一种盐酸咪达唑仑糖浆杂质A和杂质B及其用途的制作方法
一种盐酸咪达唑仑糖浆杂质a和杂质b及其用途
技术领域
[0001]
本发明属于医药领域,具体一种盐酸咪达唑仑糖浆杂质a和杂质b及其用途。
背景技术:
[0002]
盐酸咪达唑仑糖浆是一种麻醉镇静类常用药物,化学名为1-甲基-8-氯-6-(2-氟苯基)-4h-咪唑并[1,5-a][1,4]-苯并二氮杂盐酸盐,它是一种短效的苯骈二氮杂类中枢神经系统(cns)镇静剂。苯骈二氮杂类药物能可逆的与cnsγ-氨基丁酸(gaba)-苯骈二氮复合受体结合而发挥其药理作用,gaba是中枢系统重要的抑制性传导递质。盐酸咪达唑仑糖浆主要通过口服,用于小儿镇静、抗焦虑、失忆及小儿全麻诱导。是icu短期镇静主要药物之一。其具体化学结构如下所示:
[0003][0004]
为了保证用药安全,活性药物成分中的每一个杂质都必须进行安全性评估,即建立保证安全性的杂质限度。根据人用药品注册技术标准国际协调会(ich)要求,原料药或其制剂组合物中的单个杂质量如果超过0.05%,应需要报告;单个杂质量如超过0.1%,就需要进行确证;单个杂质量如超过0.15%,则需要有安全性数据支持。
[0005]
盐酸咪达唑仑在盐酸咪达唑仑糖浆(ph为2.9~3.5)中部分以开环形式(中间体i)存在。在放置过程中,中间体i与辅料柠檬酸和柠檬酸钠相互作用,生成杂质a和杂质b。该杂质在多批次糖浆中均有检出,杂质a为本品特有的降解杂质,且我们测定了杂质a的校正因子,不在0.8~1.2之间。根据指导原则规定不在该范围的杂质在计算时建议采用外标法进行计算。因此我们将杂质a作为对照品,用于杂质定位及样品检测中杂质的计算。
[0006][0007]
关于盐酸咪达唑仑糖浆中杂质的产生,发现制备盐酸咪达唑仑糖浆的过程中,通过不同的起始原料,通过不同的生产工艺,以及在盐酸咪达唑仑糖浆的储存过程中,容易产生各种杂质。因此,需要对新的杂质进行结构确认、含量控制,以满足药品制备的要求,为毒理学研究提供基础,同时也为制剂工艺控制、药品储存提供参考。
技术实现要素:
[0008]
本发明提供了盐酸咪达唑仑糖浆或其组合物中的杂质a,杂质b及其制备方法,以及作为盐酸咪达唑仑糖浆或其组合物中质量控制的参照标准品的应用,
[0009][0010]
本发明提供一种盐酸咪达唑仑糖浆或其组合物中的杂质a和杂质b,其具体结构如下所示:
[0011][0012]
本发明进一步提供一种盐酸咪达唑仑糖浆或其组合物中的杂质a和杂质b的制备方法,包括以下步骤:
[0013][0014]
将中间体i溶于非质子性溶剂,碱性条件下加入中间体ii,反应制备得到杂质a与杂质b。
[0015]
在制备杂质a、杂质b的一个实施例方案中,中间体i与中间体ii反应过程中还包括控制温度条件进行保温反应,所述温度条件选自0~40℃,优选20~30℃。
[0016]
在制备杂质a、杂质b的一个实施例方案中,所述非质子溶剂选自二甲基亚砜、二甲基甲酰胺、二甲基乙酰胺、1,4-二氧六环、n-甲基吡咯烷酮、乙腈、六甲基磷酰胺或四氢呋喃中的任意一种或多种,优选1,4-二氧六环或四氢呋喃的任意一种。
[0017]
在制备杂质a、杂质b的一个实施例方案中,所述碱性试剂选自氢氧化钠、氢氧化钾、氢氧化锶、氢氧化锂、氢氧化钡、氢氧化钙、氢氧化铯、碳酸氢钠、碳酸氢钾、碳酸钠、碳酸钾、碳酸锶、碳酸铯、硫化钠、氢化钠、丁基锂、(六氢)吡啶、喹啉、甲醇钠、乙醇钠、丙醇钠、异丙醇钠、正丁醇钠、叔丁醇钠、甲醇钾、乙醇钾、丙醇钾、异丙醇钾、正丁醇钾、叔丁醇钾、二乙胺、三正丁胺、三丙基胺、二异丙基胺、二异丙基乙胺、叔丁醇铝、三乙胺或二甲氨基吡啶中的任意一种或多种,优选三乙胺。
[0018]
本发明的一个实施例方案中,还提供一种纯化杂质a和b方法:将上述制备得到的包含杂质a和b的反应液中的溶剂蒸除,向残留物中加入水和乙酸乙酯的混合溶剂,加氨水调节ph为9~10。分液,保留水相。向水相中加入稀盐酸调节ph为3~4,用萃取剂萃取3-5次,水相保留;合并有机相,用盐水洗一次,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,得黄色油状物。
[0019]
纯化杂质a和b方法中,所述纯化过程中,加氨水调节溶液ph为8~14,优选ph为9~10;加稀盐酸调节溶液ph为1~5,优选ph为3~4。
[0020]
纯化杂质a和b的一个实施例方案中,所述萃取剂选自二氯甲烷、甲苯、乙酸乙酯或丁酮中的任意一种或多种,优选丁酮。
[0021]
纯化杂质a和b方法的一个实施例方案中,所述油状物通过制备液相分离,收集相应保留时间的目标峰流动相,除去溶剂得到杂质a。
[0022]
纯化杂质a和b方法的一个实施例方案中,所保留水相通过制备液相分离,收集相应保留时间的目标峰流动相,除去溶剂得到杂质b。
[0023]
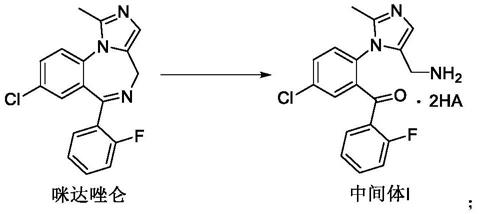
本发明进一步提供中间体i的制备方法,包括:
[0024][0025]
其中ha选自氢卤酸和硫酸,优选为盐酸;将咪达唑仑在低温条件下与浓盐酸反应,纯化后得中间体i;本发明的一个实施例方案中,所述低温条件优选-5~20℃,更优选0℃~10℃。
[0026]
制备中间体i的一个实施例方案中,所述咪达唑仑和浓盐酸的质体比(w/v)为1.0:1~4.0:1,优选1.5:1~2.5:1。
[0027]
本发明的一个实施例方案中,还提供一种纯化中间体i方法:向上述制备得到的包含中间体i的反应液中的加入异丙醇,减压蒸馏除去大部分溶剂;重复上步一次。再向反应瓶中补加适量异丙醇,室温打浆,过滤得白色固体。
[0028]
本发明进一步提供中间体ii的制备方法,包括:
[0029][0030]
将柠檬酸与乙酸酐在乙酸条件下,加热搅拌反应,经纯化得到中间体ii。
[0031]
本发明制备中间体ii的一个实施例方案中,所述柠檬酸与乙酸酐的摩尔比为1:1.5~1:3,优选1:1.8~1:2。
[0032]
本发明制备中间体ii的一个实施例方案中,所述加热条件优选20~60℃,更优选35℃~40℃。
[0033]
本发明制中间体式ii的一个实施例方案中,还提供一种纯化中间体ii方法:将上述制备得到的包含中间体ii的反应液中的溶剂蒸除,向残留物中加入氯仿,室温搅拌至析出大量固体,过滤,干燥得白色固体。
[0034]
本发明提供杂质a在盐酸咪达唑仑糖浆中的质量控制中的应用,所述杂质a作为盐酸咪达唑仑糖浆的杂质对照品,
[0035][0036]
本发明的一个实施例方案中,杂质a优选作为盐酸咪达唑仑糖浆的质量控制中的应用,所述杂质a作为盐酸咪达唑仑糖浆的杂质对照品。
[0037]
本发明进一步提供盐酸咪达唑仑糖浆的杂质a的测定方法,所述方法包括:
[0038]
1)、提供盐酸咪达唑仑糖浆的供试品、自身对照品和杂质a的标示品;
[0039]
2)、用色谱法测定供试品、对照品和标示品,确定盐酸咪达唑仑糖浆中杂质a的存在和/或量。
[0040]
本发明优选的实施例方案中,咪达唑仑或其药物组合物的杂质a的测定方法包括:
[0041]
1)、提供盐酸咪达唑仑糖浆的供试品;
[0042]
2)、提供杂质a的标示品;
[0043]
3)、通过hplc分析杂质a的标示品,确定杂质a的保留时间;
[0044]
4)、通过hplc分析盐酸咪达唑仑糖浆的供试品,确定供试品中是否含有保留时间与步骤3)保留时间基本一致的物质,从而确定盐酸咪达唑仑糖浆中杂质a的存在。
[0045]
本发明提供杂质b在盐酸咪达唑仑糖浆中的质量控制中的应用,所述杂质b作为盐酸咪达唑仑糖浆的杂质对照品,
[0046][0047]
本发明的一个实施例方案中,杂质b优选作为盐酸咪达唑仑糖浆中的质量控制中的应用,所述杂质b作为盐酸咪达唑仑糖浆的杂质对照品。
[0048]
本发明进一步提供盐酸咪达唑仑糖浆的杂质b的测定方法,所述方法包括:
[0049]
1)、提供盐酸咪达唑仑糖浆的供试品、自身对照品和杂质b的标示品;
[0050]
2)、用色谱法测定供试品、对照品和标示品,确定盐酸咪达唑仑糖浆的杂质b的存在和/或量。
[0051]
本发明优选的实施例方案中,盐酸咪达唑仑糖浆的杂质b的测定方法包括:
[0052]
1)、提供盐酸咪达唑仑糖浆的供试品;
[0053]
2)、提供杂质b的标示品;
[0054]
3)、通过hplc分析杂质b的标示品,确定杂质b的保留时间;
[0055]
4)、通过hplc分析盐酸咪达唑仑糖浆的供试品,确定供试品中是否含有保留时间与步骤3)保留时间基本一致的物质,从而确定盐酸咪达唑仑糖浆中杂质b的存在。
[0056]
本发明提供一种盐酸咪达唑仑糖浆,在所述盐酸咪达唑仑糖浆中,杂质a的含量选自0.001~0.1%(质量百分比,w/w)。
[0057]
本发明的一个实施例方案中,所述杂质a的含量选自0.001~0.05%(质量百分比,w/w)。
[0058]
本发明提供一种盐酸咪达唑仑糖浆,在所述盐酸咪达唑仑糖浆中,杂质b的含量选自0.001~0.1%(质量百分比,w/w)。
[0059]
本发明的一个实施例方案中,所述杂质b的含量选自0.001~0.05%(质量百分比,w/w)。
[0060]
本发明提供盐酸咪达唑仑糖浆在制备治疗神经精神类疾病的药物中的用途,所述盐酸咪达唑仑糖浆中包含含量选自0.001~0.1%(质量百分比,w/w)的杂质a。
[0061]
本发明提供盐酸咪达唑仑糖浆在制备治疗神经精神类疾病的药物中的用途,所述盐酸咪达唑仑糖浆中包含含量选自0.001~0.1%(质量百分比,w/w)的杂质b。
[0062]
本发明的一个实施例方案中,所述神经精神类疾病选自麻醉,疼痛,镇静,抗惊厥或癫痫中的任意一种。
[0063]
发明的有益效果
[0064]
本发明所提供的盐酸咪达唑仑糖浆中杂质a、杂质b、可用于盐酸咪达唑仑糖浆在制备或储存过程中产生的杂质的含量的检测和控制,控制盐酸咪达唑仑糖浆中的相关杂质含量符合ich药用标准,同时也为工艺条件控制和储存条件控制提供参考。
附图说明
[0065]
附图1:杂质a和杂质b在盐酸咪达唑仑糖浆中的hplc图谱。
具体实施方式
[0066]
以下将结合实施例更详细地解释本发明,本发明的实施例仅用于说明本发明的技术方案,本发明的实质和范围并不局限于此。
[0067]
实施例1中间体i的制备
[0068][0069]
将20.0g咪达唑仑和40ml纯化水加入500ml反应瓶中,搅拌条件下冰浴降温。控温0~10℃条件下,滴加10ml浓盐酸。滴毕,室温反应3小时。向反应瓶中加入200ml异丙醇,减压蒸除大部分溶剂;重复上步一次。再向反应瓶中补加150ml异丙醇,室温打浆2小时。过滤,滤饼用少量异丙醇和甲基叔丁基醚淋洗,得24.6g白色固体。
[0070]
实施例2中间体ii的制备
[0071][0072]
将100.0g柠檬酸,96.0g乙酸酐和50ml乙酸加入1000ml反应瓶中,加热搅拌,35~40℃反应20小时。减压蒸除大部分溶剂。
[0073]
向反应瓶中加入300ml氯仿,室温搅拌21小时,先析出部分油状物,随后油状物固化,析出大量白色固体。过滤,滤饼用氯仿泡洗两次,固体于45
±
5℃真空干燥24小时,得77.1g白色固体。
[0074]
实施例3杂质a和杂质b的制备
[0075][0076]
将4.2g中间体ii和50ml 1,4-二氧六环加入250ml反应瓶中,搅拌溶解。将1.0g中间体i和50ml 1,4-二氧六环加入另一100ml反应瓶中,搅拌均匀。将1.0g三乙胺加入中间体i的1,4-二氧六环悬浮液中,搅拌均匀,再将该混合物加入中间体ii的1,4-二氧六环溶液中,室温反应。tlc检测反应进程,待反应结束。
[0077]
将反应液移至250ml单口瓶中,减压蒸除有机溶剂。向残留物中加入30ml乙酸乙酯和30ml水,搅拌均匀,加氨水调节ph为9~10。分液,保留水相。向水相中加入30ml乙酸乙酯,搅拌萃取,分液,弃去有机相。合并水相,向水相中加入稀盐酸调节ph为3~4,用丁酮萃取5次(30ml/次),水相保留(杂质b粗品);合并丁酮相,用50ml盐水洗一次,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,得1.9g黄色油状物(杂质a粗品)。
[0078]
实施例4另一种杂质a和杂质b的制备方法
[0079][0080]
将8.4g中间体ii和100ml四氢呋喃加入500ml反应瓶中,搅拌溶解。将2.0g中间体i和100ml四氢呋喃加入另一250ml反应瓶中,搅拌均匀。将2.0g三乙胺加入中间体i的四氢呋喃悬浮液中,搅拌均匀,再将该混合物加入中间体ii的四氢呋喃溶液中,室温反应。tlc检测反应进程,待反应结束。
[0081]
将反应液移至500ml单口瓶中,减压蒸除有机溶剂。向残留物中加入50ml乙酸乙酯和50ml水,搅拌均匀,加氨水调节ph为9~10。分液,保留水相。向水相中加入50ml乙酸乙酯,搅拌萃取,分液,弃去有机相。合并水相,向水相中加入稀盐酸调节ph为3~4,用丁酮萃取5次(50ml/次),水相保留(杂质b粗品);合并丁酮相,用100ml盐水洗一次,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,得4.3g黄色油状物(杂质a粗品)。
[0082]
杂质a的纯化:实施例4中得到的黄色油状物(杂质a粗品)通过制备液相对杂质a样
品进行分离纯化。
[0083]
仪器:岛津lc-20ap制备液相
[0084]
色谱柱:welch xtimate c18(30
×
250mm,5μm)
[0085]
流动相:a:水(0.1%甲酸,0.5%醋酸铵)b:乙腈
[0086]
洗脱梯度:
[0087][0088]
流速:30ml/min;柱温:室温;检测波长:254nm
[0089]
按照上述色谱条件收集相应保留时间的目标峰流动相,在室温下真空蒸发去除乙腈,浓缩后水溶液进一步冷冻干燥,制备得白色粉末638.6mg。
[0090]1h-nmr(400mhz,dmso-d6)δ:1.946(s,3h),2.497(dd,j=12.6,2.0hz,2h),2.579(dd,j=15.1,5.4hz,2h),3.881(t,j=5.9hz,2h),6.584(s,1h),7.256(m,2h),7.496(td,j=7.5,1.9hz,1h),7.562(d,j=8.5hz,1h),7.606(m,1h),7.651(m,1h),7.788(d,j=2.5hz,1h),7.832(dd,j=8.5,2.5hz,1h)。
[0091]
13
c-nmr(100mhz,dmso-d6)δ12.935,32.935,42.831,42.987,72.64,116.419/116.251,124.710/124.691,124.780,126.175,129.237,130.059,130.667,131.543,132.604,132.141,134.105,135.484/135.413,139.252,144.177,160.900/158.880,171.505,171.697,173.272,190.537
[0092]
q-tof lc-ms(m/z):518.1125[m+h]
+
。
[0093]
ir(kbr)ν:3400,1666,1610,1485,1398,1298。
[0094]
杂质b的纯化:实施例4中萃取后所保留水相(杂质b粗品)通过制备液相对杂质b样品进行分离纯化。
[0095]
所使用仪器、色谱柱、流动相等条件同杂质a的纯化。进行梯度洗脱,收集相应保留时间的目标峰流动相,在室温下真空蒸发去除乙腈,浓缩后水溶液进一步冷冻干燥,制备得白色粉末332.3mg。
[0096]1h-nmr(400mhz,dmso-d6)δ:1.956(s,3h),2.408(dd,j=14.4,3.2hz,1h),2.498(dd,j=14.5,6.5hz,1h),2.560(dd,j=15.6,8.7hz,1h),2.645(dd,j=15.6,6.5hz,1h),3.858(m,2h),6.582(s,1h),7.250(m,2h),7.491(m,1h),7.542(dd,j=8.4,4.9hz,1h),7.620(m,1h),7.789(d,j=2.5hz,1h),7.850(m,1h),7.958(brs,1h)。
[0097]
13
c-nmr(100mhz,dmso-d6)δ12.934,32.575,42.995/42.960,43.560/43.370,72.450,116.463/116.292,124.682/124.652,124.736,126.154/126.106,129.305,129.886,130.704,131.985,131.515/131.488,132.603,134.177,135.523/135.452,139.202,144.572,160.938/158.917,175.253/175.199,171.240/171.186,168.883/168.807,190.435。
[0098]
q-tof lc-ms(m/z):518.1125[m+h]
+
。
[0099]
实施例5杂质a和杂质b在盐酸咪达唑仑糖浆中的hplc测定
[0100]
由于试验编码与本申请命名的差异,杂质a在图谱中的命名为杂质q,杂质b在图谱中的命名为杂质u;杂质c在图谱中的命名为杂质r;杂质d在图谱中的命名为杂质s。
[0101]
精密量取盐酸咪达唑仑糖浆5ml(约相当于咪达唑仑10mg),置20ml量瓶中,用流动相a稀释至刻度,摇匀,避光放置2小时,作为供试品溶液。取杂质a和杂质b对照品各适量,精密称定,置同一量瓶中,加流动相a溶解并定量稀释制成每1ml中约含各杂质均100μg的溶液,作为杂质贮备液;另精密称取盐酸咪达唑仑对照品适量,加杂质贮备液1、杂质贮备液2适量,用流动相a稀释制成每1ml约含盐酸咪达唑仑0.5mg、各杂质1μg的溶液,作为系统适用性溶液。按照高效液相色谱法(中国药典2015年版四部通则0512)测定。用十八烷基硅烷键合硅胶为填充剂(ymc-pack ods-a,4.6mm
×
250mm,5μm或效能相当的色谱柱);以醋酸铵溶液(用冰醋酸调节ph值至5.2)-甲醇(50:50)为流动相a;以醋酸铵溶液用冰醋酸调节ph值至5.2)-甲醇(20:80)为流动相b,按下表进行线性梯度洗脱;流速为每分钟1.0ml;柱温为30℃;检测波长为254nm。
[0102][0103]
取系统适用性溶液20μl注入液相色谱仪,记录色谱图,杂质a、杂质b、咪达唑仑依次出峰;各峰之间的分离度应符合要求,理论板数按咪达唑仑峰计算应不低于5000。精密量取供试品溶液各20μl,注入液相色谱仪,记录色谱图,见附图1。
[0104]
含量计算:
[0105]
根据面积归一化法计算各杂质在盐酸咪达唑仑糖浆供试品中质量百分比,具体实验结果见下表1:
[0106]
表1各杂质在盐酸咪达唑仑糖浆中的含量
[0107]
峰号化合物名保留时间面积高度面积(%)理论塔板数分离度1/10.434101029100.04219227
--
2杂质a11.2822957518410.123157762.573/14.1363527520230.147229657.794杂质c15.9193158828030.132444355.285杂质d16.3482904324350.121445101.406/22.37098348450.0418548019.557/22.93391597400.038759271.768/25.369118959360.050938837.349咪达唑仑27.58823833175177116099.306987616.51总计//23999646/100.000//
[0108]
通过上述实施例5的结果可知,本发明提供的杂质a和杂质b作为盐酸咪达唑仑糖浆的杂质对照品,可用于其质量控制,对盐酸咪达唑仑糖浆的质量监控具有非常重要的作用。
[0109]
尽管以上已经对本发明作了详细描述,但是本领域技术人员理解,在不偏离本发明的精神和范围的前体下,可以对本发明进行各种修改和改变。本发明的权利范围并不限于上文所作出的详细描述,而应当属于权利要求书。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1