产品和组合制剂在哺乳动物宿主中减少肿瘤的发生,降低肿瘤负担,产生肿瘤消退,和/或防止转移的发生和癌症复发。在癌症治疗中,以治疗有效量给药本发明的药物组合物、药剂盒、产品和组合制剂。
当在本文中使用时,术语“药剂盒”、“产品”或“组合制剂”在如上所定义的组合配偶体(a)和(b)可以独立地给药或利用具有不同量的组合配偶体(a)和(b)的不同固定组合给药,即同时或在不同时间点给药的意义上,特别定义了“成套药剂”。所述成套药剂的组分然后可以同时或在时间上错开,即对于所述成套药剂的任何部分来说在不同时间点并具有相等或不同的时间间隔来给药。在所述组合制剂中待给药的组合配偶体(a)与组合配偶体(b)的总量的比例可以变化。所述组合配偶体(a)和(b)可以通过相同途径或不同途径给药。
“有效量”意味着本发明的药物组合物、药剂盒、产品和组合制剂单独地或与所述药物组合物、药剂盒、产品或组合制剂的其他活性成分相组合,在包括人类的哺乳动物中预防、消除或减轻癌症的有害效应的量。应该理解,所述给药的剂量可以由本领域技术人员根据患者、病情、给药方式等改变。
术语“sting”是指干扰素基因的刺激物受体,也被称为tmem173、eris、mita、mpys、savi或net23。当在本文中使用时,术语“sting”和“sting受体”互换使用,并且包括sting的不同同工型和变体。最长的同工型,即人类sting同工型1的mrna和蛋白质序列具有ncbi参考序列[nm_198282.3]和[np_938023.1]。较短的同工型人类sting同工型2的mrna和蛋白质序列具有ncbi参考序列[nm_001301738.1]和[np_001288667.1]。
当在本文中使用时,术语“sting活化剂”是指能够活化sting途径的分子。sting途径的活化可以包括例如炎性细胞因子的刺激,所述炎性细胞因子包括干扰素,例如1型干扰素包括ifn-α、ifn-β、3型干扰素如ifn-λ、ip-10(干扰素-γ诱导的蛋白,也被称为cxcl10)、pd-l1、tnf、il-6、cxcl9、ccl4、cxcl11、nkg2d配体(mica/b)、ccl5、ccl3或ccl8。sting途径的活化也可以包括tank结合激酶(tbk)1磷酸化的刺激,干扰素调控因子(irf)活化(例如irf3活化),ip-10或其他炎性蛋白和细胞因子的分泌。sting途径的活化可以例如通过化合物刺激sting途径活化的能力来确定,正如使用干扰素刺激测定法、报告基因测定法(例如hstingwt测定法或thp1-dual测定法)、tbk1活化测定法、ip-10测定法或本领域技术人员已知的其他测定法所检测的。sting途径的活化还可以通过化合物提高被sting或sting途径活化的蛋白质的编码基因的转录水平的能力来确定。这种活化可以例如使用rnaseq测定法来检测。
sting途径的活化可以通过选自下述的一种或多种“sting测定法”来确定:干扰素刺激测定法,hstingwt测定法,thp1-dual测定法,tank结合激酶1(tbk1)测定法,干扰素-γ诱导的蛋白10(ip-10)分泌测定法或pd-l1测定法。
更具体来说,一种分子,如果能够在表达sting的细胞中刺激一种或多种sting依赖性细胞因子的生产比未处理的表达sting的细胞高至少1.1倍、1.2倍、1.3倍、1.4倍、1.5倍、1.6倍、1.7倍、1.8倍、1.9倍、2倍或更高,则所述分子是sting活化剂。优选地,所述sting依赖性细胞因子选自干扰素、1型干扰素、ifn-α、ifn-β、3型干扰素、ifn-λ、cxcl10(ip-10)、pd-l1tnf、il-6、cxcl9、ccl4、cxcl11、nkg2d配体(mica/b)、ccl5、ccl3或ccl8,更优选为ccl5或cxcl10。
偶联核酸分子
根据本发明所述的某些偶联核酸分子的另一个优点是基于下述事实,即它们可以通过仅使用寡核苷酸固相合成作为一个分子来合成,从而允许低成本和高制造规模。
本发明的偶联核酸分子包含:双链核酸组成部分,其中第一链的5’末端与互补链的3’末端通过环连接在一起;和任选的连接到所述环的促进胞吞的分子。所述双链核酸组成部分的另一个末端是游离的。
根据本发明所述的偶联核酸分子可以由它们的治疗活性所必需的多种特征来定义,例如它们的最小和最大长度、至少一个游离末端的存在和双链部分、优选为双链dna部分的存在。
所述偶联核酸分子能够活化parp-1蛋白。另一方面,所述偶联核酸分子不活化dna-pk。
本发明还涉及本发明的偶联核酸分子的可药用盐。
核酸分子
所述偶联核酸分子的长度可变,只要它足以允许parp(parp-1)蛋白的适当结合或活化并且不足以允许包含ku和dna-pkc蛋白的ku蛋白复合体的适当结合即可。正如已经显示的,为了确保与这种ku复合体的结合并允许dna-pkc活化,偶联核酸分子的长度必须大于20bp、优选为约32bp,因此所述长度至多20bp。此外,已显示为了允许parp的适当结合和活化,偶联核酸分子的长度必须大于8bp。
所述双链核酸组成部分的长度为10至20个碱基对。至多20bp的长度防止所述分子能够活化dna-pk。在特定情况下,所述双链核酸组成部分的长度为11至19个碱基对。例如,所述长度可以是11至19bp、12至19bp、13至19bp、14至19bp、15至19bp、16至19bp、12至16bp、12至17bp、12至18bp、13至16bp、13至17bp、13至18bp、14至16bp、14至17bp、14至18bp、15至16bp、15至17bp或15至18bp。在非常特定情况下,所述双链核酸组成部分的长度为16bp。“bp”意味着所述分子包含所指示长度的双链部分。
所述核酸分子的效果不依赖于其序列。因此,所述核酸分子可以被定义为包含下式
5’nnnn(n)an-∫
3’nnnn(n)an-∫
其中n是核苷酸,“a”是5至15的整数,并且两条链彼此互补。“-∫”指示所述核苷酸连接到环。在特定情况下,“a”是6至14的整数。在另一种特定情况下,“a”可以是6至14、7至14、8至14、9至14、10至14、11至14、6至13、7至13、8至13、9至13、10至13、11至13、6至12、7至12、8至12、9至12或10至12的整数。
优选地,所述核酸分子的序列是非人类起源的(即它们的核苷酸序列和/或构象不原样存在于人类细胞中)。所述偶联核酸分子优选地与已知基因、启动子、增强子、5’-或3’-上游序列、外显子、内含子等没有显著程度的序列同源性或同一性。换句话说,所述偶联核酸分子与人类基因组中的任何基因具有低于80%或70%、甚至低于60%或50%的序列同一性。确定序列同一性的方法在本领域中是公知的,并包括例如blastn2.2.25。例如,同一性百分数可以使用第37版人类基因组(humangenomebuild37)(参见grch37.p2和替代组装体)来确定。所述偶联核酸分子在严紧条件下不与人类基因组dna杂交。典型的严紧条件是允许将完全互补的核酸与部分互补的核酸区分开的条件。
此外,所述偶联核酸分子的序列优选地不含5’-cpg-3’,以避免公知的toll样受体(tlr)介导的免疫反应。
所述偶联核酸分子必须具有一个游离末端作为双链断裂的模拟物。所述游离末端可以是游离的平末端或5'-/3'-突出末端。“游离末端”在本文中是指具有5’末端和3’末端两者的核酸分子,特别是双链核酸组成部分。
例如,根据本发明所述的分子的双链核酸组成部分或核酸包含下述序列(seqidno:1)或由所述序列构成:
5’cccagcaaacaagcct-∫
3’gggtcgtttgttcgga-∫
在特定实施方式中,所述偶联核酸分子具有双链组成部分,所述双链组成部分包含与seqidno:1相同的核苷酸序列。任选地,所述偶联核酸分子具有与seqidno:1相同的核苷酸组成,但核苷酸序列不同。因此,所述偶联核酸分子包含的双链组成部分的一条链具有6个a、7个c、2个g和1个t。优选地,所述偶联核酸分子的序列不含任何5’-cpg-3’二核苷酸。或者,所述双链组成部分包含seqidno:1的至少9、10、11、12、13、14、15或16个连续核苷酸。在更特定实施方式中,所述双链组成部分由seqidno:1的9、10、11、12、13、14、15或16个连续核苷酸构成。
在另一种特定情况下,根据本发明所述的分子的双链核酸组成部分或核酸包含下述序列(seqidno:2)或由所述序列构成:
5’cagcaacaag-∫
3’gtcgttgttc-∫
在特定实施方式中,所述偶联核酸分子具有双链组成部分,所述双链组成部分包含与seqidno:2相同的核苷酸序列。任选地,所述偶联核酸分子具有与seqidno:2相同的核苷酸组成,但核苷酸序列不同。因此,所述偶联核酸分子包含的双链组成部分的一条链具有5个a、3个c和2个g,优选地,所述偶联核酸分子的序列不含任何5’-cpg-3’二核苷酸。
所述双链核酸组成部分可以包含具有修饰的磷酸二酯骨架的核苷酸,特别是为了保护它们免于降解。优选地,所述具有修饰的磷酸二酯骨架的核苷酸位于所述核酸分子的双链组成部分的游离末端处。在一种情况下,位于所述核酸分子的双链组成部分的游离末端处的核苷酸的1、2或3个核苷酸间键具有修饰的磷酸二酯骨架,优选地在两条链上都具有。或者,优选的偶联核酸分子在链的末端处具有3'-3'核苷酸键。
在特定实施方式中,所述核酸分子可以被定义为包含下式
nnnn(n)an-∫
nnnn(n)an-∫
其中下划线的核苷酸n的核苷酸间键具有修饰的磷酸二酯骨架。
例如,根据本发明所述的分子的双链核酸组成部分或核酸包含下述序列(seqidno:1)或由所述序列构成:
5’cccagcaaacaagcct-∫
3’gggtcgtttgttcgga-∫
其中下划线的核苷酸n的核苷酸间键具有修饰的磷酸二酯骨架。
在另一种特定情况下,根据本发明所述的分子的双链核酸组成部分或核酸包含下述序列(seqidno:2)或由所述序列构成:
5’cagcaacaag-∫
3’gtcgttgttc-∫
其中下划线的核苷酸n的核苷酸间键具有修饰的磷酸二酯骨架。
所述修饰的磷酸二酯骨架可以是硫代磷酸酯骨架。
当所述修饰的磷酸二酯键是硫代磷酸酯键时,所述分子可以是下述分子:
5’cscscsagcaaacaagcct-∫
3’gsgsgstcgtttgttcgga-∫
或
5’csasgscaacaag-∫
3’gstscsgttgttc-∫
在替代情况下,所述双链核酸组成部分可以在所述分子的3’末端处的两个最后核苷酸上、所述分子的5’末端处的两个最后核苷酸上或所述分子的3’末端和5’末端两者处的两个最后核苷酸上包含一个修饰的磷酸二酯键,例如硫代磷酸酯键。
例如,所述双链核酸组成部分包含选自下述的组成部分或由其构成:
5’cccagcaaacaagcct-∫
3’gggtcgtttgttcgga-∫
5’cagcaacaag-∫
3’gtcgttgttc-∫
5’cccagcaaacaagcct-∫
3’gggtcgtttgttcgga-∫
和
5’cagcaacaag-∫
3’gtcgttgttc-∫。
当所述修饰的磷酸二酯键是硫代磷酸酯键时,所述分子可以是下述分子:
5’cccagcaaacaagcct-∫
3’gsggtcgtttgttcgga-∫
5’cagcaacaag-∫
3’gstcgttgttc-∫
5’csccagcaaacaagcct-∫
3’gsggtcgtttgttcgga-∫
和
5’csagcaacaag-∫
3’gstcgttgttc-∫。
在另一种替代情况下,所述双链核酸组成部分可以在所述分子的3’末端处的三个最后核苷酸上或所述分子的5’末端处的四个最后核苷酸上包含三个修饰的磷酸二酯键,例如硫代磷酸酯键。
例如,所述双链核酸组成部分包含选自下述的组成部分或由其构成:
5’cccagcaaacaagcct-∫
3’gggtcgtttgttcgga-∫
5’cagcaacaag-∫
3’gtcgttgttc-∫
5’cccagcaaacaagcct-∫
3’gggtcgtttgttcgga-∫
和
5’cagcaacaag-∫
3’gtcgttgttc-∫。
当所述修饰的磷酸二酯键是硫代磷酸酯键时,所述分子可以是下述分子:
5’cscscsagcaaacaagcct-∫
3’gggtcgtttgttcgga-∫
5’csasgscaacaag-∫
3’gtcgttgttc-∫
5’cccagcaaacaagcct-∫
3’gsgsgstcgtttgttcgga-∫
和
5’cagcaacaag-∫
3’gstscsgttgttc-∫。
所述双链核酸组成部分基本上包含脱氧核糖核苷酸。然而,它也可以包括一些核糖核苷酸或修饰的脱氧核糖核苷酸或核糖核苷酸。在一种情况下,所述双链核酸组成部分只包含脱氧核糖核苷酸。在另一种情况下,所述双链核酸组成部分包含脱氧核糖核苷酸和相对于所述核酸分子的核苷酸总数至多30、20、15或10%的核糖核苷酸或修饰的脱氧核糖核苷酸。在特定情况下,所述双链核酸组成部分包含只含脱氧核糖核苷酸的第一链和带有核糖核苷酸或修饰的脱氧核糖核苷酸的互补链。根据一个实施方式,所述偶联核酸分子包含对应于核糖的2位的修饰。例如,所述偶联核酸分子可以包含至少一个2'-修饰的核苷酸,例如具有2'-脱氧、2'-脱氧-2'-氟、2'-o-甲基、2'-o-甲氧基乙基(2'-o-moe)、2'-o-氨基丙基(2'-o-ap)、2'-o-二甲基氨基乙基(2'-o-dmae)、2'-o-二甲基氨基丙基(2'-o-dmap)、2'-o-二甲基氨基乙氧基乙基(2'-o-dmaeoe)或2'-o-n-甲基乙酰胺基(2'-o-nma)修饰或例如2’-脱氧-2’-氟阿拉伯糖核苷酸(fana)。然而,这种2'-修饰的核苷酸优选地不位于链的5’或3’末端处。
在特定情况下,所述偶联核酸分子具有至少1、2、3或更多个2'-脱氧-2’-氟阿拉伯糖核苷酸(fana)。fana采取dna样结构,导致所述偶联核酸分子被感兴趣的蛋白质的识别未被改变。fana包括下述嘧啶2'-氟阿拉伯糖核苷和嘌呤2'-氟阿拉伯糖核苷:
9-(2-脱氧-2-氟-β-d-呋喃阿拉伯糖基)腺嘌呤(2'-fana-a);
9-(2-脱氧-2-氟-β-d-呋喃阿拉伯糖基)鸟嘌呤(2’-fana-g);
1-(2-脱氧-2-氟-β-d-呋喃阿拉伯糖基)胞嘧啶(2’-fana-c);
1-(2-脱氧-2-氟-β-d-呋喃阿拉伯糖基)尿嘧啶(2’-fana-u)。
例如,根据本发明所述的分子的双链核酸组成部分或核酸包含下述序列(seqidno:1)或由所述序列构成:
5’cccagcaaacaagcct-∫
其中u是1-(2-脱氧-2-氟-β-d-呋喃阿拉伯糖基)尿嘧啶(2’-f-ana-u)或2’-脱氧-2’-氟阿拉伯糖尿苷。具体来说,所述双链核酸组成部分包含下述序列(seqidno:1)或由所述序列构成:
5’cccagcaaacaagcct-∫
更具体来说是
5’cscscsagcaaacaagcct-∫
在另一个实例中,根据本发明所述的分子的双链核酸组成部分或核酸包含下述序列(seqidno:1)或由所述序列构成:
5’cccagcaaacaagcct-∫
其中g是2’-脱氧-2’-氟阿拉伯糖鸟苷。具体来说,所述双链核酸组成部分包含下述序列(seqidno:1)或由所述序列构成:
5’cccagcaaacaagcct-∫
更具体来说是
5’cscscsagcaaacaagcct-∫
在另一个实例中,根据本发明所述的分子的双链核酸组成部分或核酸包含下述序列(seqidno:1)或由所述序列构成:
3’gggtcgtttgttcgga-∫
其中c是2’-脱氧-2’-氟阿拉伯糖胞苷。具体来说,所述双链核酸组成部分包含下述序列(seqidno:1)或由所述序列构成:
3’gggtcgtttgttcgga-∫
更具体来说是
3’gsgsgstcgtttgttcgga-∫
环
所述环被连接到所述双链组成部分的第一链的5’末端和互补链的3’末端,并任选地连接到促进胞吞的分子。
所述环优选地包含10至100个原子、优选地15至25个原子的链。
环可以包括2至10个核苷酸,优选地3、4或5个核苷酸。非核苷酸环非穷举性地包括脱碱基核苷酸、聚醚、聚胺、聚酰胺、肽、糖类、脂类、聚烃或其他聚合化合物(例如低聚乙二醇,例如具有2至10个之间的乙二醇单元、优选地4、5、6、7或8个乙二醇单元的低聚乙二醇)。在一个实施方式中,所述环可以选自n-(5-羟甲基-6-磷酸己基)-11-(3-(6-磷酸己基硫基)琥珀酰亚胺基))十一碳酰胺、1,3-双-[5-羟基戊基酰胺基]丙基-2-(6-磷酸己基)、六聚乙二醇、四脱氧胸苷酸(t4)、1,19-双(磷酸)-8-肼基-2-羟基-4-氧杂-9-酮基-十九烷和2,19-双(磷酸)-8-肼基-1-羟基-4-氧杂-9-酮基-十九烷。
所述促进胞吞的分子任选地通过连接物偶联到所述环。可以使用本领域中已知的任何连接物将所述促进胞吞的分子共价附连到所述环。例如,wo09/126933在第38-45页提供了方便的连接物的广泛综述。非穷举性的,所述连接物可以是脂族链、聚醚、聚胺、聚酰胺、肽、糖类、脂类、聚烃或其他聚合化合物(例如低聚乙二醇,例如具有2至10个之间的乙二醇单元、优选地3、4、5、6、7或8个乙二醇单元、更优选地6个乙二醇单元的低聚乙二醇),并且并入了可以通过化学或酶的方式断裂的任何键,例如二硫键、保护的二硫键、酸不稳定键(例如腙键)、酯键、原酸酯键、磷酰胺键、可生物切割的肽键、偶氮键或醛键。此类可切割的连接物详细描述在wo2007/040469的第12-14页、wo2008/022309的第22-28页中。
所述促进胞吞的分子通过本领域技术人员已知的任何手段,任选地通过低聚乙二醇间隔物连接到所述环。
在特定实施方式中,所述在促进胞吞的分子与环之间的连接物包含c(o)-nh-(ch2-ch2-o)n或nh-c(o)-(ch2-ch2-o)n,其中n是1至10的整数,优选地n选自3、4、5和6。在非常特定实施方式中,所述连接物是co-nh-(ch2-ch2-o)4(甲酰胺基三乙二醇)。
在另一个特定实施方式中,所述在促进胞吞的分子与环分子之间的连接物是二烷基-二硫化物{例如(ch2)p-s-s-(ch2)q,其中p和q是1至10、优选地3至8的整数,例如6}。
在另一个特定实施方式中,所述环已被开发,以便与寡核苷酸固相合成相容。因此,可以在所述核酸分子的合成期间并入所述环,从而便于合成并降低其成本。
所述环可以具有选自下述结构式之一的结构:
-o-p(x)oh-o-{[(ch2)2-o]g-p(x)oh-o}r-k-o-p(x)oh-o-{[(ch2)2-o]h-p(x)oh-o-}s(i)
其中r和s独立地是整数0或1;g和h独立地是1至7的整数,并且g+h之和为4至7;
其中k是
其中i、j、k和l独立地是0至6、优选地1至3的整数;
或
-o-p(x)oh-o-[(ch2)d-c(o)-nh]b-chr-[c(o)-nh-(ch2)e]c-o-p(x)oh-o-(ii)
其中b和c独立地是0至4的整数,并且b+c之和为3至7;
d和e独立地是1至3、优选地1至2的整数;
其中r是-lf-j,
其中x是o或s,l是连接物,优选为任选地被一个或几个选自氨基、酰胺和酮基的基团中断的直链亚烷基和/或低聚乙二醇,f是0或1的整数,并且j是促进胞吞的分子或h。
当j是h时,所述分子可用作合成子,以便制备所述与促进胞吞的分子偶联的分子。或者,所述分子也可以在不偶联任何促进胞吞的分子的情况下用作药物。
在特定实施例中,所述分子可以是
在第一种情况下,所述环具有根据结构式(i)所述的结构:
-o-p(x)oh-o-{[(ch2)2-o]g-p(x)oh-o}r-k-o-p(x)oh-o-{[(ch2)2-o]h-p(x)oh-o-}s(i)
x是o或s。在结构式(i)中每次出现-o-p(x)oh-o-时,x可以在o和s之间变化。优选地,x是s。
g+h之和优选为5至7,特别是6。因此,如果r是0,h可以是5至7(其中s是1);如果g是1,h可以是4至6(其中r和s是1);如果g是2,h可以是3至5(其中r和s是1);如果g是3,h可以是2至4(其中r和s是1);如果g是4,h可以是1至3(其中r和s是1);如果g是5,h可以是1至2(其中r是1并且s是0或1);或者如果g是6或7,则s是0(其中r是1)。
优选地,i和j可以是相同的整数或者可以不同。i和j可以选自整数1、2、3、4、5或6,优选为1、2或3,更特别是1或2,特别是1。
优选地,k和l是相同的整数。在一种情况下,k和l是选自1、2或3的整数,优选为1或2,更优选为2。
因此,k可以是
在优选情况下,k是
在一种特定情况下,所述环具有结构式(i)
-o-p(x)oh-o-{[(ch2)2-o]g-p(x)oh-o}r-k-o-p(x)oh-o-{[(ch2)2-o]h-p(x)oh-o-}s(i)
其中x是s,r是1,g是6,s是0,并且k是
在特定情况下,f是1,并且l-j是-c(o)-(ch2)m-nh-[c(o)]t-[(ch2)2-o]n-(ch2)p-[c(o)]v-j或-c(o)-(ch2)m-nh-[c(o)-ch2-o]t-[(ch2)2-o]n-(ch2)p-[c(o)]v-j,其中m是0至10的整数;n是0至15的整数;p是0至4的整数;t和v是整数0或1,其中t和v中的至少一者是1。
更具体来说,f是1,并且l-j选自-c(o)-(ch2)m-nh-[(ch2)2-o]n-(ch2)p-c(o)-j、-c(o)-(ch2)m-nh-c(o)-[(ch2)2-o]n-(ch2)p-j、c(o)-(ch2)m-nh-c(o)-ch2-o-[(ch2)2-o]n-(ch2)p-j、-c(o)-(ch2)m-nh-c(o)-[(ch2)2-o]n-(ch2)p-c(o)-j和-c(o)-(ch2)m-nh-c(o)-ch2-o-[(ch2)2-o]n-(ch2)p-c(o)-j,其中m是0至10的整数;n是0至15的整数;并且p是0至3的整数。
任选地,f是1,并且l-j选自-c(o)-(ch2)5-nh-[(ch2)2-o]3-13-ch2-c(o)-j、-c(o)-(ch2)5-nh-c(o)-[(ch2)2-o]3-13-ch2-j、c(o)-(ch2)5-nh-c(o)-ch2-o-[(ch2)2-o]3-13-ch2-j、-c(o)-(ch2)5-nh-c(o)-[(ch2)2-o]3-13-ch2-c(o)-j和-c(o)-(ch2)5-nh-c(o)-ch2-o-[(ch2)2-o]3-13-ch2-c(o)-j或-c(o)-(ch2)5-nh-c(o)-j。
例如,f可以是1,并且l-j选自-c(o)-(ch2)5-nh-[(ch2)2-o]3-(ch2)2-c(o)-j、-c(o)-(ch2)5-nh-c(o)-[(ch2)2-o]3-(ch2)3-j、-c(o)-(ch2)5-nh-c(o)-ch2-o-[(ch2)2-o]5-ch2-c(o)-j、-c(o)-(ch2)5-nh-c(o)-ch2-o-[(ch2)2-o]9-ch2-c(o)-j、-c(o)-(ch2)5-nh-c(o)-ch2-o-[(ch2)2-o]13-ch2-c(o)-j或-c(o)-(ch2)5-nh-c(o)-j。
在非常特定情况下,f是1并且l-j是-c(o)-(ch2)m-nh-[(ch2)2-o]n-(ch2)p-c(o)-j,其中m是0至10、优选地4至6、特别是5的整数;n是0至6的整数;并且p是0至2的整数。在特定情况下,m是5并且n和p是0。在另一种特定情况下,m是5,n是3并且p是2。
在本公开的第二种情况下,所述环具有根据结构式(ii)所述的结构:
-o-p(x)oh-o-[(ch2)d-c(o)-nh]b-chr-[c(o)-nh-(ch2)e]c-o-p(x)oh-o-(ii)
其中x是o或s;
b和c独立地是0至4的整数,并且b+c之和为3至7;
d和e独立地是1至3的整数,优选为1至2;
其中r是–(ch2)1-5-c(o)-nh-lf-j或–(ch2)1-5-nh-c(o)-lf-j,并且
其中l是连接物,优选为直链亚烷基或低聚乙二醇,f是0或1的整数,并且j是促进胞吞的分子。
如果b和/或c是2或以上,则在每次出现[(ch2)d-c(o)-nh]或-[c(o)-nh-(ch2)e]时d和e可以不同。
在一种情况下,当d和e是2时,b+c之和为3至5,特别是4。例如,b可以是0并且c是3至5;b可以是1并且c是2至4;b可以是2并且c是1至3;或者b可以是3至5并且c是0。
在一种情况下,当d和e是1时,b+c之和为4至7,特别是5或6。例如,b可以是0并且c是3至6;b可以是1并且c是2至5;b可以是2并且c是1至4;或者b可以是3至6并且c是0。
在一种情况下,b、c、d和e被选择成使得所述环包含10至100个原子、优选地15至25个原子的链。
在非穷举性的实例名单中,所述环可以是下述结构之一:
-o-p(x)oh-o-(ch2)2-c(o)-nh-(ch2)2-c(o)-nh-chr-c(o)-nh-(ch2)2-c(o)-nh-(ch2)2-o-p(x)oh-o-
-o-p(x)oh-o-(ch2)2-c(o)-nh-chr-c(o)-nh-(ch2)2-c(o)-nh-(ch2)2-c(o)-nh-(ch2)2-o-p(x)oh-o-
-o-p(x)oh-o-chr-c(o)-nh-(ch2)2-c(o)-nh-(ch2)2-c(o)-nh-(ch2)2-c(o)-nh-(ch2)2-o-p(x)oh-o-
-o-p(x)oh-o-(ch2)2-c(o)-nh-(ch2)2-c(o)-nh-(ch2)2-c(o)-nh-chr-c(o)-nh-(ch2)2-o-p(x)oh-o-
-o-p(x)oh-o-(ch2)2-c(o)-nh-(ch2)2-c(o)-nh-(ch2)2-c(o)-nh-(ch2)2-c(o)-nh-chr-o-p(x)oh-o-
-o-p(x)oh-o-(ch2)2-c(o)-nh-(ch2)-c(o)-nh-chr-c(o)-nh-(ch2)-c(o)-nh-(ch2)2-o-p(x)oh-o-
-o-p(x)oh-o-(ch2)-c(o)-nh-(ch2)2-c(o)-nh-chr-c(o)-nh-(ch2)2-c(o)-nh-(ch2)-o-p(x)oh-o-或
-o-p(x)oh-o-(ch2)-c(o)-nh-(ch2)-c(o)-nh-chr-c(o)-nh-(ch2)-c(o)-nh-(ch2)-o-p(x)oh-o-
在特定情况下,所述环可以是下述结构:
-o-p(x)oh-o-(ch2)2-c(o)-nh-(ch2)2-c(o)-nh-chr-c(o)-nh-(ch2)2-c(o)-nh-(ch2)2-o-p(x)oh-o-
其中r是-lf-j;并且
其中l是连接物,优选为任选地被一个或几个选自氨基、酰胺和酮基的基团中断的直链亚烷基和/或低聚乙二醇,并且f是0或1的整数。
优选地,x是s。
l可以是–(ch2)1-5-c(o)-j,优选为–ch2-c(o)-j或–(ch2)2-c(o)-j。
或者,l-j可以是–(ch2)4-nh-[(ch2)2-o]n-(ch2)p-c(o)-j,其中n是0至6的整数;并且p是0至2的整数。在特定情况下,n是3并且p是2。
促进胞吞的分子
本发明的核酸分子任选地被偶联到在上述结构式中被称为j的促进胞吞的分子。因此,在第一种情况下,j是促进胞吞的分子。在替代情况下,j是氢。
所述促进胞吞的分子可以是亲脂性分子例如胆甾醇、单链或双链脂肪酸,或靶向能够进行受体介导的胞吞的细胞受体的配体例如叶酸和叶酸衍生物或转铁蛋白(goldstein等,ann.rev.cellbiol.19851:1-39;leamon&lowe,procnatlacadsciusa.1991,88:5572–5576)。脂肪酸可以是饱和或不饱和的,并且可以是c4-c28、优选地c14-c22、更优选地c18脂肪酸,例如油酸或硬脂酸。具体来说,脂肪酸可以具有十八烷基或二油酰基。脂肪酸可以作为用适合的连接物例如甘油、磷脂酰胆碱或乙醇胺等连接或通过用于附连到所述偶联核酸分子上的连接物连接在一起的双链形式存在。当在本文中使用时,术语“叶酸”意指叶酸和叶酸衍生物,包括蝶酸衍生物和类似物。适合用于本发明的叶酸的类似物和衍生物包括但不限于叶酸拮抗剂、二氢叶酸、四氢叶酸、亚叶酸、蝶酰基聚谷氨酸、1-去氮杂、3-去氮杂、5-去氮杂、8-去氮杂、10-去氮杂、1,5-去氮杂、5,10-二去氮杂、8,10-二去氮杂和5,8-二去氮杂叶酸、叶酸拮抗剂和蝶酸衍生物。其他叶酸类似物描述在us2004/242582中。
因此,所述促进胞吞的分子可以选自单链或双链脂肪酸、叶酸类和胆甾醇。更优选地,所述促进胞吞的分子选自二油酰基、十八烷基、叶酸和胆甾醇。在最优选实施方式中,所述促进胞吞的分子是胆甾醇。
因此,在一个优选实施方式中,所述偶联核酸分子(也被称为ox401)具有下述结构式:
其中核苷酸间键“s”表示硫代磷酸酯核苷酸间键。
在另一个特定实施方式中,所述偶联核酸分子(也被称为ox402)具有下述结构式:
其中核苷酸间键“s”表示硫代磷酸酯核苷酸间键。
在另一个优选实施方式中,所述偶联核酸分子具有下述结构式:
其中核苷酸间键“s”表示硫代磷酸酯核苷酸间键。
在其他优选实施方式中,所述偶联核酸分子具有下述结构式中的任一者:
-ox-406:
ox407:
ox408:
ox410:
和
ox411:
其中核苷酸间键“s”表示硫代磷酸酯核苷酸间键;斜体的u是2’-脱氧-2’-氟阿拉伯糖尿苷,斜体的g是2’-脱氧-2’-氟阿拉伯糖鸟苷;斜体的c是2’-脱氧-2’-氟阿拉伯糖胞苷。
或者,所述促进胞吞的分子也可以是生育酚、糖类例如半乳糖和甘露糖及其寡糖、肽例如rgd和铃蟾肽以及蛋白质例如整合蛋白。
σ-2受体配体
在特定情况下,选择所述促进胞吞的分子以便靶向癌细胞。因此,它被选择成在癌细胞中特异性表达或与正常细胞相比在癌细胞中过表达的受体的配体。
在这种情形中,所述促进胞吞的分子可以是σ-2受体(σ2r)的配体。
术语“σ-2受体(σ2r)”是指已被发现在恶性癌细胞(例如乳腺癌、卵巢癌、肺癌、脑癌、膀胱癌、结肠癌和黑素瘤)中高表达的σ受体亚型。所述σ-2受体是位于脂筏中的细胞色素相关蛋白,最通常与p450蛋白结合,并与pgrmc1复合体、egfr、mtor、半胱天冬酶和各种不同的离子通道偶联。
术语“σ-2受体(σ2r)配体”是指合成或非合成的激动剂化合物,其以高选择性和亲和性与σ2r结合,然后通过胞吞被内化。σ2r激动剂抑制肿瘤细胞增殖并在癌细胞中诱导凋亡。
在一种优选情况下,所述σ-2受体(σ2r)配体是氮杂双环壬烷类似物,更特别是在vangveravong等,bioorg.med.chem(2006)中描述的n-取代的-9-氮杂双环[3.3.1]壬-3α-基氨基甲酸酯类似物,其包含下述结构式:
特别是
其中n是1至20的整数。任选地,n是1至10、2至9、3至8、4至7或5至6的整数。
在第一种特定情况下,所述σ2r配体具有下述结构式
其中n是1至20的整数。任选地,n是1至10、2至9、3至8、4至7或5至6的整数。
在特定实施方式中,所述σ2r配体被称为sv119(n=6),并具有下述结构式:
在又一个特定实施方式中,所述σ2r配体被称为sw43(n=10),并具有下述结构式:
在另一个实施方式中,所述σ2r配体是n-取代的-9-氮杂双环[3.3.1]壬-3α-基氨基甲酸酯类似物,并具有下述结构式:
其中n是1至20的整数,并且m是0至10的整数。
在特定实施方式中,所述σ2r配体具有下述结构式:
所述σ2r配体经所述环,通过羧基或氨基,任选地经连接物偶联到所述核酸分子。
因此,在一个优选实施方式中,所述偶联核酸分子具有下述结构式:
其中核苷酸间键“s”表示硫代磷酸酯核苷酸间键。
因此,在另一个优选实施方式中,所述偶联核酸分子具有下述结构式:
其中核苷酸间键“s”表示硫代磷酸酯核苷酸间键。
因此,在另一个优选实施方式中,所述偶联核酸分子(也被称为ox405)具有下述结构式:
其中核苷酸间键“s”表示硫代磷酸酯核苷酸间键。
在另一个优选实施方式中,所述偶联核酸分子(也被称为ox407)具有下述结构式:
其中核苷酸间键“s”表示硫代磷酸酯核苷酸间键。
在其他优选实施方式中,所述偶联核酸分子具有下述结构式中的任一者:
上述化合物也被称为ox403
和
上述化合物也被称为ox404
其中核苷酸间键“s”表示硫代磷酸酯核苷酸间键。
所述核酸分子的治疗性用途
根据本发明所述的偶联核酸分子能够活化parp。它们导致癌细胞中微核和细胞毒性的增加。它们对癌细胞显示出特异性,这可以排除或限制副作用。此外,癌细胞中微核的特异性增加导致sting途径的早期活化。
因此,根据本发明所述的偶联核酸分子可用作药物,特别是用于治疗癌症。
因此,本发明涉及用作药物的根据本发明所述的偶联核酸分子。它还涉及包含根据本发明所述的偶联核酸分子的药物组合物,其特别是用于治疗癌症。
本文中设想的药物组合物除了所述活性成分之外还可以包含可药用载体。术语“可药用载体”意在涵盖不干扰所述活性成分的生物活性的有效性,并且对它给药到的宿主无毒的任何载体(例如支持物、物质、溶剂等)。例如,对于肠胃外给药来说,所述活性化合物可以在介质例如盐水、右旋糖溶液、血清白蛋白和林格氏溶液中配制成注射用单位剂型。
所述药物组合物可以以本领域中已知的方式配制成在制药相容的溶剂中的溶液或在适合的制药用溶剂或介质中的乳液、悬液或分散系或含有固体介质的丸剂、片剂或胶囊。适合于口服给药的本发明的剂型可以采取作为各自含有预定量的活性成分的胶囊、袋剂、片剂或含片的分立单元的形式;粉剂或颗粒剂的形式;在水性液体或非水性液体中的溶液或悬液的形式;或水包油乳液或油包水乳液的形式。适合于肠胃外给药的剂型方便地包含活性成分的无菌油性或水性制剂,其优选与接受者的血液等渗。每种此类剂型还可以含有其他制药上相容且无毒的辅助剂,例如稳定剂、抗氧化剂、粘合剂、染料、乳化剂或调味物质。因此,本发明的剂型包含与可药用载体缔合的活性成分和任选的其他治疗成分。所述载体在与剂型的其他成分相容并且对其接受者无害的意义上必须是“可接受的”。有利情况下,所述药物组合物通过适合的无菌溶液的注射或静脉内输注或作为口服剂型通过消化道施用。用于大多数这些化学治疗剂的安全有效给药的方法对于本领域技术人员来说是已知的。此外,它们的给药描述在标准文献中。
本发明中描述的药物组合物和产品、药剂盒或组合制剂可用于在对象中治疗癌症。
术语“癌症”和“癌性”指称或描述哺乳动物中通常以不受调控的细胞生长为特征的生理状况。癌症的实例包括但不限于实体肿瘤和血液系统癌症,包括癌、淋巴瘤、母细胞瘤(包括髓母细胞瘤和视网膜母细胞瘤)、肉瘤(包括脂肉瘤和滑膜细胞肉瘤)、神经内分泌肿瘤(包括类癌瘤、胃泌素瘤和胰岛细胞癌)、间皮瘤、神经鞘瘤(包括听神经瘤)、脑膜瘤、腺癌、黑素瘤和白血病或淋巴系恶性肿瘤。此类癌症的更具体实例包括鳞状细胞癌(例如上皮鳞状细胞癌)、包括小细胞肺癌、非小细胞肺癌、肺腺癌和肺鳞癌在内的肺癌、腹膜癌、肝细胞癌、包括胃肠道癌在内的胃癌、胰腺癌、胶质母细胞瘤、神经母细胞瘤、宫颈癌、卵巢癌、肝癌、膀胱癌、泌尿道癌、肝细胞瘤、乳腺癌、结肠癌、直肠癌,结直肠癌、子宫内膜或子宫癌、唾液腺癌、肾癌、前列腺癌、外阴癌、甲状腺癌、肝癌、肛门癌、阴茎癌、睾丸癌、食道癌、胆道肿瘤以及头颈癌。本文公开了其他癌症适应症。
在特定实施方式中,“癌”是指带有例如选自ercc1或atm缺陷的nad+耗竭的肿瘤细胞或带有idh突变的癌细胞。
在非常特定实施方式中,对于具有显示出nad+合成缺陷的肿瘤的患者,特别是对于具有带有nad+耗竭的肿瘤的患者来说,临床分层或选择更好的响应者是可能的。
最佳剂量的确定通常涉及本发明的治疗的治疗性益处水平相对于任何风险或有害副作用的平衡。所选的剂量水平依赖于各种不同因素,包括但不限于所述偶联核酸分子的活性、给药途径、给药时间、所述化合物的排泄速率、治疗的持续时间、组合使用的其他药物、化合物和/或材料以及患者的年龄、性别、体重、状况、总体健康和既往病史。偶联核酸分子的量和给药途径最终由医生斟酌,尽管通常剂量应在作用部位处达到实现所需效果的局部浓度。
本文中所公开的偶联核酸分子的给药途径可以是口服、肠胃外、静脉内、肿瘤内、皮下、颅内、动脉内、局部、直肠、透皮、真皮内、鼻、肌肉内、腹膜内、骨内等。在优选实施方式中,所述偶联核酸分子在待治疗的肿瘤部位附近给药或注射。
例如,所述偶联核酸分子的有效量为0.01至1000mg,例如优选为0.1至100mg。当然,剂量和方案可以由专业技术人员根据化疗和/或放疗方案来改变。
根据本发明所述的偶联核酸分子可以与另外的治疗剂相组合使用。所述另外的治疗剂可以例如是免疫调节剂如免疫检查点抑制剂、基于t-细胞的癌症免疫疗法包括过继细胞转移(act)、遗传修饰的t-细胞或工程化改造的t-细胞例如嵌合抗原受体细胞(car-t细胞)、常规的化学治疗、放射治疗或抗血管生成药剂、hdac抑制剂(例如贝利司他)或靶向免疫毒素。
与免疫调节剂/免疫检查点抑制剂(ici)的组合
正如通过sting途径的活化和pd-l1表达的提高所表明的,本发明人证实了偶联核酸分子与免疫调节剂的组合的高的抗肿瘤治疗效率,所述免疫调节剂例如免疫检查点抑制剂(ici)、优选为pd-1/pd-l1途径的抑制剂。因此,本发明提供了组合疗法,其中将本发明的偶联核酸分子与免疫调节剂例如免疫检查点抑制剂(ici)同时、在其之前或在其之后给药到患者。
因此,本发明涉及一种药物组合物,其包含本发明的偶联核酸分子和免疫调节剂,更具体来说用于治疗癌症。本发明还涉及一种产品,其包含作为组合制剂的本发明的偶联核酸分子和免疫调节剂,用于同时、分开或顺序使用,更具体来说用于治疗癌症。在优选实施方式中,所述免疫调节剂是pd-1/pd-l1途径的抑制剂。
本发明还提供了一种通过以下手段治疗癌症的方法:向需要的患者给药与一种或多种免疫调节剂(例如一种或多种共刺激分子的活化剂或免疫检查点分子的抑制剂)相组合的本发明的偶联核酸分子。在优选实施方式中,所述免疫调节剂是pd-1/pd-l1途径的抑制剂。共刺激分子的活化剂:
在某些实施方式中,所述免疫调节剂是共刺激分子的活化剂。在一个实施方式中,所述共刺激分子的激动剂选自ox40、cd2、cd27、cds、icam-1、lfa-1(cd11a/cd18)、icos(cd278)、4-1bb(cd137)、gitr、cd30、cd40、baffr、hvem、cd7、light、nkg2c、slamf7、nkp80、cd160、b7-h3或cd83配体的激动剂(例如激动性抗体或其抗原结合片段或可溶性融合体)。
免疫检查点分子的抑制剂:
在某些实施方式中,所述免疫调节剂是免疫检查点分子的抑制剂。在一个实施方式中,所述免疫调节剂是pd-1、pd-l1、pd-l2、ctla-4、tim-3、lag-3、nkg2d、nkg2l、kir、vista、btla、tigit、lair1、cd160、2b4和/或tgfrβ的抑制剂。在一个实施方式中,所述免疫检查点分子的抑制剂抑制pd-1、pd-l1、lag-3、tim-3或ctla-4或其任何组合。术语“抑制”或“抑制剂”包括给定分子例如免疫检查点抑制剂的某些参数如活性的降低。例如,该术语包括活性例如pd-1或pd-l1活性的至少5%、10%、20%、30%、40%、50%或更多的抑制。因此,抑制不必是100%。
抑制性分子的抑制可以在dna、rna或蛋白质水平上进行。在某些实施方式中,抑制性核酸(例如dsrna、sirna或shrna)可用于抑制抑制性分子的表达。在其他实施方式中,抑制性信号的抑制剂是与所述抑制性分子结合的多肽,例如可溶性配体(例如pd-1ig或ctla-4ig)或抗体或其抗原结合片段;例如与pd-1、pd-l1、pd-l2、ctla-4、tim-3、lag-3、nkg2d、nkg2l、kirvista、btla、tigit、lair1、cd160、2b4和/或tgfrβ结合的抗体或其片段(在本文中也被称为“抗体分子”)或其组合。
在一个实施方式中,所述抗体分子是完整抗体或其片段(例如fab、f(ab')2、fv或单链fv片段(scfv))。在其他实施方式中,所述抗体分子的重链恒定区(fc)选自例如igg1、igg2、igg3、igg4、igm、iga1、iga2、igd和ige的重链恒定区,特别是选自例如igg1、igg2、igg3和igg4的重链恒定区,更特别是igg1或igg4(例如人类igg1或igg4)的重链恒定区。在一个实施方式中,所述重链恒定区是人类igg1或人类igg4的重链恒定区。在一个实施方式中,所述恒定区被改变例如突变,以改良所述抗体分子的性能(例如提高或降低fc受体结合、抗体糖基化、半胱氨酸残基的数目、效应细胞功能或补体功能中的一者或多者)。在某些实施方式中,所述抗体分子采取双特异性或多特异性抗体分子的形式。
pd-1抑制剂
在某些实施方式中,本发明的偶联核酸分子与pd-1抑制剂相组合给药。在某些实施方式中,所述pd-1抑制剂选自pdr001(novartis)、纳武单抗(bristol-myerssquibb)、派姆单抗(merck&co)、匹地利珠单抗(curetech)、medi0680(medimmune)、regn2810(regeneron)、tsr-042(tesaro)、pf-06801591(pfizer)、bgb-a317(beigene)、bgb-108(beigene)、incshr1210(incyte)或amp-224(amplimmune)。
示例性pd-1抑制剂
在某些实施方式中,所述抗pd-1抗体是纳武单抗(cas登记号:946414-94-4)。纳武单抗的替代名称包括mdx-1106、mdx-1106-04、ono-4538、bms-936558或
在其他实施方式中,所述抗pd-1抗体是派姆单抗。派姆单抗(商品名keytruda,以前为lambrolizumab,也被称为merck3745、mk-3475或sch-900475)是一种与pd1结合的人源化lgg4单克隆抗体。派姆单抗被公开在例如整体通过参考并入本文的hamid,o.等,(2013)newenglandjournalofmedicine369(2):134-44、pct公布号wo2009/114335和美国专利号8,354,509中。
在某些实施方式中,所述抗pd-1抗体是匹地利珠单抗。匹地利珠单抗(ct-011;curetech)是一种与pd1结合的人源化lgg1k单克隆抗体。匹地利珠单抗和其他人源化抗pd-1单克隆抗体公开在整体通过参考并入本文的pct公布号wo2009/101611中。
其他抗pd1抗体公布在整体通过参考并入本文的美国专利号8,609,089、美国公布号2010028330和/或美国公布号20120114649中。其他抗pd1抗体包括amp514(amplimmune)。
在一个实施方式中,所述抗pd-1抗体分子是medi0680(medimmune),也被称为amp-514。medi0680和其他抗pd-1抗体公开在整体通过参考并入本文的us9,205,148和wo2012/145493中。
在一个实施方式中,所述抗pd-1抗体分子是regn2810(regeneron)。
在一个实施方式中,所述抗pd-1抗体分子是pf-06801591(pfizer)。
在一个实施方式中,所述抗pd-1抗体分子是bgb-a317或bgb-108(beigene)。
在一个实施方式中,所述抗pd-1抗体分子是incshr1210(incyte),也被称为incshr01210或shr-1210。
在一个实施方式中,所述抗pd-1抗体分子是tsr-042(tesaro),也被称为anb011。
其他已知的抗pd-1抗体包括在例如整体通过参考并入本文的wo2015/112800、wo2016/092419、wo2015/085847、wo2014/179664、wo2014/194302、wo2014/209804、wo2015/200119、us8,735,553、us7,488,802、us8,927,697、us8,993,731和us9,102,727中所描述的那些。
在一个实施方式中,所述抗pd-1抗体是与本文中描述的抗pd-1抗体之一竞争结合和/或结合到pd-1上的相同表位的抗体。
在一个实施方式中,所述pd-1抑制剂是抑制pd-1信号传导途径的肽,例如在整体通过参考并入本文的us8,907,053中所描述的。在某些实施方式中,所述pd-1抑制剂是免疫粘附素{例如包含融合到恒定区(例如免疫球蛋白序列的fc区)的pd-l1或pd-l2的细胞外或pd-1结合部分的免疫粘附素}。在某些实施方式中,所述pd-1抑制剂是amp-224(b7-dcig(amplimmune)),例如在整体通过参考并入本文的wo2010/027827和wo2011/066342中所公开的。
pd-l1抑制剂
在某些实施方式中,所述免疫检查点分子的抑制剂是pd-l1的抑制剂。在某些实施方式中,本发明的偶联核酸分子与pd-l1抑制剂相组合给药。在某些实施方式中,所述pd-l1抑制剂选自faz053(novartis)、阿特珠单抗(genentech/roche)、阿维单抗(merckserono和pfizer)、德瓦鲁单抗(medlmmune/astrazeneca)或bms-936559(bristol-myerssquibb)。
示例性pd-l1抑制剂
在一个实施方式中,所述pd-l1抑制剂是抗pd-l1抗体分子。在一个实施方式中,所述抗pd-l1抗体分子是阿维单抗(merckserono和pfizer),也被称为msb0010718c。阿维单抗和其他抗pd-l1抗体公开在整体通过参考并入本文的wo2013/079174中。
在一个实施方式中,所述抗pd-l1抗体分子是德瓦鲁单抗(medlmmune/astrazeneca),也被称为medi4736。德瓦鲁单抗和其他抗pd-l1抗体公开在整体通过参考并入本文的us8,779,108中。
在一个实施方式中,所述抗pd-l1抗体分子是bms-936559(bristol-myerssquibb),也被称为mdx-1105或12a4。bms-936559和其他抗pd-l1抗体公开在整体通过参考并入本文的us7,943,743和wo2015/081158中。
其他已知的抗pd-l1抗体包括例如在整体通过参考并入本文的wo2015/181342、wo2014/100079、wo2016/000619、wo2014/022758、wo2014/055897、wo2015/061668、wo2013/079174、wo2012/145493、wo2015/112805、wo2015/109124、wo2015/195163、us8,168,179、us8,552,154、us8,460,927和us9,175,082中所描述的那些。
在一个实施方式中,所述抗pd-l1抗体是与本文中描述的抗pd-l1抗体之一竞争结合和/或结合到pd-l1上的相同表位的抗体。
lag-3抑制剂
在某些实施方式中,所述免疫检查点分子的抑制剂是lag-3的抑制剂。在某些实施方式中,本发明的偶联核酸分子与lag-3抑制剂相组合给药。在某些实施方式中,所述lag-3抑制剂选自lag525(novartis)、bms-986016(bristol-myerssquibb)或tsr-033(tesaro)。
示例性lag-3抑制剂
在一个实施方式中,所述lag-3抑制剂是抗lag-3抗体分子。在一个实施方式中,所述lag-3抑制剂是bms-986016(bristol-myerssquibb),也被称为bms986016。bms-986016和其他抗lag-3抗体公开在整体通过参考并入本文的wo2015/116539和us9,505,839中。
在一个实施方式中,所述抗lag-3抗体分子是tsr-033(tesaro)。
在一个实施方式中,所述抗lag-3抗体分子是imp731或gsk2831781(gsk和primabiomed)。imp731和其他抗lag-3抗体公开在整体通过参考并入本文的wo2008/132601和us9,244,059中。
其他已知的抗lag-3抗体包括例如在整体通过参考并入本文的wo2008/132601、wo2010/019570、wo2014/140180、wo2015/116539、wo2015/200119、wo2016/028672、us9,244,059、us9,505,839中所描述的那些。
tim-3抑制剂
在某些实施方式中,所述免疫检查点分子的抑制剂是tim-3的抑制剂。在某些实施方式中,本发明的偶联核酸分子与tim-3抑制剂相组合给药。在某些实施方式中,所述tim-3抑制剂是mgb453(novartis)或tsr-022(tesaro)。
示例性tim-3抑制剂
在一个实施方式中,所述抗tim-3抗体分子是tsr-022(anaptysbio/tesaro)。
在一个实施方式中,所述抗tim-3抗体是ape5137或ape5121。ape5137、ape512和其他抗tim-3抗体公开在整体通过参考并入本文的wo2016/161270中。
其他已知的抗tim-3抗体包括例如在整体通过参考并入本文的wo2016/111947、wo2016/071448、wo2016/144803、us8,552,156、us8,841,418和us9,163,087中所描述的那些。
nkg2d抑制剂
在某些实施方式中,所述nkg2d/nkg2dl途径的抑制剂是nkg2d的抑制剂。在某些实施方式中,本发明的偶联核酸分子与nkg2d抑制剂相组合给药。在某些实施方式中,所述nkg2d抑制剂是抗nkg2d抗体分子例如抗nkg2d抗体nnc0142-0002(也被称为nn8555、iph2301或jnj-4500)。
示例性nkg2d抑制剂
在一个实施方式中,所述抗nkg2d抗体分子是nnc0142-0002(novonordisk),正如在整体通过参考并入本文的wo2009/077483和us7,879,985中所公开的。
在另一个实施方式中,所述抗nkg2d抗体分子是jnj-64304500(janssen),正如在整体通过参考并入本文的wo2018/035330中所公开的。
在某些实施方式中,所述抗nkg2d抗体是人类单克隆抗体16f16、16f31、ms和21f2,它们的生产、分离以及结构和功能表征如us7,879,985中所述。其他已知的抗nkg2d抗体包括例如在整体通过参考并入本文的wo2009/077483、wo2010/017103、wo2017/081190、wo2018/035330和wo2018/148447中所描述的那些。
在某些其他实施方式中,所述nkg2d抑制剂是免疫粘附素{例如包含融合到恒定区(例如免疫球蛋白序列的fc区)的nkg2dl的细胞外或nkg2d结合部分的免疫粘附素},正如在整体通过参考并入本文的wo2010/080124、wo2017/083545和wo2017/083612中所描述的。
nkg2dl抑制剂
在某些实施方式中,所述nkg2d/nkg2dl途径的抑制剂是nkg2dl的抑制剂,所述nkg2dl例如mica、micb、ulbp1、ulbp2、ulbp3、ulbp4或raet1家族的成员。在某些实施方式中,本发明的偶联核酸分子与nkg2dl抑制剂相组合给药。在某些实施方式中,所述nkg2dl抑制剂是抗nkg2dl抗体分子例如抗mica/b抗体。示例性mica/micb抑制剂
在一个实施方式中,所述抗mica/b抗体分子是iph4301(innatepharma),正如在整体通过参考并入本文的wo2017/157895中所公开的。
其他已知的抗mica/b抗体包括例如在整体通过参考并入本文的wo2014/140904和wo2018/073648中所描述的那些。
kir抑制剂
在某些实施方式中,所述免疫检查点分子的抑制剂是kir的抑制剂。在某些实施方式中,本发明的偶联核酸分子与kir抑制剂相组合给药。在某些实施方式中,所述kir抑制剂是利瑞鲁单抗(以前也被称为bms-986015或iph2102)。
示例性kir抑制剂
在一个实施方式中,所述抗kir抗体分子是利瑞鲁单抗(innatepharma/astrazeneca),正如在整体通过参考并入本文的wo2008/084106和wo2014/055648中所公开的。
其他已知的抗kir抗体包括例如在整体通过参考并入本文的wo2005/003168、wo2005/009465、wo2006/072625、wo2006/072626、wo2007/042573、wo2008/084106、wo2010/065939、wo2012/071411和wo/2012/160448中所公开的。
与常规化学治疗、放射治疗、抗血管生成药剂或组蛋白脱乙酰酶抑制剂(hdaci)的组合
本发明还提供了组合疗法,其中将本发明的偶联核酸分子在手术或放射治疗的同时、之前或之后使用,或与常规化学治疗、放射治疗或抗血管生成药剂、hdac抑制剂(例如贝利司他)或靶向免疫毒素同时、在其之前或在其之后给药到患者。
本发明还提供了一种通过以下手段治疗癌症的方法:将本发明的偶联核酸分子与常规化学治疗、放射治疗或抗血管生成药剂或hdaci或靶向免疫毒素相组合给药到需要的患者。本发明还涉及一种药物组合物,其包含本发明的偶联核酸分子和常规化学治疗、放射治疗或抗血管生成药剂或hdaci或靶向免疫毒素,更特别地用于治疗癌症。本发明还涉及一种产品,其包含作为组合制剂的本发明的偶联核酸分子和常规化学治疗、放射治疗或抗血管生成药剂、或hdaci、或靶向免疫毒素,用于同时、分开或顺序使用,更特别地用于治疗癌症。
本发明的其他方面和优点将在下面的实验部分中公开,所述实验部分应该被视为说明性的,而不是限制本申请的范围。在本说明书中引用了许多参考文献,每个这些引用的参考文献通过参考并入本文。
实施例
实施例1:示例性核酸分子的合成
实施例1-1:ox401的合成
ox401的合成是基于使用固相亚磷酰胺化学(da(bz);dc(bz);dg(ibu);dt(-))、heg和chol6亚磷酰胺的标准固相dna合成。
脱三苯甲基步骤使用甲苯中的3%dca进行,氧化使用吡啶/水9/1中的50mm碘进行,硫化使用吡啶/acn1/1中的50mmddtt进行。封端使用acn中的20%nmi以及2,6-二甲基吡啶/acn(40/60)中的20%ac2o一起来进行。切割和去保护分别使用acn中的20%二乙胺25min以除去磷酸酯/硫代磷酸酯上的氰基乙基保护基团和浓氨水在45℃下18小时来进行。
将所述粗品溶液载样到制备型aex-hplc柱(tskgelsuperq5pw20)上。然后进行纯化,使用ph12的含有20体积%乙腈的溴化钠盐梯度进行洗脱。在将级分合并后,在再生纤维素上通过tff进行脱盐。
ox401的纯度:通过aex-hplc测量为91.8%;通过esi-ms测量的分子量:11046.5da。
heg亚磷酰胺(六乙二醇亚磷酰胺)
(编号clp-9765,chemgenescorp)
chol6亚磷酰胺
(编号51230,amchemicals)
实施例1-2:ox402的合成
合成、切割和去保护步骤与ox401相同。
将粗品溶液载样到制备型aex-hplc柱上。然后进行纯化,使用ph8的含有20体积%乙腈的溴化钠盐梯度进行洗脱。在将级分合并后,在稳定化纤维素上通过sec进行脱盐。
ox402的纯度:通过aex-hplc测量为92.2%;通过esi-ms测量的分子量:7340.7da。
实施例1-3:骨架=ox499的合成
ox499的合成是基于与ox401相同的方案,区别在于使用dc(ac)代替dc(bz)并使用nh2-c6亚磷酰胺。
切割和去保护分别使用acn中的20%二乙胺和ama(nh3,甲胺)来进行。
将所述粗品溶液首先在制备型aex-hplc柱上在ph12下纯化,然后通过rp-hplc在ph7下纯化。在将级分合并后,在稳定化纤维素上通过sec进行脱盐。
ox499的纯度:通过aex-hplc测量为95.7%;通过esi-ms测量的分子量:10637.0da。
实施例1-4:ox403的合成
将sv119(0.123mmol)首先与9个单元的活化peg(1.2当量)偶联,然后与ox499偶联。最终的偶联化合物ox403使用rp柱纯化。
实施例1-5:ox404的合成
合成遵照与ox403相同的合成途径进行。
实施例1-6:ox406的合成
合成、切割、去保护和纯化(aex-hplc柱)步骤与ox401一致。
ox406的纯度:通过aex-hplc测量为96.5%;通过esi-ms测量的分子量:11054.3da。
实施例1-7:ox407的合成
合成、切割、去保护和纯化(aex-hplc柱)步骤与ox401一致。
ox407的纯度:通过aex-hplc测量为95.7%;通过esi-ms测量的分子量:10966.2da。
实施例1-8:ox408的合成
合成、切割、去保护和纯化(aex-hplc柱)步骤与ox401一致。
ox408的纯度:通过aex-hplc测量为88.4%;通过esi-ms测量的分子量:10982.2da。
实施例1-9:(ox410)的合成
合成、切割、去保护和纯化步骤与ox401一致。
将粗品溶液首先在制备型aex-hplc柱上纯化,然后通过rp-hplc柱纯化。
ox410的纯度:通过aex-hplc测量为83.6%;通过esi-ms测量的分子量:11051.3da。
实施例1-10:(ox411)的合成
合成、切割、去保护和纯化步骤与ox401一致。
将粗品溶液首先在制备型aex-hplc柱上纯化,然后通过rp-hplc柱纯化。
ox411的纯化:通过aex-hplc测量为83.1%;通过esi-ms测量的分子量:11051.3da。
实施例2:ox401超活化parp但不活化dna-pk
材料和方法
细胞培养
三阴性乳腺癌细胞系mda-mb-231购自atcc,并按照供应商的说明书生长。简单来说,mda-mb-231细胞在增补有10%胎牛血清(fbs)的l15leibovitz培养基中生长,并维持在37℃和0%co2的加湿气氛中。
elisa抗par化
使用夹心elisa来检测聚(adp-核糖)(par)聚合物。将细胞在增补有1mmpmsf(苯甲磺酰氟,sigma)的组织蛋白提取(t-per)缓冲液(thermoscientific)中煮沸。然后在elisa测定之前将细胞提取物在superblock缓冲液(thermoscientific)中稀释。将96孔聚苯乙烯板(thermoscientificpierce,白色不透明)用每孔100μl含有捕获抗体(小鼠抗par抗体,4μg/ml,trevigen4335)的碳酸盐缓冲液(1.5g/l碳酸钠na2co3,3g/lnahco3)在4℃包被过夜,然后将它用pbst溶液清洗。然后将所述孔用superblock在37℃外包1h。然后向65μlsuperblock添加10μl细胞提取物,并将其一式三份添加到各个孔,在4℃温育过夜,然后用pbst溶液清洗。然后添加检测抗体(兔抗par抗体,trevigen4336,在pbs/2%奶粉/1%小鼠血清中1/1000稀释)并在室温下温育1h。在清洗后,向每个孔施加第二抗体即hrp偶联的抗兔抗体(abcam,ab97085,在pbs/2%奶粉/1%小鼠血清中1/5000稀释),温育1h。为了读数,向每个孔添加75μl酶底物(supersignalpico,pierce)。立即测定化学发光读数。
elisa抗γh2ax
使用夹心elisa来检测磷酸化形式的组蛋白h2ax(γh2ax)。将细胞在增补有1mmpmsf(苯甲磺酰氟,sigma)的组织蛋白提取(t-per)缓冲液(thermoscientific)中煮沸。然后在elisa测定之前将细胞提取物在superblock缓冲液(thermoscientific)中稀释。将96孔聚苯乙烯板(thermoscientificpierce,白色不透明)用每孔100μl含有捕获抗体(小鼠抗γh2ax抗体,4μg/ml,millipore05-636)的碳酸盐缓冲液(1.5g/l碳酸钠na2co3,3g/lnahco3)在4℃包被过夜,然后将它用pbst溶液清洗。然后将所述孔用superblock在37℃外包1h。然后一式三份向每个孔施加50μl细胞提取物,并在25℃温育2h,然后用pbst溶液清洗。然后添加检测抗体(兔抗h2ax抗体,abcamab11175,在pbs/2%奶粉中1/500稀释)并在25℃温育1h。在清洗后,向每个孔施加hrp偶联的抗兔第二抗体(abcam,ab97085,在pbs/2%奶粉中1/20000稀释),在25℃温育1h。为了读数,向每个孔添加75μl酶底物(supersignalpico,pierce)。立即测定化学发光读数。
统计分析
所有统计分析使用双尾studentt检验来进行。
结果
首先,本发明人通过监测dna依赖性蛋白激酶(dna-pk)和聚(adp-核糖)聚合酶(parp)的活化分析了ox401在mda-mb-231细胞中的活性。在与模拟双链断裂的asidnatmdna组成部分相互作用后,两种酶均被活化以修饰它们的靶。用asidna处理的mda-mb-231细胞在处理后显示出组蛋白h2ax(γh2ax)的剂量依赖性磷酸化和聚(adp-核糖)(par)聚合物积累(par化),其分别由dna-pk和parp活化引起(图1a,b)。与asidnatm相比,用ox401处理的细胞不相互作用并活化dna-pk酶(图1a)。然而,ox401超活化parp酶并诱导剂量依赖性的par化,为asidnatm的两倍高(图1b)。因此,他们在mda-mb-231细胞中观察到靶点作用,其由ox401诱导的假dna损伤信号传导(par化)示出。
实施例3:ox401表现出特异性抗肿瘤活性
材料和方法
细胞培养
细胞培养使用三阴性乳腺癌细胞系mda-mb-231、组织细胞性淋巴瘤细胞系u937和非肿瘤乳腺细胞系mcf-10a进行。细胞按照供应商的说明书生长。细胞系维持在37℃下和5%co2的加湿气氛中,例外的是mda-mb-231细胞系,其维持在0%co2下。
药物处理和细胞存活率的测量
将mda-mb-231(5x103个细胞/孔)、mcf-10a(5x103个细胞/孔)和u937(2x104个细胞/孔)接种在96孔板中并在+37℃温育24小时,然后添加浓度逐渐提高的药物4至7天。在药物暴露后,使用xtt测定法(sigmaaldrich)测量细胞存活率。简单来说,将xtt溶液直接添加到含有细胞培养物的每个孔,并将所述细胞在37℃温育5小时,然后使用微孔板读板器(bmgfluostar,galaxy)读取490nm和690nm处的吸光度。细胞存活率被计算为活的处理过的细胞与活的模拟处理的细胞的比率。使用graphpadprism软件(5.04版),通过将每种细胞系的存活百分率对药物浓度对数作图,通过非线性回归模型来计算ic50(其表示50%的细胞存活时的剂量)。
结果
由于ox401与asidnatm相比只诱导parp靶点作用而不诱导dna-pk,因此我们希望确保它表现出令人感兴趣的抗肿瘤活性。将肿瘤(mda-mb-231、u937)和非肿瘤(mcf-10a)细胞用asidna(黑色)或ox401(深灰色)处理,并在处理后4天(u937)或7天(mda-mb-231和mcf-10a)使用xtt测定法测量存活率(图2)。ox401表现出比asidnatm更高的抗肿瘤活性,正如由ox401的ic50值比asidnatm低3倍所示出的(图2a)。mcf10a非肿瘤细胞对ox401不敏感,突出了ox401的肿瘤特异性(图2b)。在非肿瘤细胞中没有任何作用预测了ox401治疗在正常组织中无毒性并具有高安全性。
实施例4:ox401诱导肿瘤免疫应答
材料和方法
细胞培养
细胞培养使用三阴性乳腺癌细胞系mda-mb-231和非肿瘤乳腺细胞系mcf-10a进行。细胞按照供应商的说明书生长。将细胞系维持在37℃下和5%co2的加湿气氛中,例外的是mda-mb-231细胞系,其维持在0%co2下。
使用ox401或asidnatm的长期处理
将细胞以适合的密度接种在6孔培养板中,并在37℃温育24h,然后以5μm的浓度添加ox401或asidnatm。在处理后第7天收获细胞,将其清洗以除去药物,并再次接种在6孔培养板中恢复7天。一周处理/一周释放的时间段构成了一个处理周期。在每个处理周期后进行进一步分析(微核定量;western印迹;elisa;流式细胞术)。
western印迹分析
收获用ox401或asidnatm(5μm)处理一个周期的细胞,以适合的密度接种,然后再次处理48小时。然后将细胞在含有蛋白酶和磷酸酶抑制剂(rocheappliedscience,germany)的ripa缓冲液(150mmnacl,50mmtris碱,5mmedta,1%np-40,0.25%脱氧胆酸盐,ph7.4)中裂解。蛋白质浓度使用bca蛋白测定法(thermofisherscientific,usa)测量。将等量(15μg)的蛋白质用sds-page(12%凝胶)进行电泳,转移到硝酸纤维素膜,用含有5%脱脂奶粉的tbstween1%在室温阻断1小时,然后与第一抗体在4℃温育过夜。在用tbs/tween1%清洗后,将膜与第二抗体在室温温育1小时。结合的抗体使用增强化学发光western印迹底物试剂盒(ozyme,美国)来检测。western印迹使用下述抗体来进行:兔抗sting单克隆第一抗体(1/1,000稀释;cst-13647),小鼠抗pd-l1单克隆第一抗体(1/1,000稀释;abcamab238697),小鼠抗β-肌动蛋白单克隆第一抗体(1/10,000稀释,sigmaa1978),hrp偶联的山羊抗兔igg第二抗体(1/2,000稀释,millipore12-348)和hrp偶联的山羊抗小鼠igg第二抗体(1/2,000稀释,millipore12-349)。
用于检测细胞表面pd-l1的流式细胞术
收获用ox401或asidnatm(5μm)处理一个周期的细胞,以适合的密度接种在6孔板中,然后再次处理48小时。然后将细胞用pbs清洗,并与alexafluor488偶联的抗pd-l1单克隆抗体(cst–14772)在4℃温育1小时。然后将细胞用pbs清洗,并使用guavaeasycyte(merck)测定荧光强度。数据使用flowjo软件(treestar,加州)进行分析。
微核定量
微核由染色体断裂或纺锤体损伤引起。它们在细胞分裂后在子代细胞的核中出现,并在细胞质中形成单个或多个微核。将用ox401或asidnatm(5μm)处理一个周期的细胞在皮氏培养皿中的盖玻片上生长。然后将细胞用pfa(4%)固定,用triton(0.5%)通透化,并用dapi(0.5mg/ml)染色。微核的频率被估算为具有微核的细胞占细胞总数的百分率。每种条件分析至少1,000个细胞。
用于检测ccl5趋化因子的elisa
收获用ox401或asidnatm(5μm)处理一个周期的细胞,以适合的密度接种在6孔板中,然后再次处理48小时。然后将细胞培养物上清液以2,000xg离心10分钟以除去碎片。试剂盒(人类simplestepelisa试剂盒–abcam–ab174446)随附的96孔板条被供应为可立即使用。将50μl每种上清液与50μl抗体混合物一式两份添加到每个孔,然后在设置到400rpm的板振摇器上在室温温育1h,然后将其用1x清洗缓冲液pt清洗。然后向每个孔添加100μltmb底物,并在设置到400rpm的板振摇器上在暗处温育10min。然后向每个孔添加100μl终止溶液,在板振摇器上1分钟,并在450nm处测定吸光度。
用于检测cxcl10(ip-10)趋化因子的elisa
收获用ox401或asidnatm(5μm)处理一个周期的细胞,以适合的密度接种在6孔板中,然后再次处理48小时。然后将细胞培养物上清液以1,000xg离心10分钟以除去碎片。试剂盒(ip-10(cxcl10)人类elisa试剂盒–abcam–ab83700)随附的96孔板条被供应为可立即使用。一式两份向每个孔添加100μl每种上清液,然后在室温温育2h,然后将其用1x清洗缓冲液pt清洗。然后向每个孔添加50μl生物素化抗ip-10抗体并温育1h,然后将其用1x清洗缓冲液pt清洗。然后向每个孔添加100μl1x链亲合素-hrp溶液,温育30min,并用1x清洗缓冲液pt清洗。然后向每个孔添加100μl发色体tmb底物溶液,并在暗处温育10-20分钟。向每个孔添加100μl终止试剂,并立即测定在450nm处的吸光度。
统计分析
所有统计分析使用双尾studentt检验来进行。
结果
由于ox401是双链dna,因此我们想知道它是否可以被先天免疫途径识别。干扰素基因刺激物(sting)是一种感应外源和内源胞浆dna两者并触发i型干扰素和促炎性细胞因子应答的胞浆受体。因此,本发明人在用ox401处理的细胞中评估了sting途径的活化。有趣的是,ox401不通过sting途径被识别为外源dna,并且不触发趋化因子和干扰素细胞因子的直接诱导(数据未示出)。
由于用ox401短期处理不直接诱导抗肿瘤免疫应答,因此本发明人假设长期处理可能通过未修复的dna结构的积累间接触发sting依赖性免疫应答。与这个假设相一致,与未处理的或asidnatm处理的细胞相比,用ox401长期处理(一周处理/一周释放的一个周期)的细胞显示出具有微核的细胞的%的两倍的显著提高(图3a)。为了验证微核增加与sting途径活化之间的联系,本发明人分析了ccl5和cxcl10靶趋化因子的释放。有趣的是,与未处理的细胞相比,用ox401长期处理的细胞分泌两倍多的ccl5和1.5倍多的cxcl10(图3b)。asidna处理的细胞未显示出ccl5或cxcl10的更多分泌(图3b)。肿瘤细胞中sting途径活化的结果包括pd-l1(程序化死亡配体1)上调,可能是针对免疫系统提供保护的一种反应。本发明人在长期处理的细胞中分析了总pd-l1或细胞表面结合的pd-l1的水平。与asidnatm处理的细胞的亲代细胞相比,ox401处理的细胞显示出总水平(图3c)和膜结合的pd-l1(图3d)的很大提高。
合在一起,这些结果证实了ox401通过微核诱导的积累触发间接的sting途径活化,并为与抗pd-l1疗法的组合治疗铺平道路。
实施例6:药学性质/pk/pd实验
将ox401以2mg的剂量通过静脉内(iv)途径注射到小鼠中产生通过hplc方法所测量的8μm的最高血浆浓度(cmax)。出人意料的是,该cmax比在相同实验条件下使用asidna获得的cmax高40倍。
实施例7:ox402超活化parp–更小但活性与ox401相同
材料和方法
细胞培养
三阴性乳腺癌细胞系mda-mb-231购自atcc,并按照供应商的说明书生长。简单来说,mda-mb-231细胞在增补有10%胎牛血清(fbs)的l15leibovitz培养基中生长,并维持在37℃和0%co2的加湿气氛中。
elisa抗par化
使用夹心elisa来检测聚(adp-核糖)(par)聚合物。将细胞在增补有1mmpmsf(苯甲磺酰氟,sigma)的组织蛋白提取(t-per)缓冲液(thermoscientific)中煮沸。然后在elisa测定之前将细胞提取物在superblock缓冲液(thermoscientific)中稀释。将96孔聚苯乙烯板(thermoscientificpierce,白色不透明)用每孔100μl含有捕获抗体(小鼠抗par抗体,4μg/ml,trevigen4335)的碳酸盐缓冲液(1.5g/l碳酸钠na2co3,3g/lnahco3)在4℃包被过夜,然后将它用pbst溶液清洗。然后将所述孔用superblock在37℃外包1h。然后向65μlsuperblock添加10μl细胞提取物,并将其一式三份添加到各个孔,在4℃温育过夜,然后用pbst溶液清洗。然后添加检测抗体(兔抗par抗体,trevigen4336,在pbs/2%奶粉/1%小鼠血清中1/1000稀释)并在室温下温育1h。在清洗后,向每个孔施加第二抗体即hrp偶联的抗兔抗体(abcam,ab97085,在pbs/2%奶粉/1%小鼠血清中1/5000稀释),温育1h。为了读数,向每个孔添加75μl酶底物(supersignalpico,pierce)。立即测定化学发光读数。
结果
本发明人还分析了活化parp和诱导假损伤信号传导(par化)所需的最小序列长度。将mda-mb-231细胞用ox402这种10个碱基对(bp)的分子处理24h,并使用抗par化elisa测定法监测parp活化。用ox402处理的mda-mb-231细胞显示出由parp作用和活化引起的剂量依赖性par化(图4)。因此,10bp的分子足以劫持并活化parp。
实施例8:ox401诱导细胞内nad+耗竭
parp蛋白以高亲和性与dsb结合。在结合后,parp被自动“par化”,并通过被称为par化的添加聚(adp-核糖)(par)聚合物来活化其他靶蛋白。通过监测蛋白质par化,在用ox401处理的mda-mb-231和mrc5细胞中研究了parp活化的动力学。
材料和方法
细胞培养
细胞培养使用三阴性乳腺癌细胞系mda-mb-231和非肿瘤mrc5原代肺成纤维细胞来进行。所有细胞系购自atcc并按照供应商的说明书在37℃和5%co2的加湿气氛中生长,例外的是mda-mb-231(37℃和0%co2)。
细胞用ox401的处理和存活率的评估
将mda-mb-231或mrc5细胞以适合的密度接种在60mm直径的培养板中,并在37℃温浴过夜。然后将细胞用5μmox401处理48小时、7天和13天,然后清洗、收获并使用锥虫蓝(4%)细胞染色测定法和eve自动细胞计数仪(vwr)计数,用于进一步分析。
nad+的细胞内水平的测量
nad含量使用nad/nadh-glo测定试剂盒(promega,g9071),按照制造商的说明书来确定。所述测定法的原理在于连续的转化:首先,nad循环酶将nad+修饰成nadh,后者被还原酶利用以将底物转变成萤光素。然后萤光素酶使用萤光素产生光。因此,产生的发光与细胞中存在的nad+的量成正比。
简单来说,收获用ox401(5μm)处理48小时、7天或13天的mda-mb-231或mrc5细胞,并接种在96孔板中(5x104个细胞/孔)。然后使用1%dtab缓冲液将细胞裂解,在每个孔中添加25μl0.4mhcl,在60℃温育15min并在室温温育10min。在每个孔中添加25μl检测试剂trizma和100μlnad检测试剂。在微孔板读板器(enspiretmperkin-almer)上测量产生的发光信号。
western印迹分析
收获用ox401(5μm)处理48小时、7天或13天的mda-mb-231或mrc5细胞,并在含有蛋白酶和磷酸酶抑制剂(rocheappliedscience,germany)的ripa缓冲液(150mmnacl,50mmtris碱,5mmedta,1%np-40,0.25%脱氧胆酸盐,ph7.4)中裂解。蛋白质浓度使用bca蛋白测定法(thermofisherscientific,usa)来测量。将等量(15μg)的蛋白质用sds-page(12%凝胶)进行电泳,转移到硝酸纤维素膜,用含有5%脱脂奶粉的tbstween1%在室温阻断1小时,然后与第一抗体在4℃温育过夜。在用tbs/tween1%清洗后,将膜与第二抗体在室温温育1小时。结合的抗体使用增强化学发光western印迹底物试剂盒(ozyme,美国)来检测。western印迹使用下述抗体来进行:抗泛adp核糖结合试剂(1/1,500稀释;milliporemabe1016),小鼠抗β-肌动蛋白单克隆第一抗体(1/10,000稀释,sigmaa1978),hrp偶联的山羊抗兔igg第二抗体(1/2,000稀释,millipore12-348)和hrp偶联的山羊抗小鼠igg第二抗体(1/2,000稀释,millipore12-348)。
结果
用ox401(5μm)处理过的mda-mb-231细胞在处理后显示出par化蛋白质的积累,如处理后7天的照片所示(图5a)。由于烟酰胺腺嘌呤二核苷酸(nad+)被parp用作底物以将其靶蛋白par化,因此分析了ox401处理后的细胞内nad+水平。ox401在mda-mb-231细胞中诱导nad+的大量消耗,最大值为在处理7天后与未处理的细胞相比55%的nad+水平,其一直维持到处理后13天(图5b)。由于这种巨大的ox401诱导的nad+缺乏,我们假设肿瘤细胞在ox401处理下不能调控并维持nad+水平的体内平衡,导致细胞死亡。为了测试这一假设,我们分析了在ox401处理下mda-mb-231的细胞存活率。在ox401处理后48小时没有观察到对细胞存活率的影响,这与此时nad+水平降低非常少相符。在处理后7和13天观察到对细胞存活率的显著影响(与未处理的细胞相比,存活率分别为57%和32%),证实了nad+水平对细胞存活率的重要性(图5c)。所有这些影响均为对肿瘤细胞特异的,因为在ox401处理的mrc5非肿瘤细胞中既没有观察到nad+耗竭也没有观察到细胞死亡(图5d-f)。
合在一起,这些结果表明用ox401长期处理诱导了parp超活化和nad+消耗两者。ox401诱导nad+水平快速下降到低于与细胞存活相容的阈值,这会超过细胞nad+补充能力并触发肿瘤细胞大量死亡。
实施例10
ox401扰乱同源重组(hr)修复途径
由于ox401引诱parp并诱导假dna损伤par化信号传导,因此本发明人试验了ox401是否可以触发dna损伤的快速积累。
同源重组(hr)修复途径是一种无错修复途径,对于维持遗传稳定性和完整dna信息来说是必需的。hr是组织良好的多步骤机制,消耗大量的细胞能量。由于ox401触发高nad+消耗并因此诱导肿瘤细胞中的代谢失衡(实施例9),本发明人假设ox401可能扰乱非常依赖能量的hr修复机制。为了验证这一假设,分析了ox401处理后的hr修复效率(通过检测rad51蛋白向dsb部位的召集)。
材料和方法
细胞培养
细胞培养使用三阴性乳腺癌细胞系mda-mb-231来进行。细胞在完全l15leibovitz培养基中生长,并维持在37℃下和0%co2的加湿气氛中。
同源重组途径活性分析
对于免疫染色来说,将细胞以5x105个细胞的浓度接种在盖玻片(menzel,德国不伦瑞克)上,并在37℃温育1天。然后将细胞用奥拉帕尼(5μm)+/-ox401(5μm)处理。处理后48h,将细胞在4%聚甲醛/磷酸盐缓冲盐水(pbs1x)中固定20min,在0.5%tritonx-100中通透化10min,用2%牛血清白蛋白/pbs1x阻断,并与第一抗体在4℃温育1h。所有第二抗体以1/200的稀释度使用,在室温(rt)下45min,并将dna用4’,6-二脒基-2-苯基吲哚(dapi)染色。使用了下述抗体:小鼠抗磷酸化h2ax单克隆第一抗体(millipore,法国吉扬库尔),兔抗rad51抗体(merkmillipore,德国达姆施塔特),alexa-633偶联的山羊抗小鼠igg第二抗体(molecularprobes,eugene,美国俄勒冈州)和alexa-488偶联的山羊抗兔igg第二抗体(molecularprobes,eugene,美国俄勒冈州)。
通过流式细胞术分析药物诱导的dna损伤
将细胞用ox401(5μm)或奥拉帕尼(5μm)处理48小时,然后用冷(-20℃)70%乙醇固定并通透化至少2小时。在用pbs清洗后,将细胞用pbs中的0.5%triton在rt进一步通透化20分钟,在pbs中清洗,并与含2%bsa的pbs中的抗γ-h2ax抗体(05-636millipore)温育。在用pbs清洗后,将细胞与alexafluor488偶联的第二抗体温育。使用guavaeasycyte细胞仪(luminex)测定荧光强度。数据使用flowjo软件(treestar,加州)来分析。
结果
正如预期,正如通过流式细胞术(图6a)或通过免疫荧光检测的γh2ax灶点(图6b)测量的高的组蛋白h2ax磷酸化(γh2ax)所示,在mda-mb-231细胞中奥拉帕尼在处理后48h诱导双链断裂(dsb)的积累。作为比较,ox401不诱导γh2axdsb生物标志物的增加,并因此不触发直接dsb积累(图6a、b)。
用奥拉帕尼(5μm)处理48h的mda-mb-231细胞显示出与rad51灶点共定位的γh2ax灶点的积累,表明奥拉帕尼诱导的dsb被hr修复途径修复(图6c)。添加ox401(5μm)显著减少了由奥拉帕尼诱导的rad51灶点的形成(图6c、d),证实了ox401可能通过随着代谢失衡而来的能量耗竭而有效扰乱hr途径。
实施例11
肿瘤细胞不获得对ox401的耐药性
目前公认,癌症经历由查尔斯·达尔文在其自然选择概念中提出的进化过程。自然选择是自然界选择某些物理属性或表型传递给后代以使生物体更好地“适应”环境的过程。在靶向疗法的选择压力下,癌细胞的耐药性群体不断演化,从而产生适应由所述治疗引起的新环境的“耐药克隆”。也已确认,肿瘤细胞需要大量能量才能发展出对抗癌治疗的耐药性。由于ox401诱导nad+耗竭和代谢失衡(实施例8),因此发明人测试了细胞是否发展出对ox401的耐药性。
材料和方法
细胞培养
细胞培养使用淋巴瘤细胞系u937来进行。细胞在增补有10%fbs和1%青霉素/链霉素的完全rpmi培养基中生长,并维持在37℃下和5%co2的加湿气氛中。该细胞系根据它对ox401和他拉唑帕尼两者的高敏感性而被选择。
获得性耐药性的选择
对于用于选择耐药性的重复的处理周期来说,将u937细胞以适合的密度(2x105个细胞/ml)接种并在37℃温育24h,并以对应于相对于未处理的细胞10-20%的存活率的剂量添加药物。在2μm他拉唑帕尼或1.5μmox401下选择耐药性。在处理后第4天收获细胞,清洗,并在用0.4%锥虫蓝染色后计数(evetm计数载片,nanoentek)。在计数后,将细胞接种在适合的培养板中,并允许其恢复(无药物时段)3至7天。然后开始另一个处理/恢复周期,至多4个周期。
获得性耐药性的不可逆性——细胞存活率的测量
为了评估获得性耐药性的不可逆性,将u937亲代或耐药细胞接种在96孔板中(2x104个细胞/孔),并用浓度逐渐提高的他拉唑帕尼处理4天。在药物暴露后,使用xtt测定法(sigmaaldrich)测量细胞存活率。简单来说,将xtt溶液直接添加到含有细胞培养物的每个孔,并将细胞在37℃温育5小时,然后使用微孔板读板器(bmgfluostar,galaxy)在490nm和690nm处读取吸光度。细胞存活率被计算为活的处理过的细胞与活的模拟处理的细胞的比率。使用graphpadprism软件(5.04版),通过将每种细胞系的存活率百分数针对药物浓度的对数作图,通过非线性回归模型来计算ic50(其表示50%的细胞存活时的剂量)。
结果
对u937细胞进行了使用ox401或他拉唑帕尼的处理周期。用他拉唑帕尼处理的细胞在扩增时段中恢复,而用ox401处理的细胞在无药物扩增时段中不生长(图7a)。用他拉唑帕尼处理的细胞在处理周期中发展出获得性耐药性,细胞存活率从第一个周期后的10%演变成第四个处理周期后(处理开始后33天)超过50%的存活率(p<0.01)(图7b)。为了评估对他拉唑帕尼的不可逆耐药性状态,使耐药细胞经受剂量逐渐提高的他拉唑帕尼,以分析它们与亲代细胞相比的敏感性。对他拉唑帕尼敏感的亲代细胞显示出2μm的低ic50。tal1、tal2和tal3耐药群体显示出超过4μm的更高的ic50(图7c)。
实施例12
ox401放大抗肿瘤免疫应答
在以前的实验中(图3),本发明人显示用ox401长期处理表现出微核诱导的sting途径活化以及ccl5和cxcl10趋化因子分泌的增加。为了测试这些抗肿瘤免疫效应的效果,进行了肿瘤细胞与新鲜分离的t细胞的共培养,并评估了t细胞诱导的细胞毒性效应。
材料和方法
细胞培养
细胞培养使用三阴性乳腺癌细胞系mda-mb-231和宫颈肿瘤细胞系hela来进行。细胞购自atcc,并按照供应商的说明书生长。将细胞维持在37℃下和5%co2的加湿气氛中。
pbmc的分离
健康供体的血沉棕黄层购自efs血液中心(法国巴黎)。使用easysep直接人类pbmc分离试剂盒(19654,stemcell,法国),按照制造商的方案分离pbmc。将所述分离的pbmc在冷冻介质(10%dmso和90%fbs)中调整到5x107个细胞/ml的浓度,从其将1ml等分试样分发到冻存管中,并在液氮中在-196℃下储存直至使用。
从pbmc分离t淋巴细胞
使用easysep人类t细胞分离试剂盒(17951,stemcell,法国),按照制造商的方案从pbmc分离t淋巴细胞。将分离的t细胞以106个细胞/ml的浓度悬浮在immunocult-xft细胞扩增培养基(10981,stemcell,法国)中,并在进一步实验之前使用immunocult人类cd3/cd28/cd2t细胞活化剂(10970,stemcell,法国)活化24小时。
肿瘤和t淋巴细胞的共培养
将mda-mb-231细胞接种在12孔细胞培养板(5x104个细胞/孔)或60mm直径的细胞培养板(106个细胞/板)中,并在37℃温育24小时。在含有或不含ox401(5μm)的情况下,以4:1的效应细胞与靶细胞的比例向肿瘤细胞添加活化的t细胞。将共培养物在37℃温育48小时。在温育结束时,对每种细胞类型(贴壁肿瘤细胞或悬浮t细胞)进行计数,并收获上清液用于细胞因子释放分析。
western印迹分析
收获含有或不含t淋巴细胞的用ox401(5μm)处理的细胞,然后在含有蛋白酶和磷酸酶抑制剂(rocheappliedscience,germany)的ripa缓冲液(150mmnacl,50mmtris碱,5mmedta,1%np-40,0.25%脱氧胆酸盐,ph7.4)中裂解。蛋白质浓度使用bca蛋白测定法(thermofisherscientific,usa)测量。将等量(15μg)的蛋白质用sds-page(12%凝胶)进行电泳,转移到硝酸纤维素膜,用含有5%脱脂奶粉的tbstween1%在室温阻断1小时,然后与第一抗体在4℃温育过夜。在用tbs/tween1%清洗后,将膜与第二抗体在室温温育1小时。结合的抗体使用增强化学发光western印迹底物试剂盒(ozyme,美国)来检测。western印迹使用下述抗体来进行:兔抗sting单克隆第一抗体(1/1,000稀释;cst-13647),小鼠抗pd-l1单克隆第一抗体(1/1,000稀释;abcamab238697),小鼠抗β-肌动蛋白单克隆第一抗体(1/10,000稀释,sigmaa1978),hrp偶联的山羊抗兔igg第二抗体(1/2,000稀释,millipore12-348)和hrp偶联的山羊抗小鼠igg第二抗体(1/2,000稀释,millipore12-349)。
检测ccl5趋化因子的elisa
在存在或不存在t淋巴细胞的情况下将细胞用ox401(5μm)处理48小时。然后将细胞物培养上清液以2,000xg离心10分钟以除去碎片。试剂盒(人类simplestepelisa试剂盒–abcam–ab174446)随附的96孔板条被供应为可立即使用。将50μl每种上清液与50μl抗体混合物一式两份添加到每个孔,然后在设置到400rpm的板振摇器上在rt温育1h,然后将其用1x清洗缓冲液pt清洗。然后向每个孔添加100μltmb底物,并在设置到400rpm的板振摇器上在暗处温育10min。然后向每个孔添加100μl终止溶液,在板振摇器上1分钟,并在450nm处测定吸光度。
检测颗粒酶b的elisa
在存在或不存在t淋巴细胞的情况下将细胞用ox401(5μm)处理48小时。然后将细胞培养物上清液以2,000xg离心10分钟以除去碎片。试剂盒(人类simplestep颗粒酶belisa试剂盒–abcam–ab235635)随附的96孔板条被供应为可立即使用。将50μl每种上清液与50μl抗体混合物一式两份添加到每个孔,然后在设置到400rpm的板振摇器上在rt温育1h,然后将其用1x清洗缓冲液pt清洗。然后向每个孔添加100μltmb底物,并在设置到400rpm的板振摇器上在暗处温育10min。然后向每个孔添加100μl终止溶液,在板振摇器上1分钟,并在450nm处测定吸光度。
结果
正如由mda-mb-231肿瘤细胞存活率的降低(与不含t细胞的mda-mb-231细胞相比50%的存活率)所揭示的(图8a),新鲜活化的t细胞在共培养开始后48和72小时触发抗肿瘤细胞毒性效应。向共培养物添加ox401进一步提高了t细胞诱导的抗肿瘤细胞毒性(与不含t细胞的未用ox401处理的mda-mb-231细胞相比20%的存活率)(图8a)。有趣的是,在用ox401处理的mda-mb-231肿瘤细胞存在下,细胞毒性t细胞分泌更大量的颗粒酶b(图8b),与更高的细胞毒性效能(图8a)相符。考虑到sting途径活化对触发更高的免疫细胞召集和抗肿瘤细胞毒性的重要性,我们在存在或不存在ox401的情况下在肿瘤/免疫细胞共培养物中分析了该途径。在共培养48小时后,我们在用ox401处理的肿瘤细胞中观察到sting蛋白质水平的更高的提高(图8c)。这伴有更高的irf3蛋白磷酸化(图8c)和分泌的ccl5趋化因子的增加(图8d),表明持续的sting途径活化。
合在一起,这些发现证实了通过更高的sting途径活化,可能刺激t细胞向肿瘤细胞附近更好的召集,抗肿瘤细胞毒性t细胞被ox401高度增强。
实施例13:结合动力学(kon)和相互作用强度(kd)
材料和方法
通过spr技术,使用来自于gehealthcarelifesciences的biacoret100仪器,表征了根据本发明所述的不同分子与人类聚[adp-核糖]聚合酶1蛋白(parp-1)(115kda)的相互作用。parp1-his被捕获在固定在羧甲基化芯片表面上的抗his抗体上。
结果
结合动力学(kon)以及相互作用强度(kd)报告在图9中。
在3’和/或5’链上的前三个核苷酸上具有修饰的磷酸二酯骨架例如硫代磷酸酯键(ox401)或硫代磷酸酯键和fana修饰两者(ox410、ox411)的ox401、ox410和ox411,对parp-1具有相近的亲和性(kd)和结合动力学(kon)。在3’和/或5’链上的前三个核苷酸上具有硫代磷酸酯键的ox402与上面提到的分子相比对parp-1具有相近的结合亲和性,但对parp-1具有更低的结合动力学。
使用在3’-链中带有两个fana修饰的ox406,结合强度似乎更高。
将硫代磷酸酯修饰的数目减少到单个核苷酸(ox407和ox408)显著提高了相互作用强度(kd值降低)和结合动力学(kon)。
从这组实验清楚地显示,3’和/或5’链上前三个核苷酸的化学修饰通过调节相互作用强度(kd)和结合动力学(kon),对偶联核酸分子于parp的相互作用具有强烈影响,证实了它们是非常令人感兴趣的dna修复途径抑制剂以及因此在癌症疗法中发挥作用的潜在候选物。
序列表
<110>欧恩科斯欧公司(onxeo)
<120>新的偶联核酸分子及其用途
<130>b2921pc
<160>2
<170>patentinversion3.5
<210>1
<211>16
<212>dna
<213>人工
<220>
<223>核酸分子
<400>1
cccagcaaacaagcct16
<210>2
<211>10
<212>dna
<213>人工
<220>
<223>核酸分子
<400>2
cagcaacaag10
- 还没有人留言评论。精彩留言会获得点赞!