靶向CYP4V2基因突变位点的核酸分子及其用途的制作方法
本申请涉及生物医药领域,具体的涉及一种用于治疗cyp4v2基因突变疾病的grna和供体核酸分子。
背景技术:
结晶样视网膜变性((bietticrystallinedystrophy,bcd),又名为结晶样视网膜色素变性、结晶样角膜视网膜变性(bietticrystallinecorneoretinaldystrophy)、结晶样视网膜病变(bietticrystallineretinopathy)、bietti视网膜变性(bietti'sretinaldystrophy)(omim210370))是一种致盲性的常染色体隐性遗传性视网膜变性疾病。cyp4v2基因是目前发现的bcd致病基因之一(li等人,amjhumgenet.74:817-826,200)。cyp4v2(细胞色素p450,家族4,亚家族v,多肽2,(0mim608614),同义词:cyp4ah1)属于细胞色素p450超家族,是亚铁血红素—硫醇盐蛋白细胞色素p450亚家族4(cyp4)的成员。
目前,有多种治疗该病的方法,例如基因替代治疗方案,即使用病毒转染系统或者其他转染系统(如aav、lentivirus、逆转录病毒),将cyp4v2野生型基因转染到基因突变的细胞中,使基因突变细胞可以表达野生型cyp4v2。这种做法仅可以部分恢复突变细胞功能,并且疗效有限。原因为突变型的基因产物仍然存在于细胞中,这些突变蛋白会与正常基因产物存在竞争性抑制作用。因此,更加安全有效的治疗方法亟待被发现。
公开于该背景技术部分的信息仅仅旨在增加对本申请的总体背景的理解,而不应当被视为承认或以任何形式暗示该信息构成已为本领域一般技术人员所公知的现有技术。
技术实现要素:
本申请提供了一种特异性靶向细胞色素p450家族4亚家族v多肽2(cyp4v2)的grna,其特异性结合所述cyp4v2基因的6号外显子和7号外显子之间的内含子区,所述grna对所述cyp4v2的6号外显子和7号外显子之间的内含子区具有比较好的切割效果,使得原先的cyp4v2基因产物不存在于细胞中。本申请还提供了一种供体核酸分子,其包含了cyp4v2基因的6号内含子和11号外显子之间的核苷酸序列,所述供体核酸分子可以在内源性cyp4v2被所述grna剪切后,在基因突变细胞中修复cyp4v27-11号外显子,产生了具有正常功能的cyp4v2蛋白,具有很好修复效果。本申请提供了一种包含所述grna和/或所述供体核酸分子的载体,能够使cyp4v2突变的细胞表达正确的细胞色素p450家族4亚家族v多肽2,具有良好的基因编辑修复效率。
一方面,本申请提供了一种特异性靶向细胞色素p450家族4亚家族v多肽2(cyp4v2)基因的grna,其特异性结合所述cyp4v2基因的6号外显子和7号外显子之间的内含子区。
在某些实施方式中,所述的grna特异性结合seqidno:41所示的核苷酸序列。
在某些实施方式中,所述的grna包含seqidno:1-4中任一项所示的核苷酸序列。
在某些实施方式中,所述的grna包含5’-(x)n-seqidno:1-4-骨架序列-3’,其中x为选自a、u、c和g中任一个的碱基,且n为0-15中的任一整数。
在某些实施方式中,所述grna为单链向导rna(sgrna)。
另一方面,本申请提供了一种或多种分离的核酸分子,其编码所述的特异性靶向cyp4v2基因的grna。
另一方面,本申请提供了供体核酸分子,其包含cyp4v2基因的6号内含子和11号外显子之间的核苷酸序列。
在某些实施方式中,所述的供体核酸分子包含seqidno:39所示的核苷酸序列。
另一方面,本申请提供了载体,其包含所述的分离的核酸分子和/或所述的供体核酸分子。
在某些实施方式中,所述的载体,所述分离的核酸分子和所述供体核酸分子位于同一载体中。
在某些实施方式中,所述的载体为病毒载体。
另一方面,本申请提供了细胞,其包含所述的分离的核酸分子、所述的供体核酸分子和/或所述的载体。
在某些实施方式中,所述的细胞包括hek293细胞、肾上皮细胞和/或诱导性多能干细胞。
在某些实施方式中,所述的细胞经修饰后具备分化能力。
在某些实施方式中,所述的细胞可分化为3d-视网膜类器官。
另一方面,本申请提供了药物组合物,其包含所述的grna,所述的一种或多种分离的核酸分子,所述的供体核酸分子,所述的载体,以及药学上可接受的载剂。
另一方面,本申请提供了试剂盒,其包含所述的grna,所述的一种或多种分离的核酸分子,所述的供体核酸分子,所述的载体。
另一方面,本申请提供了所述的grna,所述的一种或多种分离的核酸分子,所述的供体核酸分子,和/或所述的载体在制备治疗疾病的药物中的应用,其中所述疾病包括cyp4v2基因中的突变所导致的疾病。
在某些实施方式中,所述突变位于cyp4v2基因6号外显子和7号外显子之间的内含子之后。
在某些实施方式中,所述疾病包括结晶样视网膜变性。
另一方面,本申请提供了一种治疗结晶样视网膜变性的方法,所述方法包括以下的步骤:向有需要的受试者导入所述的grna,所述的一种或多种分离的核酸分子,所述的供体核酸分子,和/或所述的载体。
在某些实施方式中,根据所述导入获得了正常功能的cyp4v2蛋白。
在某些实施方式中,所述导入包括注射。
在某些实施方式中,所述导入包括视网膜下腔注射。
另一方面,本申请提供了一种调节细胞中cyp4v2基因表达的方法,其包括向细胞导入所述的grna,所述的一种或多种分离的核酸分子,和/或所述的载体。
本领域技术人员能够从下文的详细描述中容易地洞察到本申请的其它方面和优势。下文的详细描述中仅显示和描述了本申请的示例性实施方式。如本领域技术人员将认识到的,本申请的内容使得本领域技术人员能够对所公开的具体实施方式进行改动而不脱离本申请所涉及发明的精神和范围。相应地,本申请的附图和说明书中的描述仅仅是示例性的,而非为限制性的。
附图说明
本申请所涉及的发明的具体特征如所附权利要求书所显示。通过参考下文中详细描述的示例性实施方式和附图能够更好地理解本申请所涉及发明的特点和优势。对附图简要说明书如下:
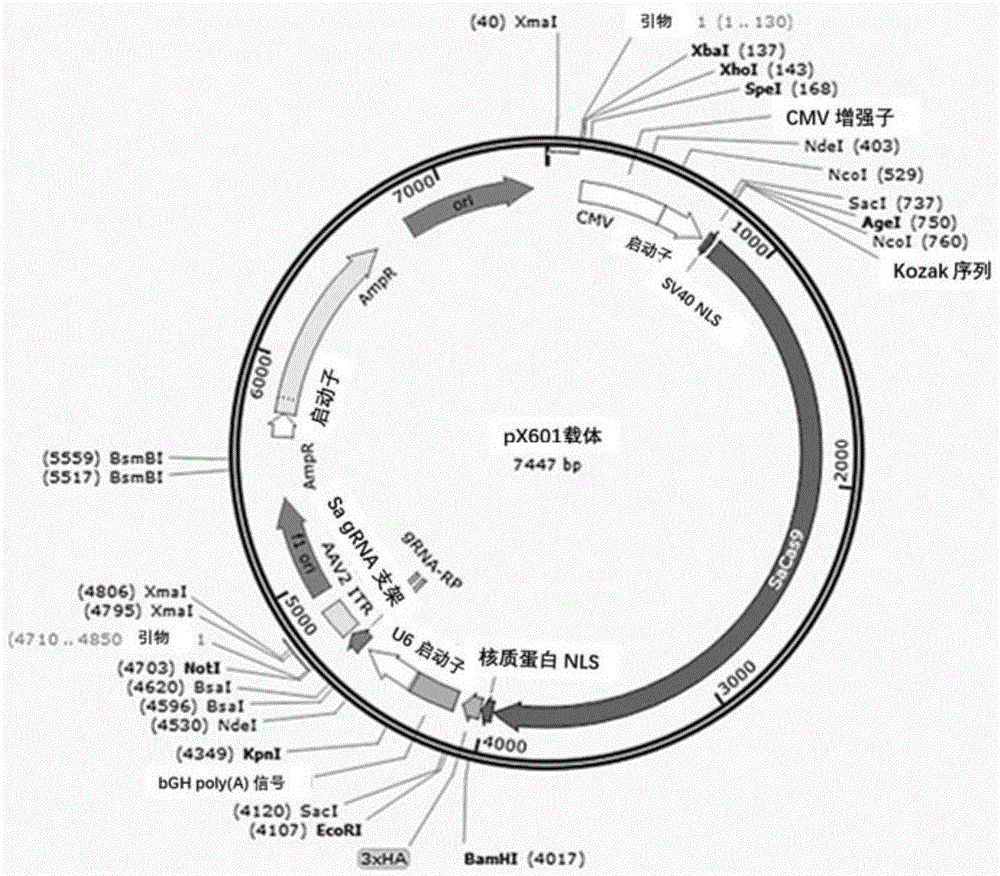
图1显示的是本申请所述px601质粒图谱。
图2显示的是本申请所述cyp4v2-hiti-sgrna1-7切割后的片段电泳图,说明sgrna1-4具有良好的切割效果。
图3a-3d显示的是本申请所述cyp4v2sgrna1-4切割后含切割位点的片段测序结果,说明sgrna1-4具有良好的切割效应。
图4显示的是本申请所述minigene片段的图谱。
图5显示的是本申请所述pmd-19t-mcs质粒图谱。
图6a-6c显示的是本申请所述sgrna1-4对剪切的影响,说明sgrna1-4不影响mrna的剪切。
图7显示的是本申请所述donor筛选实验转染细胞的结果,明了含有donor的载体转染细胞成功了。
具体实施方式
以下由特定的具体实施例说明本申请发明的实施方式,熟悉此技术的人士可由本说明书所公开的内容容易地了解本申请发明的其他优点及效果。
术语定义
在本申请中,术语“3d-视网膜类器官”通常是指一种具有三维结构、能够自我更新、自我组织并显示视网膜基本功能(例如,感受光)的人工培育的视网膜。3d-视网膜类器官可以由原代组织或干细胞(例如,多功能干细胞)分化而成,具有视网膜中所有接收光线并向大脑发出信号所必需的细胞。
在本申请中,术语“分离的核酸分子”是其与存在于所述核酸的天然来源中的其他核酸分子相分离。这种分离的核酸分子从其通常的或天然的环境中移出的或分离的,或者生产所述分子的方式使其不存在于其通常的或天然的环境中,其与通常的或天然的环境中的多肽、肽、脂质、糖类、其他的多核苷酸或其它材料分离。本申请中的分离的核酸分子可编码rna,例如,可编码特异性靶向cyp4v2基因的grna。
在本申请中,术语“供体核酸分子”通常是指向受体(例如,接收核酸分子)提供异源核酸序列的核酸分子。
在本申请中,术语“结晶样视网膜变性”通常是指一类常染色体隐性遗传眼病。其主要症状包括角膜中的晶体(透明覆盖物),沉积在视网膜的光敏组织中的细小、黄色或白色晶体状的沉积物,以及视网膜、脉络膜毛细血管和脉络膜的进行性萎缩。结晶样视网膜变性可以包括由cyp4v2基因突变引起的疾病。
在本申请中,术语“试剂盒”通常是指一起被包装在容器、接受器或其它容器中的两种或更多种组分,其中一种对应于本申请所述grna、所述一种或多种分离的核酸分子、所述的供体核酸分子和/或所述的载体,药物组合物或细胞。因此,试剂盒可以被描述为足以实现特定目标的一组产品和/或器具,其可以作为单个单元销售。
在本申请中,术语“细胞”指其如本领域一般公认的意义。该术语以其通常的生物学含义使用,并且不指完整多细胞生物,例如具体地不指人类。细胞可以存在于生物内,例如鸟类、植物和哺乳动物,例如人、牛、羊、猿、猴、猪、狗和猫。细胞可以是原核的(例如,细菌细胞)或真核的(例如,哺乳动物或植物细胞)。细胞可以具有体细胞或种系起源、全能或多能、分裂或非分裂。细胞还可以衍生自或可以包含配子或胚胎、干细胞、或完全分化的细胞。
在本申请中,术语“药物组合物”通常是指合适于向有需要的受试者施用的组合物。例如,本申请所述的药物组合物,其可以包含本申请所述的grna、本申请所述一种或多种分离的核酸分子、本申请所述的供体核酸分子和/或本申请所述的载体,以及药学上可接受的的载剂。术语“受试者”或“个体”或“动物”或“患者”在本申请中可互换用于指施用本申请的药物组合物需要的受试者,例如哺乳动物受试者。动物受试者包括人类、非人类灵长类、狗、猫、豚鼠、兔子、大鼠、小鼠、马、黄牛、乳牛等等,例如小鼠。在某些实施方式中,药物组合物可以包含用于视网膜下、非肠道、透皮、内腔内、动脉内、膜内和/或鼻内给药或直接注射入组织的组合物。例如,所述药物组合物通过视网膜下腔注射向受试者给药。
在本申请中,术语“诱导性多能干细胞”通常是指成熟的、不具有分化能力的体细胞在某些条件下恢复到全能性的状态的细胞。所述全能性是指具有的分化成机体所有类型细胞和形成完全胚胎或进一步发育成新个体的能力。例如,在本申请中,所述的诱导多能干细胞是肾上皮细胞经重编程培养基培养后获得的,其具有分化为视网膜细胞的能力。
在本申请中,术语“载体”通常是指能够转运与它连接的另一核酸的核酸分子。一类载体是“质粒”,其指其他dna区段可以连接入其中的环状双链dna环。例如,本申请所述构建的pmd-19t-mcs质粒。另一类载体是“病毒载体”,其中其他dna区段可以连接入病毒基因组。例如,本申请所述构建的aav病毒载体。
在本申请中,术语“cyp4v2”通常是指一种蛋白质,其为细胞色素p450家族4亚家族v成员2。术语“细胞色素p450”,也称作cytochromep450或cyp450,通常是指一类亚铁血红素蛋白家族,属于单氧酶的一类,参与内源性物质或包括药物、环境化合物在内的外源性物质的代谢。根据氨基酸序列的同源程度,其成员又依次分为家族、亚家族和酶个体三级。细胞色素p450酶系统可以缩写为cyp,其中家族以阿拉伯数字表示,亚家族以大写英文字母表示,酶个体以阿拉伯数字表示,例如本申请中的cyp4v2。人cyp4v2基因(hgnc:23198;ncbiid:285440)全长19.28kb,位于4q35,具有11个外显子,在脂肪酸代谢中发挥重要作用(kumars.,bioinformation,2011,7:360-365)。本申请所述的cyp4v2还可包括其功能性变体、片段、同源物等。cyp4v2几乎在所有组织中表达,但是在视网膜和视网膜色素上皮中以高水平表达,而角膜,组织中以稍低的水平表达。cyp4v2基因的突变可能与结晶样视网膜变性和/后视网膜色素变性有关。
在本申请中,术语“grna”通常是指向导rna(guiderna),一种rna分子。在自然界中,crrna和tracrrna通常作为两个独立的rna分子存在,组成grna。术语“crrna”也称为crisprrna,通常是指与所靶向的目标dna互补的一段核苷酸序列,术语“tracrrna”通常是指可与cas核酸酶结合的支架型rna。crrna和tracrna也可以融合成为单链,此时grna也可称为单链向导rna(singleguiderna,sgrna),sgrna已成为本领域技术人员在crispr技术中使用的grna的最常见的形式,因此术语“sgrna”和“grna”在本文中可具有相同的含义。sgrna可以人工合成,也可以在体外或体内由dna模板制备。sgrna可以结合cas核酸酶,也可以靶向目标dna,其可引导cas核酸酶切割与grna互补的dna位点。
在本申请中,术语“hek293细胞”通常是指“人胚胎肾细胞293”,是衍生自人胚胎肾细胞的细胞系,具有易培养,转染效率高的特点,在本领域,是很常用的研究外源基因的细胞株。
在本申请中,术语“肾上皮细胞”通常是指在人尿液中收集到的肾脏的上皮细胞。在本申请中,其是诱导多能干细胞的来源。在本领域,利用尿液中的肾上皮细胞诱导多能干细胞符合成本效益,通用,适合于应用各个年龄、性别和种族。这项技术使得获取大量病人样本相较于其它现有方式容易和经济得多。
在本申请中,术语“视网膜下腔注射”通常是指将需要导入的物质导入到感光细胞和视网膜色素上皮(rpe)层之间。在视网膜下腔注射期间,将注射的材料(例如,本申请所述的grna,本申请所述的一种或多种分离的核酸分子,本申请所述的载体,以及药学上可接受的的载剂)并在其间创建空间。
除了本文提到的特定蛋白质和核酸分子之外,本申请还可包括其功能性变体、衍生物、类似物、同源物及其片段。
术语“功能性变体”指与天然存在序列具有基本上同一的氨基酸序列或由基本上同一的核苷酸序列编码并能够具有天然存在序列的一种或多种活性的多肽。在本申请的上下文中,任何给定序列的变体是指其中残基的特定序列(无论是氨基酸或核苷酸残基)已经经过修饰而使得所述多肽或多核苷酸基本上保留至少一种内源功能的序列。可以通过天然存在的蛋白质和/或多核苷酸中存在的至少一个氨基酸残基和/或核苷酸残基的添加、缺失、取代、修饰、替换和/或变异来获得变体序列,只要保持原来的功能活性即可。
在本申请中,术语“衍生物”通常是指本申请的多肽或多核苷酸而言包括自/对序列的一个(或多个)氨基酸残基的任何取代、变异、修饰、替换、缺失和/或添加,只要所得的多肽或多核苷酸基本上保留其至少一种内源功能。
在本申请中,术语“类似物”通常对多肽或多核苷酸而言,包括多肽或多核苷酸的任何模拟物,即拥有该模拟物模拟的多肽或多核苷酸的至少一种内源功能的化学化合物。
通常,可以进行氨基酸取代,例如至少1个(例如,1、2、3、4、5、6、7、8、9、10或20个以上)氨基酸取代,只要经修饰的序列基本上保持需要的活性或能力。氨基酸取代可包括使用非天然存在的类似物。
用于本申请的蛋白质或多肽也可以具有氨基酸残基的缺失、插入或取代,所述氨基酸残基产生沉默的变化并导致功能上等同的蛋白质。可以根据残基的极性、电荷、溶解性、疏水性、亲水性和/或两性性质的相似性进行有意的氨基酸取代,只要保留内源性功能即可。例如,带负电荷的氨基酸包括天冬氨酸和谷氨酸;带正电荷的氨基酸包括赖氨酸和精氨酸;并且含具有相似亲水性值的不带电极性头基的氨基酸包括天冬酰胺、谷氨酰胺、丝氨酸、苏氨酸和酪氨酸。
在本申请中,术语“同源物”通常是指与比较的氨基酸序列和比较的核苷酸序列具有一定同源性的氨基酸序列或核苷酸序列。术语“同源性”可以等同于序列“同一性”。同源序列可以包括与主题序列是至少80%、85%、90%、99.1%、99.2%、99.3%、99.4%、99.5%、99.6%、99.7%、99.8%或99.9%相同的氨基酸序列。通常,同源物将包含与主题氨基酸序列相同的活性位点等。同源性可以根据相似性(即具有相似化学性质/功能的氨基酸残基)来考虑,也可以在序列同一性方面表达同源性。在本申请中,提及的氨基酸序列或核苷酸序列的seqidno中的任一项具有百分比同一性的序列是指在所提及的seqidno的整个长度上具有所述百分比同一性的序列。
为了确定序列同一性,可进行序列比对,其可通过本领域技术人员了解的各种方式进行,例如,使用blast、blast-2、align、needle或megalign(dnastar)软件等。本领域技术人员能够确定用于比对的适当参数,包括在所比较的全长序列中实现最优比对所需要的任何算法。
在本申请中,术语“和/或”应理解为意指可选项中的任一项或可选项的两项。
在本申请中,术语“包含”或“包括”通常是指包括明确指定的特征,但不排除其他要素。
在本申请中,术语“约”通常是指在指定数值以上或以下0.5%-10%的范围内变动,例如在指定数值以上或以下0.5%、1%、1.5%、2%、2.5%、3%、3.5%、4%、4.5%、5%、5.5%、6%、6.5%、7%、7.5%、8%、8.5%、9%、9.5%、或10%的范围内变动。
发明详述
grna
一方面,本申请提供一种特异性靶向细胞色素p450家族4亚家族v多肽2的基因(cyp4v2基因)的grna,其特异性结合所述cyp4v2基因的第6个外显子与第7个外显子之间的内含子区。
在某些情形中,所述grna可特异性结合seqidno:41所示的核苷酸序列。在某些情形中,所述grna可特异性结合与seqidno:41所示的核苷酸序列具有至少70%(例如,至少75%,至少80%,至少85%,至少90%,至少91%,至少92%,至少93%,至少94%,至少95%,至少96%,至少97%,至少98%,至少99%,或至少100%)序列同一性的核苷酸序列。在本申请中,所述“同一性”是指不同的核苷酸序列,其碱基顺序是一致的。
在某些情形中,所述grna可特异性结合与seqidno:41所示的核苷酸序列互补的核苷酸序列。在某些情形中,所述grna可特异性结合与seqidno:41所示的核苷酸序列具有至少70%(例如,至少75%,至少80%,至少85%,至少90%,至少91%,至少92%,至少93%,至少94%,至少95%,至少96%,至少97%,至少98%,至少99%,或至少100%)序列同一性的核苷酸序列互补的核苷酸序列。
本申请所述的grna可以与目标靶核酸(例如,所述cyp4v2基因的第6个外显子与第7个外显子之间的内含子区)序列结合。grna可以通过杂交(即碱基配对)以序列特异性的方式与目标靶核酸相互作用。sgrna的核苷酸序列可以根据目标靶核酸的序列而变化。
在本申请中,所述grna可包括seqidno:1-4中任一项所示的核苷酸序列。本申请中,所述grna可包括与seqidno:1-4中任一项所示的核苷酸序列具有至少70%(例如,至少75%,至少80%,至少85%,至少90%,至少91%,至少92%,至少93%,至少94%,至少95%,至少96%,至少97%,至少98%,至少99%,或至少100%)序列同一性的核苷酸序列。
在本申请中,所述grna自5’端至3’端可包含(x)n、seqidno:1-4中任一项所示的核苷酸序列和骨架序列,其中x为选自a、u、c和g中任一个的碱基,且n为0-15中的任一整数。在本申请中,所述grna可包含5’-(x)n-seqidno:1-4任一项所示的核苷酸序列-骨架序列-3’,其中x为选自a、u、c和g中任一个的碱基,且n为0-15中的任一整数。
例如,本申请所述骨架序列通常是指grna中,除识别或杂交靶序列的部分的其他部分,可包括sgrna中grna配对序列与转录终止子之间的序列。骨架序列一般不会因为靶序列的变化而变化,也不影响grna对靶序列的识别。因此,骨架序列可以是现有技术中任何可行的序列。骨架序列的结构可参见如文献nowaketal.nucleicacidsresearch2016.44:9555-9564中的figure1(图1)中a和b,figure3(图3)中a、b、c,以及figure4(图4)中a、b、c、d、e中所记载的除spacer序列之外的部分。
在某些情形中,所述grna可以为单链或双链向导rna。例如,所述grna可以为单链向导rna(例如,sgrna)。
本申请提供了一种或多种分离的核酸分子,所述分离的核酸分子可编码上文所述的特异性靶向cyp4v2基因的grna。例如,所述分离的核酸分子可包含seqidno:1-7中任一项所示的核苷酸序列。
在本申请中,所述grna序列可以设计成与cas核酸酶可识别的pam序列临近处的靶核酸杂交。所述grna可以与靶序列完全互补或不完全互补。grna与其相应的靶序列之间的互补程度至少为50%(例如,至少为约55%、约60%、约65%、约70%、约75%、约80%、约85%、约90%、约95%、约98%、或更多)。所述“cas核酸酶”通常是指能够使用crispr序列(例如,grna)作为向导,从而识别和切割特定的dna链。例如,cas9核酸酶,csn1或csx12。cas9核酸酶通常包括ruvc核酸酶结构域和hnh核酸酶结构域,分别切割双链dna分子的两条不同的链。已经在不同的细菌物种如嗜热链球菌(s.thermophiles)、无害利斯特氏菌(listeriainnocua)(gasiunas,barrangouetal.2012;jinek,chylinskietal.2012)和化脓性链球菌(s.pyogenes)(deltcheva,chylinskietal.2011)中描述了cas9核酸酶。例如,化脓链球菌(streptococcuspyogenes)cas9蛋白,其氨基酸序列参见swissprot数据库登录号q99zw2;脑膜炎奈瑟氏菌(neisseriameningitides)cas9蛋白,其氨基酸序列见uniprot数据库编号a1iq68;嗜热链球菌(streptococcusthermophilus)cas9蛋白,其氨基酸序列见uniprot数据库编号q03lf7;金黃色葡萄球菌(staphylococcusaureus)cas9蛋白(例如,本申请所述载体中的sacas),其氨基酸序列见uniprot数据库编号j7rua5。cas核酸酶通常可以在dna中识别特定的pam序列。例如,所述pam可包含seqidno:8-14中任一项所述的核苷酸序列。
本申请所述的grna和/或分离的核酸分子可以使用载体递送。在本申请中,所述载体(例如px601)可以包含或者不包含编码cas核酸酶的核酸。在本申请中,cas核酸酶可以作为一种或多种多肽单独地递送。或者,编码所述cas核酸酶的核酸分子,与一种或多种引导rna,或一种或多种crrna以及tracrrna,单独地递送,或者一起预复合地递送。例如,所述本申请的核酸分子(例如,编码所述特异性靶向rpgr基因的sgrna的分离的核酸分子)和编码cas9核酸酶的核酸分子可以位于同一载体(例如,质粒)中。所述载体可包括本领域已知的病毒或非病毒载体。
非病毒递送载体可以包括但不限于纳米颗粒、脂质体、核糖核蛋白、带正电荷的肽、小分子rna缀合物、适体-rna嵌合体和rna融合蛋白复合物。
在本申请中,所述分离的核酸分子和/或所述编码dna核酸内切酶的核酸分子可以通过质粒递送。
在某些情形中,所述载体可以是病毒载体,例如,aav、慢病毒、逆转录病毒、腺病毒、疱疹病毒和肝炎病毒。用于产生包含核酸分子(例如,本申请所述分离的核酸分子)作为载体基因组一部分的病毒载体的方法是本领域公知的,并且本领域技术人员可无需进行过多的实验进行。在另一些情形中,所述载体可以是包装了本申请所述核酸分子的重组aav病毒粒子。产生所述重组aav的方法,可包括将本申请所述核酸分子引入包装细胞系,将表达aav的rep和cap基因的包装质粒引入细胞系中,以及从包装细胞系上清中收集重组的aav。包装的细胞系可以有各种类型的细胞。例如,可以使用的包装细胞系包括但不限于hek293细胞,hela细胞和vero细胞。
供体核酸分子和载体
另一方面,本申请还提供了供体核酸分子。在本申请中,术语“供体核酸分子”通常是指向受体(例如,接收核酸分子)提供异源核酸序列的核酸分子。在某些情形下,所述供体核酸分子被导入到受体细胞中,可以修复被所述分离的核酸分子切割后的dna片段(例如,断裂后的双链dna)。在另外一些情形下,dna断裂后可以通过供体核酸分子来修复。修复的方式包括但不限于依赖于dna同源性的同源重组(homologousrecombination,hr)修复和非同源末端连接(non-homologousendjoining,nhej)的修复方式。hr利用同源序列或供体序列(例如,所述靶向载体)作为模板,在断点处插入特定的dna序列。同源序列可以在内源基因组中,例如姐妹染色单体(sisterchromatid)。或者,所述供体可以是外源核酸,例如质粒、单链寡核苷酸、双链寡核苷酸、双链寡核苷酸或病毒。例如,所述供体可以包含本申请所述供体核酸分子。这些外源核酸可以包含与cas核酸酶切割的基因座具有高度同源性的区域,此外还可包含额外的序列或序列变化(包括可掺入切割的靶基因座的缺失)。nhej直接连接双链断裂所导致的dna末端,有时会丢失或添加核苷酸序列,这可能会破坏或增强基因表达。其中,基于nhej的微同源介导的末端连接还(microhomology-mediatedendjoining,mmej)、同源非依赖性靶向整合(homology-independenttargetedintegration,hiti)和hr介导的末端连接(homology-mediatedendjoining,hmej)。例如,在本申请中,通过使用hiti修复方式将供体核酸分子连接到经所述分离的核酸分子(例如,编码所述特异性靶向cyp4v2基因的sgrna的分离的核酸分子)切割后的dna片段(例如cyp4v2基因片段)上。
本申请所述供体核酸分子,供体核酸分子可以是野生型的人的核苷酸序列或者含有不同数量内含子和外显子的基因片段。在某些情形下,所述供体核酸分子可以包含或者不包含内含子(0个、1个、2个、3个、4个、5个、6个、7个、8个、9个、10个或11个)。在某些情形下,其可包含cyp4v2基因的6号内含子和11号外显子之间的核苷酸序列。例如,其可以包含6号内含子到11号外显子之间一个或一个以上(例如,2个、3个、4个、5个或6个)的外显子的核苷酸序列,如包含了7号外显子、8号外显子、9号外显子、10号外显子和/或11号外显子中的一个或多个的核苷酸序列。例如,在本申请中,所述供体核酸分子可以包含cyp4v27号到11号外显子。例如,所述的供体核酸分子包含seqidno:38所示的核苷酸序列。在某些情形中,所述供体核酸可以包含与seqidno:38所示核苷酸序列具有至少70%(例如,至少75%,至少80%,至少85%,至少90%,至少91%,至少92%,至少93%,至少94%,至少95%,至少96%,至少97%,至少98%,至少99%,或至少100%)序列同一性的核苷酸序列。本申请所述可包含cyp4v2核苷酸序列的“核酸分子”与本申请所述特异性靶向cyp4v2的grna或“分离的核酸分子”不同。
另一方面,本申请提供了载体,其包含所述的分离的核酸分子(例如,编码所述特异性靶向cyp4v2基因的sgrna的分离的核酸分子)和/或所述的供体核酸分子(例如,编码所述人cyp4v2基因的核苷酸分子)。
在某些情形中,所述分离的核酸分子和所述供体核酸分子可以位于不同的载体上。在另外一些情形中,所述分离的核酸分子和所述供体核酸分子可以位于同一载体中。在本申请中,包含所述分离的核酸分子的载体和包含所述供体核酸分子的载体同时导入细胞后,cas核酸酶可以同步切割供体核酸分子和细胞基因组dna,将供体核酸分子整合到细胞基因组的准确位置上(例如,cyp4v2基因片段),在某些情形下,这一方法的整合效率是十分高的。
在某些实施方式中,所述的载体为病毒载体。例如,aav、慢病毒、逆转录病毒、腺病毒、疱疹病毒和肝炎病毒。用于产生包含核酸分子(例如,本申请所述分离的核酸分子)作为载体基因组一部分的病毒载体的方法是本领域公知的,并且本领域技术人员可无需进行过多的实验。
细胞、药物组合物、用途和方法
本申请提供了细胞,所述细胞可包含所述分离的核酸分子和/或所述供体核酸分子。本申请所述的细胞可表达sgrna和cas核酸酶,具有很好的dna切割效果。本申请所述细胞还可以表达具备正常功能的cyp4v2蛋白。所述细胞可以包括哺乳动物细胞,例如,来自人的细胞。例如,所述细胞可包括cos细胞、cos-1细胞、中国仓鼠卵巢(cho)细胞、hela细胞、hek293细胞、ns0细胞或骨髓瘤细胞、干细胞(例如,多能干细胞和/或全能干细胞)、和/或上皮细胞(例如,肾上皮细胞和/或视网膜上皮细胞)。在本申请中,所述的细胞可包括hek293细胞和/或尿液肾上皮细胞。本申请中,所述的细胞可以经修饰后具备分化能力。所述分化能力可包括分化成身体任何细胞类型的能力:神经元、星形胶质细胞、少突胶质细胞、视网膜上皮细胞、表皮、毛发和角质形成细胞、肝细胞、胰岛β细胞、肠上皮细胞、肺泡细胞、造血细胞、内皮细胞、心肌细胞、平滑肌细胞、骨骼肌细胞、肾细胞、脂肪细胞、软骨细胞和/或骨细胞。例如,所述细胞可被重编程为具有关键重编程基因(例如,oct4、klf4、sox2、cmyc、nanog和/或lin28)过表达的诱导多能干细胞(ipsc)。
本申请所述细胞可用于评价基因编辑治疗所需物质(例如,sgrna和供体核酸分子)的有效性和安全性。
本申请提供了药物组合物,所述药物组合物包含所述的grna,所述的一种或多种分离的核酸分子,所述的供体核酸分子,所述的载体,以及药学上可接受的载剂。所述载剂应当是无毒的,并且不应干扰活性成分的功效。
本申请提供了试剂盒。所述的试剂盒通常包含一起被包装在容器、接受器或其它容器中的两种或更多种组分。例如,本申请中所述的grna,所述的一种或多种分离的核酸分子,所述的供体核酸分子,所述的载体。
本申请所述的药物组合物可通过各种方法导入,例如,包括但不限于,玻璃体内注射(例如,前部、中间或后部玻璃体注射)、结膜下注射、前房内注射、经由颞侧注射到前房中、基质内注射、注射到脉络膜下间隙中、角膜内注射、视网膜下注射和眼内注射局部地投予眼睛。所述导入可包括视网膜下注射,视网膜下注射为注射到视网膜下空间,即感觉神经性视网膜下面。在视网膜下注射期间,将注射的材料(例如,所述的靶向载体,所述的grna,和/或所述的质粒)直接导入感光细胞和视网膜色素上皮(rpe)层之间,并在其间创建空间。
本申请提供了所述grna,所述的一种或多种分离的核酸分子,所述的供体核酸分子,和/或任一项所述的载体在制备治疗疾病的药物中的应用。其中,所述疾病可包括cyp4v2基因突变所导致的疾病。其中,所述突变位于cyp4v2基因6号外显子和7号外显子之间的内含子之后。例如,所述疾病可包括结晶样视网膜变性。
本申请提供了一种治疗结晶样视网膜变性的方法,所述方法包括以下的步骤:向有需要的受试者导入所述的grna(例如,特异性靶向cyp4v2基因的sgrna),所述的一种或多种分离的核酸分子(编码所述特异性靶向cyp4v2基因的sgrna的分离的核酸分子),所述的供体核酸分子(编码所述人cyp4v2基因的核苷酸分子),和/或所述的载体。其中,所述导入使受试者获得了正常功能的cyp4v2蛋白。
本申请所述的方法可包括离体的方法。在某些情形中,可以获得受试者特异性的诱导多能干细胞(ipsc)。然后,可以将诱导多能干细胞分化为任何类型的细胞,例如感光细胞或者视网膜祖细胞。在本申请中,可以是3d视网膜类器官。接下来,可以使用本申请所述的方法编辑这些3d视网膜类器官细胞的基因组dna。例如,该方法可以包括在3d视网膜类器官细胞的cyp4v2基因的突变位点内或附近进行编辑,使得其不编码具有突变的cyp4v2蛋白。最后,可以将3d视网膜类器官细胞植入受试者体内。
在另一些情形中,可以从受试者中分离出感光细胞或视网膜祖细胞。接下来,可以使用本申请所述的方法编辑这些感光细胞或视网膜祖细胞的基因组dna。例如,该方法可以包括在感光细胞或视网膜祖细胞的cyp4v2基因的突变位点内或附近进行编辑,使得其不具有突变的cyp4v2。最后,可以将经基因编辑的感光细胞或视网膜祖细胞植入受试者体内。
所述方法可包括在给药前对治疗剂进行全面分析。例如,对校正细胞的整个基因组进行测序,以确保没有脱靶效应(如果有的话)可以处于与对受试者的最小风险相关的基因组位置。此外,可以在植入之前分离特定细胞的群,包括克隆细胞群。
本申请所述的方法可包括使用定点核酸酶在基因组中精确的靶标位置切割dna,从而在基因组内特定位置产生单链或双链dna断裂的方法。此类断裂可以通过内源性细胞过程进行定期修复,例如同源重组、非同源末端连接。
本申请所述的方法可包括在目标基因座中靠近靶序列的位置创建一个或两个dna断裂,两个dna断裂可以为双链断裂或两个单链断裂。所述断裂可通过定点(site-directed)多肽来实现。定点多肽(例如dna核酸内切酶)可以在核酸(例如基因组dna)中引入双链断裂或单链断裂。双链断裂可以刺激细胞的内源性dna修复途径,例如,hr、nhej。
利用外源供体模板,可以在同源的侧翼区域之间引入另外的核酸序列(例如,所述靶向载体)或修饰(例如单碱基或多碱基改变或缺失),从而也可以将另外的或改变的核酸序列纳入目标基因座,外源供体可以由质粒载体递送,例如,aav载体和/或ta克隆载体(例如,zt4载体)。
本申请提供了一种调节细胞中cyp4v2基因表达的方法,其包括向细胞导入所述的grna,所述的一种或多种分离的核酸分子,和/或中任一项所述的载体。
本申请中所述方法可以是将所述grna导入到所述细胞中。例如,所述grna靶向受体细胞基因组的cyp4v2基因片段,在核酸酶的帮助下将其剪切,从而产生cyp4v2基因翻译为蛋白的机会变少、翻译的蛋白不能执行正常功能的效果。
本申请中所述方法可以是将所述的一种或多种分离的核酸分子导入到所述细胞中。例如,编码所述特异性靶向cyp4v2基因的sgrna的分离的核酸分子,使得所述的cyp4v2基因片段被破坏,从而产生cyp4v2基因翻译为蛋白的机会变少、翻译的蛋白不能执行正常功能的效果。
本申请中所述方法可以是将所述载体导入到所述细胞中。所述载体包含所述的分离的核酸分子和/或所述的供体核酸分子。在某些情形下,所述载体含有所述分离的核酸分子和所述的供体核酸分子,使得所述细胞中cyp4v2基因为所述供体核酸分子替代,从而产生了cyp4v2基因翻译为蛋白的机会的变化(例如,由不存在cyp4v2蛋白的表达转变为达到正常表达量)、翻译的蛋白功能由异常转为正常的效果。在某些情形下,所述载体含有所述分离的核酸分子,使得所述的cyp4v2基因片段被破坏,从而产生cyp4v2基因翻译为蛋白的机会变少、翻译的蛋白不能执行正常功能的效果。在另一些情形下,所述载体含有所述供体核酸分子,使得所述细胞内含有更多的cyp4v2基因片段,可以转录并翻译出更多的cyp4v2蛋白。
不欲被任何理论所限,下文中的实施例仅仅是为了阐释本申请的核酸分子、制备方法和用途等,而不用于限制本申请发明的范围。
实施例
实施例1设计sgrna
一、sgrna的筛选
根据cyp4v2基因的dna序列,在cyp4v2的外显子6与外显子7之间的内含子区设计pam序列为nngrrt和nngrr(金黄色葡萄球菌,staphylococcusaureus,sa;sacas9),长度为21bp的sgrna。
根据评分,一共设计了7个(nngrrt设计5个,nngrr设计2个)sgrna,其序列见表1。
表1.cyp4v2-hiti法benchling在线设计sgrna
二、sgrna合成
根据酶切位点bbs1序列,在设计的sgrna上下游添加bbs1酶切位点;并在各sgrna上下游400bp内设计对应引物。设计相应的寡核苷酸序列及引物见表2。
表2sgrna的设计
三、sgrna载体构建与质粒提取的具体步骤如下
1、将合成的sgrna按照以下步骤退火
将上述合成的各sgrna正向引物(f)和反向引物(r)分别稀释成50μmol,取sgrna(f、r),各5μl,配成sgrna混合物1-7。将t4多聚核苷酸激酶(pnk)及10×t4连接缓冲液于冰上融化备用。配制如下反应体系:
将上述配制好的反应体系置于pcr仪上,运行如下反应程序
回收反应产物。
2、酶切载体步骤如下
构建sgrna载体所用质粒为px601载体(addgene,61591)。质粒图谱如图1所示。
使用bsai酶切释放sgrna结合位点,在1.5mlpcr管中配制如下酶切反应体系:
然后酶切1-2h(或过夜酶切),回收纯化,测定浓度,稀释到50ng/μl。
3、将sgrna连接到步骤2回收的载体上
使用步骤2的回收载体和退火的sgrna配制如下连接体系(200μlpcr管):
将连接反应体系置于37℃,连接约1-2h,完成sgrna载体构建。
4、质粒转化
(1)取出transstbl3化学感受态细胞(北京全式金生物技术有限公司cd-521-02)置于冰上解冻;
(2)取1μl连接产物至于50μl感受态细胞中,冰上孵育半小时,42度热激90sec,冰上2min;
(3)加入无抗培养基500μl于37℃,200rpm摇1h;
(4)800rpm/min离心5min,弃上清,剩约100μl,使用含氨苄抗生素的lb琼脂培养基筛选阳性克隆;
(5)第二天挑筛选出的阳性克隆菌(培养基500μl)摇3-4小时,取200μl送测序。
5.质粒提取(按照omega去内毒素质粒大提试剂盒进行)
(1)将测序正确的px601-sacas9-cyp4v2-sgrna1-7共七个质粒进行过夜大摇(50-200ml),37℃摇床培养12-16h,以扩增质粒,第二天进行提取(摇菌时间摇小于16h)。
(2)取50-200ml的菌液,于室温下4000×g离心10min,收集菌体。
(3)弃去培养基。往沉淀中加入10ml溶液i/rna酶a混和液,通过移液枪吹打或者漩涡振荡使细胞完全重新悬浮。
(4)加入10ml溶液ii,盖上盖子,轻轻上下颠倒离心管8-10次以获得澄清裂解物。如有必要,可把裂解液置于室温静置2-3min。
(5)加入5ml预冷的n3缓冲液,盖好盖子,并温和地上下颠倒离心管10次,直至形成白色絮状沉淀,可在室温下静置孵育2min。
(6)准备一个针筒过滤器,拉出针筒中的活塞,将针筒竖直放在一个合适的试管架上,在注射器下端出口处放置一个收集管,针筒开口朝上。立即将裂解液倒入过滤器的针筒中。细胞裂解液在针筒中停留5min。此时白色絮状物会漂浮于裂解液表面。细胞裂解液可能已从过滤注射器口流出。用新的50ml试管收集细胞裂解液。小心轻轻地将注射器活塞插入针筒中,慢慢推动活塞以使裂解液流入到收集试管中。
(7)加入0.1倍体积的etr溶液(蓝色)至已流出的过滤裂解液中,颠倒试管10次,然后于冰浴中静置10min。
(8)将上述裂解液于42℃下水浴5min。裂解液又将再次出现浑浊。此时于25℃,4000×g离心5min,etr溶液将在试管底部形成蓝色分层。
(9)将上清液移至另一新的50ml试管中,加入0.5倍体积室温的无水乙醇,轻轻颠倒试管6-7次,室温放置1-2min。
(10)将一个
(11)将
(12)将
(13)将
(14)将
(15)用最高速(不超过6000×g)空甩10min,以干燥
(16)(选做)进一步风干
a)把
(17)把
(18)4000×g离心5min,以洗脱出dna。
(19)弃除柱子,把dna产物保存于-20℃。
(20)再次将提取的质粒px601-sacas9-cyp4v2-sgrna1至px601-sacas9-cyp4v2-sgrna7送测序以确保构建的质粒正确。
四、在293t细胞中验证sgrna有效性
1、293t细胞培养
(1)冻存细胞的复苏
a)将恒温水浴锅温度调至37℃,将冻存细胞从液氮中取出,用镊子夹住盖子,在水中快速晃动。
b)将冻存液转移到15ml刻度离心管中,缓慢地加入10ml的细胞培养液,并轻轻的晃动混匀液体。拧紧盖子,过火,1000rpm/min,离心3分钟。
c)过火,加入适量培养液,轻轻吹打底部的细胞沉淀,然后将细胞转移至培养瓶中放到培养箱中培养。293t细胞使用的培养基为添加10%胎牛血清和100u/ml双抗的高糖dmem,于37℃,5%co2培养。
(2)细胞传代
a)倒置显微镜下观察细胞的形态和密度,当细胞在培养瓶中的汇合度到达80%-90%时,开始对细胞进行传代。
b)将细胞培养瓶中的旧培养液洗出来,用pbs清洗3次。向培养瓶中加入500μl的含edta的胰蛋白酶,放入培养箱中孵育1min左右,待细胞间隙变大,细胞变圆时,立即向培养瓶中加入1ml的培养液终止消化,并用吸管轻轻吹打细胞,待细胞全部从瓶底飘起后,将培养瓶中的液体转移到离心管中,1000rpm/min离心2min。
c)弃掉上清,再向离心管中加入2ml培养基使沉淀的细胞重新悬浮。将细胞悬浮液分装到4个新培养瓶中,每个加入4ml的培养液,轻摇培养瓶,使细胞混合均匀铺满培养瓶,放入细胞培养箱中进行培养。
d)转染之前1天,每孔中分别加入密度为80%、细胞舒展、细胞间隙均匀的293t细胞100w,次日细胞长到80-90%汇合度时进行质粒转染。
2、采用pei(聚乙烯亚胺)法转染293t细胞
(1)取1.5ml的ep管,每管中加入250μl的dmem培养基(无血清),依次加入1.5μgpx601-cyp4v2-sgrna1质粒和1μg的plent-gfp质粒(共转染,用以粗略标志px601-cyp4v2-sgrna1质粒是否转入),充分涡旋混匀后,每管加入7.5μl的pei转染试剂,涡旋混匀,室温放置20min后进行转染。以同样方法在6孔板的其他孔分别加入px601-cyp4v2-sgrna2至px601-cyp4v2-sgrna7质粒转染体系、px601空质粒转染体系、空白对照。
(2)逐滴加入培养基中(六孔板培养基加无血清dmem=2ml/孔),孵箱培养,12-18h之内半量换液。次日,荧光显微镜下观察gfp的表达情况,以评估转染效率,转染效率良好的情况下继续培养转染后的质粒。
(3)抗性细胞筛选
a)配液:10mg/ml(母液)稀释为1μg/ml的嘌呤霉素,筛选细胞
b)换液:转染后两日,在每个孔中(包括阴性对照组)加入3ml含嘌呤霉素的293t细胞培养基,开始筛选转染阳性的细胞,以后每日观察细胞的存活情况,每2日换液,换液时加上相应量的嘌呤霉素。待到阴性对照的孔的细胞完全死亡,而实验组和对照组的细胞有存活的(说明转染成功),停止抗生素筛选,改用正常培养基。
3、293t细胞基因组dna的提取
(1)待6孔板细胞长到80-90%汇合度后,传代到6cm培养皿培养。
(2)待细胞长到80-90%汇合度后,收细胞,准备提取基因组dna,整个过程在7-10天左右。基因组的提取使用南京诺唯赞细胞提取试剂盒,实验步骤如下:
a)400×g离心5min收集细胞,弃上清液。加入220μlpbs(磷酸盐缓冲液)、10mlrnase溶液和20μlpk工作液至样品中,重悬细胞。室温静置15min以上;
b)加入250μlgb缓冲液至细胞重悬液中,涡旋混匀,65℃水浴15-30min,过柱纯化;
c)加入250μl无水乙醇至消化液中,涡旋混匀15-20sec;
d)将gdna吸附柱置于2ml收集管中。将上一步所得混合液(包括沉淀)转移至吸附柱中。12000×g离心1min。若出现堵柱现象,在14000×g下离心3-5min。若混合液超过750μl需分次过柱;
e)弃滤液,将吸附柱置于收集管中。加入500μl清洗缓冲液a至吸附柱中。12000×g离心1min;
f)弃滤液,将吸附柱置于收集管中。加入650μl清洗缓冲液b至吸附柱中。12000×g离心1min;
g)重复步骤4;
h)弃滤液,将吸附柱置于收集管中。12000×g空管离心2min;
i)将吸附柱置于新的1.5ml离心管。加入30-100μl预热至70℃的洗脱缓冲液至吸附柱的膜中央,室温放置3min。12000×g离心1min;
注意:对于dna含量丰富的组织,可再加入30-100μl洗脱缓冲液重复洗脱;
j)弃掉吸附柱,将dna保存于2-8℃,测浓度并记录,长期保存需放置于-20℃。
4、t7e1酶切实验
(1)用提取上述转染了px601-sgrna1至px601-sgrna7质粒的293t细胞的基因组dna为模板,围绕靶点附近,利用前述每条sgrna对应设计合成的上下游引物对提取的基因组dna进行扩增,进行dna片段pcr(共7组,分别命名为j1-j7)。
(2)使用液体回收试剂盒(omegagelextractionkit(200)d2500-02)对上述pcr产物进行液体dna回收;dna回收步骤如下:
a)将pcr反应产物中加入等体积的膜结合液,切胶回收的需要每1mg加入1μl的膜结合液,50-60℃加热7min,直至所有的凝胶溶解完全,涡旋混匀,过柱回收;
b)将上述液体降入到回收柱中,10000×g离心1min,去滤液;
c)加入700μl的清洗缓冲液,>13000×g离心1min,去滤液;
d)重复步骤c);
e)空管>13000×g离心10min;
f)将离心柱转移到新的1.5ml的ep管中,做好标记,加入20-30μl的洗脱缓冲液或ddh2o,室温放置2min;
g)>13000×g离心1min,弃掉吸附柱,将dna保存于2-8℃,测浓度并记录,长期保存需放置于-20℃。
(3)t7e1酶切实验进行sgrna效率验证
将上述获得的pcr回收或切胶回收产物进行t7e1酶切反应
a)t7e1酶切退火体系(19.5μl):
b)t7e1酶切退火程序:
95℃2min
95℃to85℃温度以2℃/s下降
85℃to25℃温度以0.1℃/s下降
16℃∞。
c)t7e1酶切反应体系
试剂体积(μl)
退火产物9.75或9.5
t7e1酶0.25或0.5
将所述体系在37℃孵育20min
d)酶切产物跑胶
配胶:2.5%凝胶,加双倍染料
跑胶程序:电压140v,20min到30min
e)查看跑胶结果。
酶切结果如图2所示,泳道1是标志物的电泳结果,泳道2是阴性对照(即不加sgrna)的电泳结果,泳道3是sgrna1切割后的片段长度的结果,泳道4是sgrna5切割后的片段长度的结果,泳道5是阴性对照的电泳结果,泳道6是sgrna2切割后的片段长度的结果,泳道7是sgrna6切割后的片段长度的结果,泳道8是sgrna7切割后的片段长度的结果,泳道9是阴性对照的电泳结果,泳道10是sgrna3切割后的片段长度的结果,泳道11是sgrna4切割后的片段长度的结果。
各片段的扩增引物、片段长度及切割后片段长度见表3。
表3.cyp4v2-hiti法sgrna切割后片段长度表
5、基因组dna的包含切割位点的pcr片段的基因测序,套峰检验
将上述j1~j7的dna扩增片段测序,测序峰图如图3a-3d所示,可以发现sgrna1.2.3.4.有较明显的切割效应。
实施例2验证sgrna靶点对剪切的影响
一、4个sgrna对应pmd19-tminigene质粒,及其对照阴性质粒和阳性质粒设计。
按照donorhiti方法插入后会残留pam+3bpsgrna/18bpsgrna片段特点,设计minigene1.2.3.4,分别对应sgrna1.2.3.4.,minigene片段如图4所示。
二、minigene质粒构建步骤如下
1、合成minigenedna片段及阳性对照(正常野生型内含子,不影响剪切)和阴性对照(患者突变内含子,影响剪切)dna片段。
2、酶切质粒载体
质粒载体为pmd-19t-mcs,质粒图谱见图5。
(1)用kpni酶和mlui双酶切质粒
在cutsmart缓冲液中,将kpni酶与mlui酶与所述minigene孵育,37℃,30min。配制2.5%的电泳凝胶,加双倍染料,对酶切产物进行电泳,电压设置为140v,时间为20min到30min。电泳结束后,切胶纯化。
(2)对酶切后线性化载体进行胶回收
使用液体回收试剂盒(omegagelextractionkit(200)d2500-02)对上述pcr产物进行液体dna回收;dna回收步骤如下:
a)将pcr反应产物中加入等体积的膜结合液,切胶回收的需要每1mg加入1μl的膜结合液,50-60℃加热7min,直至所有的凝胶溶解完全,涡旋混匀,过柱回收;
b)将上述液体转入回收柱,10000×g离心1min,去滤液;
c)加入700μl的清洗缓冲液,>13000×g离心1min,去滤液;
d)重复步骤c);
e)空管>13000×g离心10min;
f)将离心柱转移到新的1.5ml的ep管中,做好标记,加入20-30μl的洗脱缓冲液或ddh2o,室温放置2min;
g)>13000×g离心1min,弃掉吸附柱,将dna保存于2-8℃,测浓度并记录,长期保存需放置于-20℃。
(3)连接
使用上一步的回收载体和合成的minigene1-6dna片段及对应阳性对照配制如下连接体系(在200μlpcr管中配制):
将上一步的连接反应体系置于37℃连接约1-2h,完成sgrna载体构建。
(4)质粒转化
a)取出transstbl3化学感受态细胞置于冰上解冻;
b)取1μl步骤3的连接产物至于50μl感受态细胞中,冰上孵育半小时,42度热激90sec,冰上2min;
c)加入无抗培养基500μl于37℃,200rpm摇菌1h,然后于800rpm/min离心5min,弃上清,剩约100μl,使用含氨苄抗生素的lb琼脂培养基筛选阳性克隆;
d)第二天挑筛选出的阳性克隆菌(培养基500μl)摇3-4小时,取200μl送测序。
(5)质粒提取
将测序正确minigene1-4与阳性、阴性对照共6个质粒进行过夜大摇(50-200ml),37℃摇床培养12-16h,以扩增质粒,第二天进行提取(摇菌时间摇小于16h)。质粒提取按照omega去内毒素质粒大提试剂盒进行。
三、minigene质粒转染293t细胞
1、培养293t细胞
2、采用pei(聚乙烯亚胺)法转染293t细胞
(1)取1.5ml的ep管,按以上顺序进行编号,每管中加入250μl的dmem培养基(无血清),依次按上述表格加入1.5μgminigene1-4质粒和阳性、阴性对照质粒,充分涡旋混匀后,每管加入7.5μl的pei转染试剂,涡旋混匀,室温放置20min后进行转染。6孔板的另设一个孔做空白对照。
(2)逐滴加入培养基中(六孔板培养基加无血清dmem=2ml/孔),孵箱培养,12-18h之内半量换液。次日,荧光显微镜下观察gfp的表达情况,以评估转染效率,转染效率良好的情况下继续培养转染后的质粒。
(3)抗性细胞筛选
a)配液:10mg/ml(母液)稀释为1μg/ml的嘌呤霉素,筛选细胞
b)换液:转染后两日,在每个孔中(包括阴性对照组)加入3ml含嘌呤霉素的293t细胞培养基,开始筛选转染阳性的细胞,以后每日观察细胞的存活情况,每2日换液,换液时加上相应量的嘌呤霉素。待到阴性对照的孔的细胞完全死亡,而实验组和对照组的细胞有存活的(说明转染成功),停止抗生素筛选,改用正常培养基。
3、293t细胞基因组dna的提取
(1)待6孔板细胞长到80-90%汇合度后,传代到6cm培养皿培养。
(2)待细胞长到80-90%汇合度后,收细胞,准备提取基因组dna,整个过程在7-10天左右。基因组的提取使用南京诺唯赞细胞提取试剂盒。
(3)转染后293t细胞rna水平验证
1)rna提取
rna提取采用tiangenrnapreppure培养细胞/细菌总rna提取试剂盒(dp430),实验步骤如下:
a)收集细胞
i.悬浮细胞的收集(收集细胞数量请不要超过1×107):估计细胞数量,300×g离心5min,将细胞收集到离心管中,仔细吸除所有培养基上清。
ii.单层贴壁细胞的收集(收集细胞数量请不要超过1×107),吸除培养基,用pbs洗涤细胞,吸除pbs,向细胞中加入含有0.10-0.25%胰蛋白酶的pbs处理细胞,当细胞脱离容器壁时,加入含有血清的培养基,失活胰蛋白酶,将细胞溶液转移至无rna酶的离心管中,300×g离心5min,收集细胞沉淀,仔细吸除所有上清。
b)裂解处理
轻弹离心管底部,使细胞沉淀松散,根据细胞数量加入适量裂解液rl(使用前请先检查是否已加入β-巯基乙醇),旋涡震荡。
c)将所有溶液转移至过滤柱cs上(过滤柱cs放在收集管中),12000rpm(~13400×g)离心2min,收集滤液。向滤液中加入1倍体积70%乙醇(通常为350μl或600μl),混匀(此时可能会出现沉淀),得到的溶液和沉淀一起转入吸附柱cr3中(吸附柱cr3放入收集管中),12000rpm(~13400×g)离心30-60sec,倒掉收集管中的废液,将吸附柱cr3放回收集管中。
d)向吸附柱cr3中加入350μl去蛋白液rw1,12000rpm(~13400×g)离心30-60sec,倒掉收集管中的废液,将吸附柱cr3放回收集管中。
e)dnasei工作液的配制:取10μldnasei储存液放入新的无rna酶的离心管中,加入70μlrdd缓冲液,轻柔混匀。
f)向吸附柱cr3中央加入80μl的dnasei工作液,室温放置15min。
g)向吸附柱cr3中加入350μl去蛋白液rw1,12000rpm(~13400×g)离心30-60sec,倒掉收集管中的废液,将吸附柱cr3放回收集管中。向吸附柱cr3中加入500μl漂洗液rw(使用前请先检查是否已加入乙醇),室温静置2min,12000rpm(~13,400×g)离心30-60sec,倒掉收集管中的废液,将吸附柱cr3放回收集管中,重复。
h)12000rpm(~13,400×g)离心2min,倒掉废液。将吸附柱cr3置于室温放置数分钟,以彻底晾干吸附材料中残余的漂洗液。
i)将吸附柱cr3转入一个新的无rna酶的离心管中,加入30-100μl无rna酶的ddh2o室温放置2min,12000rpm(~13400×g)离心2min,得到rna溶液。
2)反转录cdna
反转录cdna采用trantranscript一步法移除gdna和合成cdna超级混合物试剂盒(at311),实验步骤如下:
a)cdna合成和gdna去除。如下加入各组分,轻轻混匀
b)反转录程序如下:
42℃30min
85℃5sec
c)收集样品
3)对cdna进行pcr
4)pcr产物测序验证
(5)转染293t细胞后在蛋白水平验证
1)蛋白质提取
a)获得293t细胞并裂解。细胞经pbs洗涤后,以6孔板为例,加入100-200μlripa裂解液,根据需要加入蛋白酶抑制剂cocktail、磷酸酶抑制剂pmsf和rna酶),刮下细胞至ep管中。置于冰上,每5min涡旋一次,重复三次。
b)将裂解好的细胞在4℃,12000rpm离心10min。取50μl上清作为input对照,加10μl6×加样缓冲液煮沸5min。煮后的样品可短期保存于4℃,或长期保存于-20℃。
2)westernblot(蛋白杂交实验)验证
a)玻璃板对齐后放入架子中,把两侧的夹子夹紧。要使短玻璃靠外,长玻璃靠内。然后垂直卡在架子上准备灌胶。操作时要使两侧玻璃对齐,以免漏胶,灌胶前加水检漏。配胶时最后一步加入temed,然后立即摇匀,马上灌胶,灌胶时枪头沿角落加入,以免产生气泡,胶面至距离短玻璃1-2cm即可。加入无水乙醇压胶。
b)当无水乙醇和胶之间有明显的分界线时,说明胶已凝,再等3min使胶充分凝固,倒去胶上层无水乙醇并用纸吸干。按照说明书上比例制备浓缩胶,插入梳子,待胶凝固即可。注意:插入梳子时要使梳子保持水平。
c)上样。
d)电泳:小胶的每个泳道上样量通常不超过20μg总蛋白。尽量保持每个样品中的离子强度一致,包括标记物(marker)和没有样品的空白泳道。电泳时,跑浓缩胶的电压为80v,跑分离胶的电压为120v,电压越大分离越快,电泳的时间一般1-2h,至溴酚蓝即将跑出胶即可终止电泳。
e)转膜
i蛋白从胶到膜放置顺序是:夹子的黑面-海绵垫-滤纸-胶-膜-滤纸-海绵垫-夹子的白面。然后将整个装置全部沉浸在缓冲液里。其中pvdf膜需先经甲醇浸泡,最后放置在电转液中。
ii将夹子放入转移槽中,要使夹子的黑色面对槽的黑色面,夹子的白色面对槽的红色面。
iii需要在电转槽中加入冰用来降温,采用120v电压,200ma恒流进行转膜。蛋白分子越大,电压/电流越大,所需转膜时间越长。
f)封闭
取膜,然后用含5%脱脂牛奶的tbs溶液封闭1小时以上。
注意:接触胶的那面膜要朝上。
g)抗体的孵育与检测
孵育一抗:用含2.5%脱脂牛奶的tbst稀释抗体比例为1:1000,室温1.5-2h,或者4℃过夜。洗膜:用0.1%tbst洗涤3次,每次10min。孵育二抗:含2.5%脱脂牛奶的tbst溶液以适当比例稀释室温2h。洗膜:用0.1%tbst洗涤3次,每次10min,再用tbs洗10min,用滤纸在膜的侧面适当吸干膜上水分。将膜放置在浸有ecl显色液混合液(1ml)的保鲜膜上,将保鲜膜折叠。最后用凝胶成像系统检测。
westernblot的结果如图6a-6b所示。图6a是β-actin(β-肌动蛋白)在各组细胞中的表达情况,泳道1是标记物marker,泳道2是minigene1,泳道3是minigene2,泳道4是minigene3,泳道5是minigene4,泳道6是阴性对照,泳道7是阳性对照,在各组中,β-actin均有表达且表达量基本一致,表明上样量相同,结果具有可比性。图6b是绿色荧光蛋白(greenfluorescentproteins,gfp)在各组细胞中的表达情况,各泳道代表的组别与图6a一致,结果显示在各组中,minigene1-4表达与阳性对照(正常人内含子片段,不影响剪切)一致,均有gfp的表达,阴性对照(患者内含子片段,影响剪切)表达较弱。图6c是标签蛋白(flag)在各组细胞中的表达情况,各泳道代表的组别与图6a一致,结果显示在阴性对照组没有flag的表达,而在各实验组中和阳性对照组中均有表达。因此,结果显示sgrna1-4对剪切无影响。
实施例3donor的设计及筛选
一、载体构建:
将cyp4v2野生型基因的6号内含子和7-11号外显子之间的序列作为donor序列;将donor序列、egfp报告基因一起构建到px601载体(sgrna1-4的px601-sgrna1至px601-sgrna4载体)上,得到px601-donor(1-4)-egfp载体;sgrna载体用实施例1中所述的px601-sgrna(1-4)载体。px601质粒图谱如图1所示。
其中,6号内含子的长度根据sgrna切割位点进行调整,7-11号外显子donor序列如seqidno:38所示;egfp序列如seqidno:39所示。
二、采用pei法转染ipsc
1、具体步骤如下:
以sgrna1组为例:
(1)取4个6孔板,接种ipsc细胞,待细胞密度长到80%时进行转染;
(2)第一组(空白对照组):1.5ml管中加入1.5μg的px601质粒;
第二组(实验组):1.5ml管中加入1.5μg的上述构建的px601-sgrna1质粒(提供sacas9)、1.5μgpx601-donor-egfp质粒;
2个管中分别再加入250μl无血清dmem培养基,混匀后加入12μg的pei(1mg/ml),立即充分混匀,室温孵育20min;
(3)将混合液逐滴加入培养基中,孵箱培养,24小时半量换液。
sgrna2-4组的实验方法同sgrna1组。
三、抗性细胞筛选
配液:10mg/ml(母液)稀释为1μg/ml的嘌呤霉素;
换液:每孔加入含1μg/ml的嘌呤霉素的培养基3ml,每天换液并持续观察细胞死亡状况。每个实验组及空白组转染效果是相近的,因此选取转染代表图,转染效果如图7所示,图7中的左图是空白对照,右图是转染后的细胞,可以看到细胞表达了荧光蛋白,说明转染效果是比较好的。
四、提取细胞dna,测序后,验证序列修复结果
基因组的提取使用南京诺唯赞细胞提取试剂盒。根据测序结果可知,px601-sgrna-donor1.2.3.4载体修复有效。
实例4使用患者3d(三维)视网膜组织体外验证sgrna的基因编辑效率
一、患者肾上皮细胞的提取和培养
使用北京赛贝公司提供的肾上皮细胞分离和培养试剂盒进行,实验步骤如下:
1、将
2、照上紫外灯,准备好12孔板、50ml离心管、15ml离心管、电动移液器、移液管、吸管、5ml枪及枪头、1ml枪及枪头,打开37℃水浴锅。
3、取尿:戴手套、消毒、最好中段尿、并用封口膜封口。
4、取明胶750μl/孔,包被皿底(3个孔)不少于半小时,置于37℃。
5、取75%酒精消毒尿瓶外表面,分装至50ml锥形底离心管中,封口后400×g10min。
6、取出
7、最小速度移动移液管,沿着上液面,缓慢吸取上清至1ml。
8、重悬至15ml离心管中,加入10ml洗涤液,混匀,200×g10min。
9、取出12孔板,吸去明胶,用洗涤液清洗一次(500μl),每孔加入750μl
10、取出15ml离心管,剩余0.2ml细胞团。
11、
12、观察:
d1:观察有无污染;
d2:补充分离培养基——女性:500μl/孔;男性:250μl/孔;
d4:若无贴壁:半量换液,每两天换液,缓慢沿壁加入1ml分离完全培养基。
13、直到出现贴壁:细胞出现贴壁(3~7天或9~10天)后,选择
二、ipscs的诱导
将患者来源的(c.802-8_810del17bpinsgc)肾上皮细胞诱导成ipscs,步骤如下:
1、体细胞汇合度达到70-90%,即可进行消化传代,将细胞接种于96孔板中;接种密度控制在5000—15000个/孔,可根据细胞情况设置3个密度梯度,每个梯度设置3个复孔;细胞接种当天记为第-1天。
2、第0天:镜下观察细胞的汇合度以及状态,选择不同梯度的复孔进行消化计数,选择细胞量达到10000-20000个的孔进行重编程。请按下表配置重编程培养基a:
重编程培养基a体积
体细胞培养基10ml
重编程添加剂ⅰ10μl
3、先将重编程添加剂ⅱ离心,再将97μl重编程培养基a到加入到重编程添加剂ⅱ管中,混匀配成重编程培养基b,将100μl重编程培养基b加入选定的符合条件的一个96孔中,将培养板放回培养箱。
4、第1-2天:镜下观察,并拍照记录细胞的形态变化。若细胞形态变化明显即可撤去重编程培养基b,换为重编程培养基a继续培养;若形态变化不明显,可不换液。
5、第3天:若细胞形态在前两天就已经发生了明显的形变,且细胞生长速度较快,可以进行胰酶消化传代。根据细胞状态和细胞量将细胞传至六孔板的2-6个孔,加入重编程培养基c,尽量形成单细胞贴壁。请按下表配置重编程培养基c:
重编程培养基c体积
重编程培养基a9.8ml
重编程添加剂ⅲ5μl
6、第4天:观察细胞的贴壁情况,若大部分细胞贴壁良好,则更换新鲜的
7、第5天:镜下观察,若有小簇克隆(4个细胞以上的克隆团块)形成,可将体细胞培养基换为
8、第6-8天:镜下观察,若小簇克隆变大,一个克隆团块有10个以上的细胞,可直接将
9、第9-20天:镜下观察,并拍照记录细胞形态变化。每天更换经过室温平衡的新鲜
10、第21天:镜下观察,若单个细胞克隆能填满整个10倍镜视野,可用1ml注射器针头(或其他器具如玻璃针)切割克隆,并将其挑取至提前包被基质胶的48孔板中(若克隆状态较好,细胞厚实且生长较快,可直接挑取到24孔板中)。
11、克隆挑出后用
三、患者的3d视网膜的诱导
具体步骤如下:
四、aav8病毒的构建和包被
1、质粒扩增:构建好的aav载体、包装质粒和辅助质粒需经过大量去内毒素抽提,使用qiagen大抽试剂盒进行质粒的大量抽提,步骤同前;
2、aav8-293t细胞转染:转染当天观察细胞密度,达到80-90%,即可将载体质粒、包装质粒和辅助质粒进行转染;
3、aav8病毒收毒:病毒颗粒同时存在于包装细胞和培养上清中。可以将细胞和培养上清都收集下来以获得最好的收率;
4、aav8纯化后,-80℃长期保存。
五、利用3d视网膜组织,在体外验证sgrna基因编辑效率
1、使用aav8感染3d视网膜组织:观察组织状态,当3d视网膜组织生长状态良好时,即可将包载有目的质粒的aav8病毒进行感染。
2、验证基因编辑效率。
(1)t7e1酶切实验
步骤如实施例1中的t7e1酶切实验所示。
(2)topopcr克隆
为了将px601-cyp4v2-sgrna,pmd19-t-donor的编辑效率量化并将实验组和对照组的切割效率进行统计学分析,我们使用invitrogen的zero
a)使用3d视网膜组织感染aav病毒后获得的dna作为扩增模板,使用invitrogen的platinumsuperfitm系列的dna聚合酶进行pcr反应,反应体系如下(50μl体系):
b)touchdownpcr(降落pcr)程序:
具体步骤同实施例一中步骤三的sgrna退火的pcr步骤进行。
c)topopcr克隆反应如下
配好上述体系,轻轻混匀,室温放置5min;
置于冰上,去除感受态细胞,准备转化。
d)topopcr克隆反应转化感受态细胞
取50或100μl的感受态细胞,加入2μl的上述topopcr克隆反应液体,冰上放置5-30min;42℃热激30sec,不要晃动;立刻将反应转至冰上,加入250μl的s.o.c.培养基;37℃,200rpm,摇晃复苏1h;取出相应数量的lb培养板(加入25μg/ml博来霉素),取100μl上述菌液进行涂板,置于37℃孵箱过夜培养。
e)挑菌送sanger测序。
次日早上,取出菌板,每个板子上挑出80个菌;37℃,200rpm,摇菌3-4h;每个菌样取200μl送桑格测序。
f)分析sanger测序结果并进行统计学分析。
实施例5在人源化小鼠模型上验证基因编辑效率
一、人源化小鼠的构建:
人源化小鼠的构建工作由北京百奥塞图公司完成。
1、人源化小鼠的制作方案
针对人的突变位点,我们委托北京百奥赛图基因生物技术有限公司构建了cyp4v2人源化的小鼠模型。
2、技术内容:
(1)设计构建识别靶序列的sgrna;
(2)构建导致靶基因切割的crispr/cas9载体;
(3)sgrna/cas9的活性检测;
(4)设计构建基因敲进的打靶载体,按照方案中设计的替换6-8号外显子(包括内含子序列);
(5)体外转录获得sgrna/cas9mrna;
(6)小鼠受精卵注射sgrna/cas9mrna和打靶载体;
(7)cyp4v2基因敲入f0代小鼠检测及扩繁;
(8)cyp4v2基因敲入f1代杂合子小鼠的获得及基因型鉴定(southernblottingdna杂交验证,包含外源及内源探针各一个)。
3、技术方法:
(1)小鼠基因组靶序列的扩增和测序,设计并构建针对靶序列的crispr/cas9载体质粒,并进行活性检测:
(2)小鼠基因组dna提取,扩增识别靶序列的sgrna;
(3)设计并构建导致目的序列切割的crispr/cas9载体质粒;
(4)利用百奥赛图自主开发的检测试剂盒对sgrna/cas9进行活性检测;
(5)选择高活性的sgrna/cas9靶位点序列信息,设计并构建cyp4v2基因敲进的打靶载体;
(6)小鼠受精卵原核注射sgrna/cas9mrna和打靶载体,并获得f0/f1代阳性小鼠,该项实验过程主要包括如下内容:
sgrna/cas9mrna的体外转录;
收集小鼠受精卵;
小鼠受精卵注射rna和打靶载体;
受精卵移植到代孕小鼠输卵管;
cyp4v2基因人源化点突变的f0代小鼠基因型鉴定及扩繁;
cyp4v2基因人源化点突变的f1代小鼠的获得及基因型鉴定(包含pcr检测和southernblotting杂交验证)。
二、饲养和繁育
获得两种人源化小鼠之后,让f1代杂合的小鼠进行内交,尽快获得足够数量的f2代或f3代人源化纯合的小鼠,用作aav病毒注射。
三、小鼠视网膜下腔注射aav病毒
将px601-sgrna(提供sacas9)和px601-donor-egfp载体包装成腺相关病毒,注射cyp4v2突变模型小鼠的视网膜,以验证所设计的sgrna和donor在体内的编辑和修复效率。以px601-sgrna1+px601-donor1-egfp为例,具体步骤如下;sgrna2-4组实验方法同此。
1、实验设计
(1)空白对照组:生理盐水。
(2)实验组:px601-sgrna1(提供sacas9)和px601-donor-egfp。
2、aav(腺相关病毒)血清型选择
选择对视网膜偏嗜性较好的aav2/8血清型。
3、病毒包装
将px601-donor1-egfp,px601-sgrna1载体分别和aav2/8及aav-helper包装成腺相关病毒(aav)。
4、小鼠注射实验
取20只cyp4v2突变模型小鼠,每组5只分4组进行实验。
实验计划如下:
(1)空白对照组:小鼠每只眼睛注射2μl生理盐水。
(2)实验组:将包装的px601-sgrna1和px601-donor1-egfp病毒分别按照1:1的比例等量混合,小鼠每只眼睛注射2μl(1e10vg)。
(3)一个月后检测治疗效果。
实验步骤如下:
(4)注射前30分钟用1%阿托品散瞳;麻醉前再次散瞳。
(5)按照80mg/kg氯胺酮和8mg/kg甲苯噻嗪安的比例,对小鼠进行腹腔内注射,麻醉小鼠后,将小鼠放置在眼外科手术显微镜的动物实验平台前方,再在小鼠眼睛上滴一滴0.5%的丙美卡因进行局部麻醉。以100:1的浓度在aav病毒里加入荧光素钠原液,低速离心混匀。
(6)用胰岛素针在小鼠眼睛睫状体平坦部预扎一个小孔,用微量注射器的针头穿过该小孔后进入小鼠眼睛玻璃体腔,这时在小鼠眼睛上滴加适量2%羟甲基纤维素,从而在镜下能清晰见到小鼠眼底,再继续将针头避开玻璃体插入对侧周边的视网膜下,缓慢推入带有荧光素钠的aav病毒,每只眼睛注射量为1μl,以荧光素钠为指示剂,判断是否注射入视网膜下腔。
(7)术后观察小鼠有无异常,给予新霉素眼膏预防感染。
四、评价基因编辑治疗的效果:
实验结果发现各实验组治疗效果基本一致,因此选取px601-sgrna+px601-donor-egfp病毒组为代表来说明基因编辑治疗效果。
主要通过以下方式说明基因编辑治疗效果:
通过erg(视网膜电生理)评价治疗后小鼠功能改变,在治疗后,突变小鼠模型的视网膜功能有改善;
取材切片后,通过he染色观察小鼠视网膜形态学变化,形态接近野生型小鼠的视网膜状态;
通过免疫荧光染色观察基因编辑治疗载体是否在视网膜相应位置表达,基因编辑治疗载体在视网膜相应位置表达;
通过一系列视网膜特殊marker的染色标记观察视网膜组织形态变化,发现治疗后,小鼠的视网膜组织形态有改善。
五、评估aav8-px601-cyp4v2-sgrna在人源化小鼠中的编辑效率
验证方法见实施例4中第五步“视网膜组织体外验证sgrna基因编辑效率”,利用t7e1酶切实验和topopcr克隆实验来验证sgrna的基因编辑效率,结果发现sgrna可以取得比较好的基因编辑效率。
最后应说明的是:以上实施例仅用以说明本申请的技术方案,而非对其限制;尽管参照前述实施例对本申请进行了详细的说明,领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本申请各实施例技术方案的精神和范围。
序列表
<110>北京大学第三医院(北京大学第三临床医学院);北京中因科技有限公司
<120>靶向cyp4v2基因突变位点的核酸分子及其用途
<130>0138-pa-005cn
<160>54
<170>patentinversion3.5
<210>1
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna1
<400>1
ctgggctctaggaattccacc21
<210>2
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna2
<400>2
cataggctccatagtcctaca21
<210>3
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna3
<400>3
cagaaatcgcaagcatagagg21
<210>4
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna4
<400>4
gcagtctttccaacacaagaa21
<210>5
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna5
<400>5
agtgtgatcacctggttatag21
<210>6
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna6
<400>6
aaaagttctggaaatgaacgg21
<210>7
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna7
<400>7
tgtatatggtccgtacctgaa21
<210>8
<211>6
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna1-pam
<400>8
aagaat6
<210>9
<211>6
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna2-pam
<400>9
ccgggt6
<210>10
<211>6
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna3-pam
<400>10
gtgaat6
<210>11
<211>6
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna4-pam
<400>11
tagaat6
<210>12
<211>6
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna5-pam
<400>12
gagaat6
<210>13
<211>5
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna6-pam
<400>13
tgggg5
<210>14
<211>5
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sgrna7-pam
<400>14
aggaa5
<210>15
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna1-f
<400>15
caccgctgggctctaggaattccacc26
<210>16
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna1-r
<400>16
aaacggtggaattcctagagcccagc26
<210>17
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna2-f
<400>17
caccgcataggctccatagtcctaca26
<210>18
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna2-r
<400>18
aaactgtaggactatggagcctatgc26
<210>19
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna3-f
<400>19
caccgcagaaatcgcaagcatagagg26
<210>20
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna3-r
<400>20
aaaccctctatgcttgcgatttctgc26
<210>21
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna4-f
<400>21
caccggcagtctttccaacacaagaa26
<210>22
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna4-r
<400>22
aaacttcttgtgttggaaagactgcc26
<210>23
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna5-f
<400>23
caccgagtgtgatcacctggttatag26
<210>24
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna5-r
<400>24
aaacctataaccaggtgatcacactc26
<210>25
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna6-f
<400>25
caccgaaaagttctggaaatgaacgg26
<210>26
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna6-r
<400>26
aaacccgttcatttccagaacttttc26
<210>27
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna7-f
<400>27
caccgtgtatatggtccgtacctgaa26
<210>28
<211>26
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna7-r
<400>28
aaacttcaggtacggaccatatacac26
<210>29
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna1-p1-f
<400>29
tggagttatgtccttgtggtg21
<210>30
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna1-p1-r
<400>30
cctgctactaagtggcctgaa21
<210>31
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna2/6-p2.6-f
<400>31
cgtcattcccacgattgcct20
<210>32
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna2/6-p2.6-r
<400>32
tggtggttagcacttagcgag21
<210>33
<211>22
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna3/4-p3.4-f
<400>33
aagcatggcagtgtttgagttg22
<210>34
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna3/4-p3.4-r
<400>34
cgttcatttcattggcccgt20
<210>35
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna5-p5-f
<400>35
cggcagtcattttcaaaggca21
<210>36
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna5-p5-r
<400>36
caggcctcagtaggcaattct21
<210>37
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna7-p7-f
<400>37
accaacagtgtaagtccctga21
<210>38
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>cyp4v2sgrna7-p7-r
<400>38
acacagcaccctgtttgttc20
<210>39
<211>774
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>donor
<400>39
gtcatcgctgaacgggccaatgaaatgaacgccaatgaagactgtagaggtgatggcagg60
ggctctgccccctccaaaaataaacgcagggcctttcttgacttgcttttaagtgtgact120
gatgacgaagggaacaggctaagtcatgaagatattcgagaagaagttgacaccttcatg180
tttgaggggcacgatacaactgcagctgcaataaactggtccttatacctgttgggttct240
aacccagaagtccagaaaaaagtggatcatgaattggatgacgtgtttgggaagtctgac300
cgtcccgctacagtagaagacctgaagaaacttcggtatctggaatgtgttattaaggag360
acccttcgcctttttccttctgttcctttatttgcccgtagtgttagtgaagattgtgaa420
gtggcaggttacagagttctaaaaggcactgaagccgtcatcattccctatgcattgcac480
agagatccgagatacttccccaaccccgaggagttccagcctgagcggttcttccccgag540
aatgcacaagggcgccatccatatgcctacgtgcccttctctgctggccccaggaactgt600
ataggtcaaaagtttgctgtgatggaagaaaagaccattctttcgtgcatcctgaggcac660
ttttggatagaatccaaccagaaaagagaagagcttggtctagaaggacagttgattctt720
cgtccaagtaatggcatctggatcaagttgaagaggagaaatgcagatgaacgc774
<210>40
<211>717
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>egfp
<400>40
gtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggc60
gacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggc120
aagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctc180
gtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcag240
cacgacttcttcaagtccgctatgcccgaaggctacgtccaggagcgcaccatcttcttc300
aaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtg360
aaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaag420
ctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggc480
atcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgac540
cactaccagcagaacgcccccatcggcgacggccccgtgctgctgcccgacaaccactac600
ctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctg660
ctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaag714
<210>41
<211>2073
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>intron
<400>41
gtaagtccctgacttttacaattgtggtaaaatagacataacataaaatttccctttata60
accattttaactgtacagtttggtggtattaagtgcattcacgatgttgtgcaaccatcc120
ccaccgttcatttccagaacttttggtaagtccatgatgttgatgttttgttaacatacc180
cggtgtaggactatggagcctatgtctcagaaaataaaacttgaataataatagaaaaca240
atttttcatataaaaaattatacttaagtataaaaatgtatacttcaattatgtagtcaa300
caaatattaattaagtactcgctaagtgctaaccaccataccaaatgttggaaatgtagt360
aatgagtaggacatgtgtatatggtccgtacctgaaaggaagttattctagtaggagagg420
tgatctatcaacacataattacaacatgtgatatgagctgtgaacacttatgaacaaaca480
gggtgctgtgtaaaagaataaaggaacaaagatctatgtataggagttttctggaaaatg540
tttggattcggcagtcattttcaaaggcagagggcattgatagcagtatcttaacatgga600
aaacattaaaactaactagatattagtattctatttccaattcaaaaataaccagaagat660
agtgatgttgttttgaatataggatgtcaatctttgtgttaataatgtgttttgaaaaag720
caagacttaattgaaaatatacatcaaattataatttcagtgtattaaaaaactgcctgt780
ttaaatatgtcctttctttgctgtaaattttggttaaaatctattggagttatgtccttg840
tggtgaagtacaccctacccccaagagagcaaatgatgaataaatcagtagatgttccat900
gaatgcaatgttggctgagctggccacagtggagtgtgatcacctggttataggagaata960
gccagcaggttatatttcataattatatttttccttaaattttgtgcattaatatttaat1020
agcaataattaaatgaattccagactgaatagacaattttattcattgaataaacattga1080
gaattgcctactgaggcctgggctctaggaattccaccaagaataaaaaaagacatggtg1140
ttttgccctcaaattgcttagaatctattcaggccacttagtagcaggtttggtcaatta1200
cctggtaggaaaagaaggttgaaatgtacaaccaaattaagtgtattggccttcattctt1260
ttatccatccaagaaatatctagtgagtatttgttaaatgccaagcagtgctctagaggt1320
tggaaatgtatcaatgaacaaagacatgcacaagcatggcagtgtttgagttgtaagaga1380
aagatgctaaataataagtataacgtaagtataatatcacattataataataagtatgac1440
agaagtatcacataatgtcaggtggcaagggaaatggagaaaaagcattgttaggaggcg1500
tcgcagtgctagggacaggggtagcagggatgagggaggtctctctgatcgtgtgacaat1560
gagcagagatctggaagaagggagggggcagccgtgcaggcctctgaggaaagcatttca1620
ggcagcagaaatcgcaagcatagagggtgaattcactaatctgcttgctgtttcttttct1680
cctctacaacatgtaattaagtctataattagactgttgtataatgattgcattcactct1740
accacttaaactttttctcattagcgcaatccaatctctagggtactatctagtagacta1800
tgattctattcttgtgttggaaagactgcaaaaatagcacttgaaaattaggaatttgcg1860
tgacattaaatttgtttttaaaagtttcaccagataatccccaaaatattaatgaggctt1920
tactgtattttcacaagagcctatgttgtcgaaatgttgaaataggcttagaaaaataaa1980
tgaaagaaactagcatattttataagaaaatgtgttaactagggtgcatccaagtccaaa2040
cagaagcatgtgattatcattcaaatcatacag2073
<210>42
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p1.5-f
<400>42
gtgaagtacaccctaccccca21
<210>43
<211>22
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p1.5-r
<400>43
ccaggtaattgaccaaacctgc22
<210>44
<211>22
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p2.6.7-f
<400>44
ggatgggaacacaaaaagagcc22
<210>45
<211>23
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p2.6.7-r
<400>45
acacagcaccctgtttgttcata23
<210>46
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p3.4-f
<400>46
tgcacaagcatggcagtgtt20
<210>47
<211>23
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p3.4-r
<400>47
catttcattggcccgttcagc21
<210>48
<211>21
<212>rna
<213>人工序列(artificialsequence)
<220>
<223>sgrna1-rna
<400>48
cugggcucuaggaauuccacc21
<210>49
<211>21
<212>rna
<213>人工序列(artificialsequence)
<220>
<223>sgrna2-rna
<400>49
cauaggcuccauaguccuaca21
<210>50
<211>21
<212>rna
<213>人工序列(artificialsequence)
<220>
<223>sgrna3-rna
<400>50
cagaaaucgcaagcauagagg21
<210>51
<211>21
<212>rna
<213>人工序列(artificialsequence)
<220>
<223>sgrna4-rna
<400>51
gcagucuuuccaacacaagaa21
<210>52
<211>21
<212>rna
<213>人工序列(artificialsequence)
<220>
<223>sgrna5-rna
<400>52
agugugaucaccugguuauag21
<210>53
<211>21
<212>rna
<213>人工序列(artificialsequence)
<220>
<223>sgrna6-rna
<400>53
aaaaguucuggaaaugaacgg21
<210>54
<211>21
<212>rna
<213>人工序列(artificialsequence)
<220>
<223>sgrna7-rna
<400>54
uguauaugguccguaccugaa21
- 还没有人留言评论。精彩留言会获得点赞!