一种法匹拉韦和/或其衍生物的精制方法与流程
本发明属于药物合成领域,具体涉及一种法匹拉韦和/或其衍生物的精制方法。
背景技术:
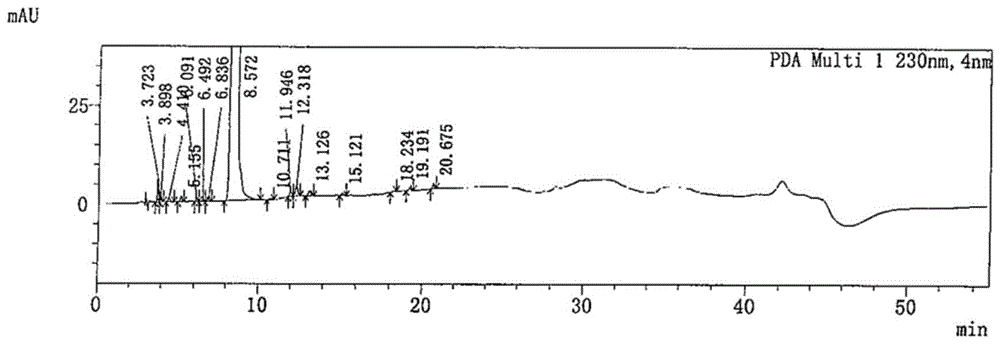
:法匹拉韦(favipiravir)为rna依赖的rna聚合酶(rdrp)抑制剂类广谱抗病毒药物,该药物对人类应对可能出现的烈性病毒性传染病和可能的生物恐怖袭击具有重要的现实意义。日本在其国家药品质量标准中拟定法匹拉韦为白色至淡黄色粉末。但法匹拉韦的制备中需要多步连续反应(或放大生产),易得到颜色较重(如黄色、灰色、棕色、黑色等)的固体,而不符合药品质量标准要求。cn102307865b仅公开了制得固体产物,但没有公开制得固体的颜色。cn104496917a公开制得黄色固体,不符合药品质量标准。cn1418220a公开了将粗品经硅胶柱层析纯化精制制得浅黄色固体的方法,但该方法存在成本高、不适宜工业化生产、产品性状非粉末、不符合药品质量标准等缺陷。cn106478528a公开了通过色谱柱纯化分离得到淡黄色固体,存在成本高、不适宜工业化生产等缺陷。cn101809003a公开了淡黄白固体的制备方法,该方法存在下述缺陷:一是淡黄白固体是经6-氟-3-羟基-2-氰基吡嗪的二丙胺盐特定中间体制备方法下的产物,并不是常规纯化操作;二是有机胺难以去除造成溶剂残留,增加药品质量控制的难度;三是产品性状非粉末,不符合原研药品拟定质量标准。cn107226794a和cn106866553a公开了乙醇重结晶制得类白色固体的方法,但当粗品颜色较重时,采用醇类包括乙醇重结晶难以得到白色到淡黄色固体,故存在适用范围受限问题。综上,现有技术有关法匹拉韦的纯化方法存在以下缺陷:一是柱层析纯化不适宜工业化生产;二是采用常规重结晶纯化难以得到符合质量标准颜色、性状的产品,即使重结晶过程中使用活性炭进行脱色仍难以符合药品质量标准;三是即使实验室小试阶段产品接近淡黄色,放大生产中极大可能颜色不达标,造成不可预估的损失。故亟需一种简便易行、适宜工业化大生产的脱色、精制方法,以便得到符合药品质量标准的产品。技术实现要素:本发明的目的在于提供一种法匹拉韦和/或其衍生物的精制方法,包括如下步骤:将待精制的法匹拉韦和/或其衍生物溶于有机溶剂a中,经硅胶过滤后,干燥,纯化,即得。本发明的优选技术方案中,所述方法中待精制的法匹拉韦:硅胶的重量比为1:1-1:6,优选为1:2-1:5,更优选为1:3-1:4。本发明的优选技术方案中,所述硅胶的粒径选自100-200目、200-300目、300-400目的任一种或其组合。本发明的优选技术方案中,所述有机溶剂a选自乙酸乙酯、二氯甲烷、四氢呋喃、异丙醇、乙醇、甲醇、丙酮、石油醚的任一种或其组合。本发明的优选技术方案中,当有机溶剂a为二氯甲烷/乙酸乙酯组合时,二氯甲烷:乙酸乙酯的重量比为2:1-10:1,优选为3:1-9:1,更优选为4:1-8:1,最优选为5:1-7:1。本发明的优选技术方案中,溶解温度为20℃-40℃,优选为25℃-35℃。本发明的优选技术方案中,所述纯化选自重结晶、打浆的任一种或其组合。本发明的优选技术方案中,所述重结晶包括以下步骤:将待精制的法匹拉韦溶于有机溶剂b中,冷却析晶,分离固体后,干燥,即得。本发明的优选技术方案中,所述有机溶剂b选自乙酸乙酯、二氯甲烷、四氢呋喃、异丙醇、乙醇、甲醇、丙酮、石油醚的任一种或其组合。本发明的优选技术方案中,所述重结晶步骤中的溶解温度为重结晶溶剂体系沸点温度以下10℃至沸点温度。本发明的优选技术方案中,所述重结晶步骤的冷却温度为-10℃至30℃,优选为-5℃至25℃。本发明的优选技术方案中,所述重结晶步骤的冷却选自自然冷却降温、强制冷却降温的任一种或其组合。本发明的优选技术方案中,所述重结晶步骤的析晶选自静置析晶、搅拌析晶的任一种。本发明的优选技术方案中,所述重结晶步骤的分离选自过滤、离心的任一种或其组合。本发明的优选技术方案中,所述重结晶步骤中的干燥选自真空干燥或减压干燥。本发明的优选技术方案中,所述重结晶步骤中的干燥温度为40℃-70℃,优选为50℃-60℃。本发明的优选技术方案中,所述重结晶步骤中加入活性炭。本发明的优选技术方案中,所述打浆包括以下步骤:将待精制的法匹拉韦分散于有机溶剂c中,搅拌,分离固体后,干燥,即得。本发明的优选技术方案中,所述有机溶剂c选自二氯甲烷或二氯甲烷与乙酸乙酯、四氢呋喃、异丙醇、乙醇、甲醇、丙酮、石油醚的任一种或其组合。本发明的优选技术方案中,所述打浆步骤中的分散温度为室温至体系沸点温度。本发明的优选技术方案中,所述打浆步骤中的分离选自过滤、离心的任一种或其组合。本发明的优选技术方案中,所述打浆步骤中的干燥选自真空干燥或减压干燥的任一种。本发明的优选技术方案中,所述打浆步骤中的干燥温度为40℃-70℃,优选为50℃-60℃。本发明的另一目的在于提供一种法匹拉韦组合物,所述药物组合物含有法匹拉韦以及含量不高于0.1%的杂质a和含量不高于0.1%的杂质b,本发明的优选技术方案中,所述杂质a的含量不高于0.08%,优选不高于0.06%。本发明的优选技术方案中,所述杂质b的含量不高于0.05%,优选为未检出。本发明的优选技术方案中,所述法匹拉韦的纯度不低于99.9%。本发明的另一目的在于提供法匹拉韦和/或其衍生物用于制备病毒rna聚合酶抑制剂的组合物中的应用。本发明的另一目的在于提供法匹拉韦和/或其衍生物用于制备病毒致命性的突变诱发剂的组合物中的应用。本发明的另一目的在于提供法匹拉韦和/或其衍生物用于制备预防和/或治疗新型冠状病毒(2019-ncov)感染的药物中的应用。本发明的优选技术方案中,所述的治疗2019-ncov感染选自降低2019-ncov复制的能力、降低2019-ncov负荷、增加病毒清除率中的任一种或其组合。本发明的优选技术方案中,所述的预防2019-ncov感染选自2019-ncov易感性降低、持续感染的能力降低中的任一种或其组合。本发明的优选技术方案中,所述预防和/或治疗2019-ncov感染选自改善2019-ncov感染引起的发热、乏力、干咳、鼻塞、流涕、单纯性感染、轻症肺炎、重症肺炎、急性呼吸窘迫综合征、脓毒症、脓毒症休克、代谢性酸中毒、凝血功能障碍中的任一种症状或其组合症状。本发明的另一目的在于提供法匹拉韦和/或其衍生物用于制备预防和/或治疗流感病毒感染的药物中的应用。本发明的优选技术方案中,所述的治疗流感病毒选自新型流感病毒、再发型流感病毒的任一种或其组合。本发明的优选技术方案中,所述的治疗流感病毒感染选自降低流感病毒复制的能力、降低流感病毒负荷、增加病毒清除率的任一种或其组合。本发明的优选技术方案中,所述预防流感病毒感染选自流感病毒易感性降低、持续感染的能力降低的任一种或其组合。本发明的优选技术方案中,所述流感病毒选自a型流感病毒、b型流感病毒、c型流感病毒、高致病性禽流感病毒、对现有抗流感药物耐药病毒株的任一种或其组合。本发明的优选技术方案中,所述a型流感病毒选自禽流感、甲型h1n1流感感染的任一种或其组合。本发明的优选技术方案中,对现有抗流感药物耐药病毒选自对盐酸金刚烷胺、磷酸奥司他韦、扎那米韦的任一种或其组合表现为耐药。本发明的优选技术方案中,所述药物可用于免疫缺陷型患者的抗病毒治疗。本发明的优选技术方案中,所述流感病毒感染选自流感病毒导致的急性呼吸道感染。本发明的另一目的在于提供法匹拉韦和/或其衍生物用于制备预防和/或治疗埃博拉病毒感染的药物中的应用。本发明的优选技术方案中,所述的法匹拉韦衍生物选自法匹拉韦的药学上可接受的盐、酯、异构体、溶剂化物、中间体的任一种。除非另有说明,本发明涉及液体与液体之间的百分比时,所述的百分比为体积/体积百分比;本发明涉及液体与固体之间的百分比时,所述百分比为体积/重量百分比;本发明涉及固体与液体之间的百分比时,所述百分比为重量/体积百分比;其余为重量/重量百分比。与现有技术相比,本发明具有下述有益技术效果:1、本发明的精制方法解决了法匹拉韦难以脱色的问题,所得产品为白色或类白色粉末状产品,符合药品质量标准的相关要求。2、本发明的精制方法制得的法匹拉韦的纯度在99.9%以上,且将粗品中含有15个杂质经精制后仅存在两个杂质,即杂质a和杂质b。并且,杂质b在多个批次中均未检出,而杂质a含量仅约0.05%。3、本发明的精制方法适用于其他产品及中间体的纯化脱色,而且脱色程度大,纯化效果好,且操作简便,绿色环保,成本低,适于工业化生产。附图说明图1法匹拉韦粗品hplc检测图谱;图2对比例4产品hplc检测图谱;图3实施例1产品hplc检测图谱;图4实施例2产品hplc检测图谱;图5实施例3产品hplc检测图谱;图6实施例4产品hplc检测图谱;图7法匹拉韦对vero细胞的细胞毒性的mts测定结果;图8法匹拉韦在vero细胞上抑制病毒感染的定量rt-pcr检测结果。具体实施方式下面列举一部分具体实施例对本发明进行说明,有必要在此指出的是以下具体实施例只用于对本发明作进一步说明,不代表对本发明保护范围的限制。其他人根据本发明做出的一些非本质的修改和调整仍属于本发明的保护范围。对比例1向反应瓶中加入5g法匹拉韦粗品(浅棕色固体)和100ml异丙醇,升温至溶解澄清,加入活性炭,保温30min,热滤,滤液降温至0-10℃,搅拌析晶,过滤,干燥,所得产品为淡黄色粉末。收率为68.8%。对比例2向反应瓶中加入5g法匹拉韦粗品(浅棕色固体)和50ml乙酸乙酯,升温至溶解澄清,加入活性炭,保温30min,热滤,滤液降温至0-10℃,搅拌析晶,过滤,干燥,所得产品为淡黄色粉末。收率为62.6%。对比例3向反应瓶中加入5g法匹拉韦粗品(浅棕色固体)和330ml乙酸乙酯/二氯甲烷=1:5(w/w),升温至溶解澄清,加入活性炭,保温30min,热滤,滤液降温0-10℃搅拌析晶,过滤,干燥,所得产品为淡黄色粉末,收率为66.5%。对比例4将5.00g法匹拉韦粗品(浅棕色固体,纯度:99.427%,15个杂质,杂质a:0.048%,杂质b:0.105%)加入1l反应釜中,加入180ml二氯甲烷/乙酸乙酯=2:1(w/w)混合溶剂搅拌溶解,将10g200-300目硅胶均匀铺在布氏漏斗中,过滤,并用560ml二氯甲烷/乙酸乙酯=2:1混合溶剂淋洗滤饼,合并滤液,40-60℃减压浓缩,浓缩完毕,60℃真空干燥,真空度≥0.08mpa,干燥结束,得到淡黄色粉末3.38g,收率67.6%,纯度为99.825%(4个杂质,杂质a:0.042%,杂质b:0.013%)。对比例5精制溶剂的选择序号1:分别将5.0g粗品和100ml异丙醇加到反应瓶中,加热回流至溶解澄清。过滤,滤液降温至0~25℃,保温析晶1小时。过滤,滤饼经60℃真空干燥得到产品。序号2:分别将5.0g粗品和55ml乙酸乙酯加到反应瓶中,加热回流至溶解澄清。过滤,滤液降温至0~25℃,保温析晶1小时。过滤,滤饼经60℃真空干燥得到产品。序号3:分别将5.0g粗品和35ml丙酮加到反应瓶中,加热回流至溶解澄清。过滤,滤液降温至0~25℃,保温析晶1小时。过滤,滤饼经60℃真空干燥得到产品。序号4:5.0g粗品和125ml二氯甲烷加到反应瓶中,加热至回流,混合物搅拌打浆,过滤,滤饼用二氯甲烷淋洗,滤饼经60℃真空干燥得到产品。结果见表1。表1序号精制溶剂精制方式精制收率外观法匹拉韦------棕色1异丙醇重结晶67.5%浅黄色2乙酸乙酯重结晶66.4%浅黄色3丙酮重结晶50.4%浅黄色4二氯甲烷热打浆70.8%黄色由表1可见,采用乙酸乙酯、异丙醇、丙酮重结晶、二氯甲烷打浆可明显改善产品颜色。然而,重结晶、打浆仅能使棕色固体变为浅黄色或黄色,并不能得到白色或类白色产品,放大生产时难以达到质量标准要求。实施例1法匹拉韦的精制取5.00g法匹拉韦粗品(浅棕色固体,纯度:99.427%,15个杂质,杂质a:0.048%,杂质b:0.105%)加入1l单口瓶中,加入330ml二氯甲烷/乙酸乙酯=5:1(w/w)混合溶剂搅拌溶解,随后用25.00g200-300目硅胶均匀铺在布氏漏斗中,进行过滤,并且用相应的混合溶剂40ml进行淋洗,收集滤液,减压浓缩蒸干,随后加入45ml乙酸乙酯,加热搅拌至体系回流,加入0.50g活性炭,搅拌加热1h,热过滤,滤饼用5ml乙酸乙酯淋洗,滤液冷却至0-10℃,保温搅拌析晶2h,滤饼用5ml预冷的乙酸乙酯淋洗,滤饼60℃真空干燥,真空度≥0.08mpa,干燥,得类白色粉末3.41g,收率68.2%,纯度为99.945%(2个杂质,杂质a:0.045%,杂质b:未检出)。实施例2法匹拉韦的精制取5.00g法匹拉韦粗品(浅棕色固体,纯度:99.427%,15个杂质,杂质a:0.048%,杂质b:0.105%,)加入1l单口瓶中,加入600ml二氯甲烷/乙酸乙酯=10:1(w/w)混合溶剂搅拌溶解,随后用25.00g200-300目硅胶均匀铺在布氏漏斗中,进行过滤,并且用相应的混合溶剂40ml进行淋洗,收集滤液,减压浓缩蒸干,随后加入45ml乙酸乙酯,加热搅拌至体系回流,加入0.50g活性炭,搅拌加热1h,热过滤,滤饼用5ml乙酸乙酯淋洗,滤液冷却至0-10℃,保温搅拌析晶2h,滤饼用5ml预冷的乙酸乙酯淋洗,滤饼60℃真空干燥,真空度≥0.08mpa,干燥,得类白色粉末3.40g,收率68.0%,纯度为99.949%(1个杂质,杂质a:0.051%,杂质b:未检出)。实施例3法匹拉韦的精制取5.00g法匹拉韦粗品(浅棕色固体,纯度:99.427%,15个杂质,杂质a:0.048%,杂质b:0.105%,)加入3l单口瓶中,加入2800ml二氯甲烷溶剂搅拌溶解,随后用25.00g200-300目硅胶均匀铺在布氏漏斗中,进行过滤并且用相应的溶剂40ml进行淋洗,收集滤液,减压浓缩蒸干,随后加入45ml乙酸乙酯,加热搅拌至体系回流,加入0.50g活性炭,搅拌加热1h,热过滤,滤饼用5ml乙酸乙酯淋洗,滤液冷却至0-10℃,保温搅拌析晶2h,滤饼用5ml预冷的乙酸乙酯淋洗,滤饼60℃真空干燥,真空度≥0.08mpa,干燥,得白色粉末3.46g,收率69.2%,纯度为99.936%(2个杂质,杂质a:0.054%,杂质b:未检出)。实施例4法匹拉韦的精制取5.00g法匹拉韦粗品(浅棕色固体,纯度:99.427%,15个杂质,杂质a:0.048%,杂质b:0.105%,)加入1l单口瓶中,加入330ml二氯甲烷/乙酸乙酯=5:1(w/w)混合溶剂搅拌溶解,随后用15.00g200-300目硅胶均匀铺在布氏漏斗中,进行过滤,并且用相应的溶剂40ml进行淋洗,收集滤液,减压浓缩蒸干,随后加入45ml乙酸乙酯,加热搅拌至体系回流,加入0.50g活性炭,搅拌加热1h,热过滤,滤饼用5ml乙酸乙酯淋洗,滤液冷却至0—10℃,保温搅拌析晶2h滤饼用5ml预冷的乙酸乙酯淋洗滤饼60℃真空干燥,真空度≥0.08mpa,干燥,得类白色粉末3.66g,收率73.2%,纯度为99.951%(1个杂质,杂质a:0.049%,杂质b:未检出)。实施例5法匹拉韦对新型冠状病毒(2019-ncovs)抑制作用研究1.实验材料法匹拉韦(由实施例4得到);vero细胞购自美国标准生物品收藏中心(atcc),由军事医学研究院微生物流行病研究所病毒学研究室保存;新型冠状病毒北京分离株(2019-ncovbetacov/beijing/amms01/2020)由军事医学研究院微生物流行病研究所病毒学研究室分离并保存;培养vero细胞的培养基为含10%胎牛血清(fbs),1%双抗(pen-strep),10mmhepes的dmem液体培养基;根据病毒全长基因组序列,利用oligo7及seqbuilder软件在其保守区域设计荧光定量rt-pcr引物与探针,由上海生物工程有限公司合成,page级,纯度>98%,见表2。表2新冠病毒定量rt-pcr检测引物和探针引物名称位置引物序列5′-3′cov-实时-f322312-22333tcctggtgattcttcttcaggtcov-实时-r322436-22455tctgagagagggtcaagtgccov-实时-p322334-22354fam-agctgcagcaccagctgtcca-bhq12.实验方法(1)vero细胞培养在75cm2培养瓶中加入dmem完全培养基12ml,于5%co2、饱和湿度的细胞培养箱中37℃培养,3天传代一次。传代时移除旧培养基,用pbs洗细胞1次,加入2ml0.25%胰酶-edta在培养箱中消化3min。光学显微镜下观察到细胞变圆,弃去胰酶后,再加入9ml培养基终止消化,用移液管将细胞吹吸成单细胞,按1:3的比例取出细胞液加入新的培养瓶,再在新的培养瓶中补加新的培养基至12ml,混匀,于37℃、5%co2、饱和湿度的细胞培养箱中继续培养。(2)病毒扩大培养实验前一天,将vero细胞按1:3的比例接种至t75的培养瓶中,待细胞密度长至90%以上时供实验用。向t75培养瓶中加入moi=0.1的新型冠状病毒液,在37℃、5%co2的培养箱中孵育1小时。向培养瓶中加入15ml2%fbs的dmem培养基,在37℃、5%co2的培养箱中培养3天。将病毒液收集至50ml离心管,在4℃离心机6000rpm离心10min,收集上清,分装并保存于-80℃。(3)病毒ticd50测定提前一天将vero细胞以10000/孔浓度接种96孔板。用含2%fbs的dmem培养基10倍梯度稀释病毒液(10-2~10-9)。将96孔板中的原细胞培养基吸弃,向96孔板中加入200μl/孔的病毒液,设4个复孔。于37℃、5%co2细胞培养箱中孵育。每天观察细胞病变(cpe),持续观察4天。用reed-muench法计算病毒半数感染剂量(tcid50)。(4)药物细胞毒性试验提前一天将vero细胞以10000/孔浓度接种96孔板。待细胞长满单层,96孔板弃去培养液,加入含不同浓度药物(起始浓度为400μm,以3倍为稀释梯度将待测药物进行倍比稀释,共8个浓度)的2%fbsdmem维持液100μl/孔,每个浓度测3个复孔。并设立培养液对照和正常细胞对照组。继续培养,每天观察细胞状态。加药后72小时,加入20μlmts溶液,37℃,5%co2条件孵育1小时,测定od490值。采用公式计算药物对细胞毒性:最后,利用graphpadprism7软件对数据进行s拟合分析,计算出药物的cc50。(5)法匹拉韦对新型冠状病毒的抑制作用提前一天将vero细胞以10000/孔浓度接种96孔板。待细胞长满单层,96孔板弃去培养液,加入用2%fbs的dmem维持液稀释至100tcid50的新型冠状病毒液,100μl/孔,放置37℃、5%co2孵箱培养2小时。弃病毒液,加入用2%fbs的dmem维持液配制成400、200、100、50、25和12.5μm的法匹拉韦溶液,200μl/孔。每个药物3复孔。并设立病毒对照和正常细胞对照组。放置37℃、5%co2孵箱培养,每天观察细胞病变(cpe)。在感染后2天,每个浓度药物处理组孔取100μl细胞培养上清,提取核酸,使用定量rt-pcr检测病毒rna拷贝数。通过拟合剂量-反应曲线计算药物50%有效浓度(ec50)。a.病毒核酸提取采用qiagen公司的qiaampviralrnaminikit提取细胞上清中的病毒核酸。1)生物安全柜内吸取560μl包含载体rna的avl缓冲液(核酸提取裂解液)至1.5ml的离心管中;2)吸取100μl不同浓度药物处理的细胞培养上清(枪头弃于含75%乙醇消毒液收集器中),加入上述的avl缓冲液中,脉冲涡旋式混匀15秒;3)室温孵育10分钟,简单离心使离心管顶端液体落到底部;4)在样品中加入560μl无水乙醇,混匀15秒,简单离心;5)小心将630μl液体加入未浸湿的qiaamp小柱中,盖好盖,离心8000rpm/min、1min,弃去收集管,将柱子置于一新的2ml收集管上;打开qiaamp小柱的盖子,重复步骤5,直至样品全部离心;6)打开盖子,向柱中加入500μlaw1缓冲液,盖好盖,离心8000rpm/min、1min,弃去收集管,将柱子置于一新的2ml收集管上;7)打开盖子,加入500μlaw2缓冲液,盖好盖,离心14000rpm/min、3min;8)将柱子置于一新的2ml收集管上,空离1min;将柱子置于一新的1.5ml离心管上,加入60μlave洗脱缓冲液,室温静置1min,离心8000rpm/min、1min。离心液即为病毒rna,立即检测或-80℃以下保存。b.荧光定量rt-pcr检测病毒核酸载量采用takara公司的onesteprt-pcrkit(rr064a)进行定量rt-pcr检测。20μl反应体系见表3。表3荧光定量rt-pcr检测体系试剂体积(μl)2×onesteprt-pcrbufferiii10takaraextaphs(5u/μl)0.4primescriptrtenzymemixii0.4pcrforwardprimer(10μm)0.4pcrreverseprimer(10μm)0.4taqmanprobe0.4rna2rnasefreedh2o6将20μl反应液加入罗氏适配器96孔板(lightcycler480multiwellplate96,04729692001)中,3000rpm/min,低速离心30s后放入roche公司的lightcycler480ⅱ定量pcr仪中进行扩增,反应条件:42℃反转录5min,95℃预变性10s;95℃变性5s,60℃退火20s,扩增40个循环,每个循环反应结束时收集荧光信号。依据病毒rna拷贝数和ct值的换算公式:rnacopies/ml=ct*(-0.3)+13.17,计算病毒rna载量。利用graphpadprism7软件,通过拟合剂量-反应曲线计算50%有效浓度(ec50)。(6)选择指数si=cc50/ec503.实验结果(1)病毒tcid50滴定用倒置显微镜观察新型冠状病毒对vero细胞的致细胞病变(cpe)作用,采用reed-muench法计算病毒半数感染剂量(tcid50)。见表4。由表4结果可知,该病毒在vero细胞上的tcid50滴度为log105.4/ml。表4新型冠状病毒在vero细胞上的tcid50滴度病毒稀释度cpe-cpe+累计cpe↑累计存活↓病变率(%)10-20413010010-3049010010-4045010010-531132510-64007010-740011010-840015010-9400190(2)药物对细胞毒性试验(mts法)采用mts法测定了不同浓度的法匹拉韦对vero细胞的细胞毒性,并计算药物的半数毒性浓度(cc50)。结果见附图7。由图7结果可知,法匹拉韦对vero细胞的cc50>400μm。(3)法匹拉韦对新型冠状病毒的抑制作用(ec50)采用核酸定量法测定法匹拉韦在细胞水平抑制新型冠状病毒效果,并计算药物的ec50。结果见附图8。由图8结果可知,法匹拉韦在细胞水平对2019-ncov新型冠状病毒具有抑制作用,其ec50为67μm。(4)法匹拉韦在vero细胞上的抑制病毒结果根据cc50和ec50,计算出法匹拉韦的选择指数si>5.97。结果见表5。表5法匹拉韦对vero细胞的细胞毒性以及病毒抑制结果细胞系化合物细胞毒cc50(μm)ec50(μm)sivero法匹拉韦>40067>5.97由表5结果可知,法匹拉韦在vero细胞对2019-ncov新型冠状病毒具有的抑制作用,其ec50值为67μm,其选择指数si>5.97。4.实验结论本发明方法得到的法匹拉韦可有效抑制新型冠状病毒(ec50=67),且对正常细胞的没有毒性作用。以上对本发明具体实施方式的描述并不限制本发明,本领域技术人员可以根据本发明作出各种改变或变形,只要不脱离本发明的精神,均应属于本发明权利要求保护的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1