使用机器学习对蛋白质和其它序列定义的生物分子进行进化数据驱动设计的方法和设备
使用机器学习对蛋白质和其它序列定义的生物分子进行进化数据驱动设计的方法和设备
1.相关申请
2.本技术要求美国临时申请第63/020,083号(于2020年5月5日提交)和美国临时申请第62/900,420号(于2019年9月13日提交)的权益。前述临时申请中的每一篇的整个内容通过引用并入本文。
技术领域
3.本公开涉及用于设计序列定义的分子(诸如蛋白质)的数据驱动、基于进化的方法,并且更具体地涉及将无监督序列模型与监督功能性模型相结合以设计具有所需功能性的蛋白质的迭代方法。
背景技术:
4.本文所提供的背景描述是出于总体上呈现本公开背景的目的。就此背景技术部分中描述的著作而言,当前署名的发明人的著作以及在提交时可能未以其它方式具有作为现有技术的资格的描述的各方面既不明确地也不隐含地被认作是针对本公开的现有技术。
5.蛋白质是参与各种生物过程的分子机器,包括对生命至关重要的那些过程。例如,它们能够在体内催化微秒级的生物化学反应,否则这些反应需要数年时间。蛋白质参与运输(血液蛋白质血红蛋白将氧气从肺部运输到组织)、能动性(鞭毛提供精子能动性)、信息处理(蛋白质构成细胞中的信号转导途径)和正常身体机能的细胞调节信号(如激素胰岛素)基础。提供宿主免疫的抗体是蛋白质,负责肌肉收缩和细胞间运输的分子马达(如驱动蛋白和肌球蛋白)也是如此。当光线到达眼睛时,眼睛中被称为视紫质的膜蛋白感知入射光子,进而激活下游蛋白质级联,以最终告诉大脑眼睛看到了什么。因此,蛋白质执行高度多样化和专门化的功能。
6.尽管蛋白质表现出一系列非凡的特性,但所有蛋白质都是仅由20个称为氨基酸的单元构成的聚合物。每个蛋白质都是氨基酸的线性排列,称为蛋白质序列。在其天然状态下,蛋白质分子本身会扭曲、转动和折叠,通常形成一个不规则的三维小球状体。氨基酸的精确三维排列(称为蛋白质结构)以及氨基酸之间的相互作用产生了蛋白质的功能。蛋白质功能模型可以用于导出鉴定蛋白质分子中的所有原子相互作用的功能特性(即,其能量结构)。存在两种独立的方法(一种基于蛋白质结构,另一种基于进化统计量)来理解蛋白质的能量结构。
7.结构导向的观点是有价值的。例如,结合位点残基的作用可以通过诱变来测试,基于这样的想法,如果关键氨基酸被置换为具有较低平均功能活性的残基(例如丙氨酸),则预测蛋白质表现出其发挥作用的能力会降低。例如,使用这种方法,已经证明了构成蛋白质和配体界面的氨基酸的重要性。
8.然而,上述空间邻近原则并没有完全捕获生物化学功能的决定因素。例如,氨基酸可以在结构中以复杂的协同方式相互作用,即使在远处也能影响结合位点的功能,并且单
独的结构并不能提供理解此类协同作用如何安排的通用模型。因此,需要其它蛋白质设计方法。特别地,蛋白质设计的优化空间太大且太复杂而无法仅使用基于结构的方法来驾驭。
9.蛋白质设计的目标是鉴定具有某些所需特性的新型分子。这可以被视为一个优化问题,其中执行搜索以获得最大化给定的定量需要的蛋白质。然而,蛋白质空间的优化极具挑战性,因为搜索空间很大,是离散的,并且大部分都充满了非结构化、非功能性序列。制备和测试新蛋白质既昂贵又耗时,而且潜在候选物的数量非常庞大。
10.因此,需要改进的技术来更有效和稳健地搜索根据给定的定量需要优化的蛋白质。
附图说明
11.当结合附图考虑时通过参考以下详细描述来提供对本公开的更完整的理解,其中:
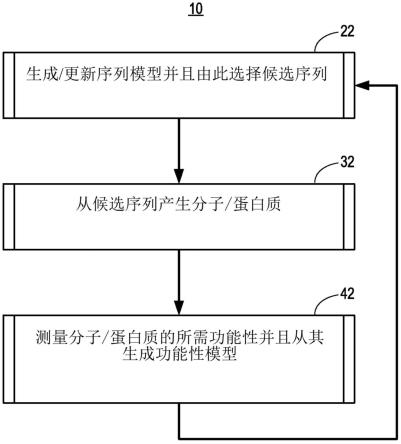
12.图1a显示了根据一种实施方式用于设计序列定义的分子的方法10的流程图;
13.图1b显示了根据一种实施方式对蛋白质实施的方法10的更详细流程图;
14.图1c显示了根据一种实施方式为序列定义的分子生成合成序列的计算模型的示意图;
15.图1d显示了根据一种实施方式用于对序列定义的分子进行数据驱动设计的方法10的示意图;
16.图1e显示了展示由本文所述的系统和/或方法产生的示例性设计或候选蛋白质的图,其中此类蛋白质可以以一种或多种替代形式提供(例如,作为最终产品)和/或可以应用于各种行业;
17.图2显示了根据一种实施方式在迭代搜索中遍历以设计蛋白质的路径的实例;
18.图3a显示了根据一种实施方式在具有无监督学习部分和监督学习部分的实施方式中的方法10的另一种实施方式的流程图;
19.图3b显示了根据一种实施方式在方法10的监督学习部分中的迭代循环的流程图;
20.图3c显示了根据一种实施方式以无监督学习部分和监督学习部分的形式表示的方法10的示意图;
21.图4显示了根据一种实施方式的变分自编码器(vae)的示意图;
22.图5显示了根据一种实施方式vae与由高斯过程回归定义的函数组合使用以寻找帕累托(pareto)最优候选氨基酸序列的示意图;
23.图6a显示了根据一种实施方式的基因合成的第一种实施方式的示意图;
24.图6b显示了根据一种实施方式的基因合成的第二种实施方式的示意图;
25.图7显示了在遗传序列中的密码子与氨基酸之间的映射;
26.图8a显示了根据一种实施方式的将直接耦合分析(dca)模型用于序列模型的方法10的示意图;
27.图8b显示了细菌和真菌中莽草酸途径的一部分,其导致芳香族氨基酸酪氨酸和苯丙氨酸的生物合成;
28.图8c显示了包括具有两个功能活性位点的二聚体(例如,条目800c1、800c2和800c3)的大肠杆菌分支酸变位酶(cm)的原子结构;
29.图9a显示了根据一种实施方式从玻尔兹曼机直接耦合分析(bmdca)模型(即,概括经验一阶统计msa)中采样的序列的一阶统计量;
30.图9b显示了根据一种实施方式从概括经验二阶统计量msa的bmdca模型中采样的序列的二阶统计量;
31.图9c显示了根据一种实施方式从概括经验三阶统计量msa的bmdca模型中采样的序列的三阶统计量;
32.图9d显示了根据一种实施方式在msa中的所有自然cm序列(例如条目900d1的阴影圆圈)和源自bmdca模型的序列(例如条目900d2的阴影圆圈)之间的距离矩阵的前两个主要成分;
33.图9e显示了cm的定量高通量功能测定,其中cm变体文库在缺乏分支酸变位酶的大肠杆菌菌株中表达,在选择性条件下作为混合群体生长,然后进行下一代测序以计数输入和选择群体中每个cm等位基因的频率;
34.图9f显示了在大致五个对数级范围内在计算的相对富集度(“re”)和催化功率1n(kc/km)之间的近似线性关系图;
35.图10a显示了根据一种实施方式自然cm序列的数量随着统计能量变化的的直方图;
36.图10b显示了根据一种实施方式的自然cm序列随着r.e得分变化的直方图;
37.图10c显示了根据一种实施方式在温度t=0.33时bmdca生成的序列随着统计能量变化的直方图;
38.图10d显示了根据一种实施方式在温度t=0.66时bmdca生成的序列随着统计能量变化的直方图;
39.图10e显示了根据一种实施方式在温度t=i.0时bmdca生成的序列随着统计能量变化的直方图;
40.图10f显示了根据一种实施方式在温度t=0.33时bmdca生成的序列随着r.e.得分变化的直方图;
41.图10g显示了根据一种实施方式在温度t=0.66时bmdca生成的序列随着r.e.得分变化的直方图;
42.图10h显示了根据一种实施方式在温度t=1.0时bmdca生成的序列随着r.e.得分变化的直方图;
43.图10i显示了根据一种实施方式仅使用一阶统计量生成的序列随着统计能量变化的直方图;
44.图10j显示了根据一种实施方式仅使用一阶统计量生成的序列随着r.e得分变化的直方图;
45.图11a显示了具有功能序列(例如条目1100a1的阴影条)和非功能序列(例如条目1100a2的阴影条)的所有合成cm序列的散点图,显示了在bmdca统计能量和催化功能之间的关系;
46.图11b显示了具有功能序列(例如条目1100b1的阴影圆圈或条目)和非功能序列(例如条目1100b2的阴影圆圈或条目)的、msa中的自然cm序列的序列变异随着前两个主要成分变化的散点图。
47.图11c显示了具有功能序列(例如条目1100c1的阴影圆圈或条目)和非功能序列(例如条目1100c2的阴影圆圈或条目)的、从bmdca模型来源的序列的序列变异随着前两个主要成分变化的散点图。
48.图11d显示了在edca<40或者没有从自然cm序列的功能互补模式来源的额外统计条件(p(x=1|σ))的情况下合成序列数量的直方图;
49.图11e显示了在edca<40或者有从自然cm序列的功能互补模式来源的额外统计条件(p(x=1|σ))的情况下合成序列数量的直方图;
50.图11f显示了具有对低统计能量贡献最大的位置(例如条目1100f2的阴影球体)和对大肠杆菌特定功能有贡献的位置(例如条目1100f1的阴影球体)的大肠杆菌cm结构;
51.图12显示了根据一种实施方式的蛋白质优化系统的示意图;
52.图13显示了根据一种实施方式的用于训练深度学习(dl)网络的方法的流程图;并且
53.图14显示了根据一种实施方式的人工神经网络的实例。
54.本文所述的附图描绘了本文所公开的系统和方法的各个方面。应当理解,每个附图描绘了所公开系统和方法的特定方面的实施例,并且附图中的每个附图旨在符合其一个或多个可能的实施例。此外,只要有可能,本文的描述参考本文附图中所包含的附图标记,其中多个附图中描绘的特征由一致的附图标记表示。
技术实现要素:
55.根据一个实施方案的方面,提供了一种设计具有所需功能性的蛋白质的方法。该方法包括以下步骤:(i)使用机器学习模型确定合成蛋白质的候选氨基酸序列,该机器学习模型已被训练来学习蛋白质的训练数据集氨基酸序列中的隐式模式,该机器学习模型表达在潜在空间中学习的隐式模式;和(ii)执行迭代循环。循环的每次迭代包含、包括以下步骤:(i)合成候选基因并产生与各个候选氨基酸序列对应的候选蛋白质,每个候选基因编码对应的候选氨基酸序列,(ii)通过使用一种或多种测定测量候选蛋白质的值评估候选蛋白质分别表现出所需功能性的程度,和(iii)当迭代循环的一个或多个停止标准未得到满足时,从测量值计算潜在空间中的适合度函数,并且使用适合度函数与机器学习模型的组合来选择用于后续迭代的候选氨基酸序列。
56.根据另一个实施方案的方面,提供了一种设计具有所需功能性的蛋白质的系统。该系统包括(i)基因合成系统、(ii)测定系统和(iii)处理电路系统。基因合成系统被配置为合成与编码候选氨基酸序列的各个候选基因序列对应的候选基因。测定系统被配置为测量与各个候选氨基酸序列对应的候选蛋白质的值,所测量的值提供所需功能性的标记。处理电路系统被配置为(i)使用机器学习模型确定合成蛋白质的候选氨基酸序列,该机器学习模型已被训练来学习蛋白质的训练数据集中的隐式模式,该机器学习模型表达在潜在空间中学习的隐式模式;和(ii)执行迭代循环。循环的每次迭代包括以下步骤:(a)发送待合成的候选氨基酸序列,(b)接收通过使用一种或多种测定测量候选蛋白质而生成的测量值,和(c)当迭代循环的一个或多个停止条件未得到满足时,根据所测量的值计算潜在空间中的适合度函数,并且使用适合度函数与机器学习模型的组合来选择用于后续迭代的候选氨基酸序列。
57.根据第三个实施方案的方面,提供了一种包括可执行指令的非暂时性计算机可读存储介质,其中所述指令在由电路系统执行时导致该电路系统执行以下步骤:(i)使用机器学习模型确定合成蛋白质的候选氨基酸序列,该机器学习模型已被训练来学习蛋白质的训练数据集氨基酸序列中的隐式模式,该机器学习模型表达在潜在空间中学习的隐式模式;和(ii)执行迭代循环。循环的每次迭代包含、包括以下步骤:(i)合成候选基因并产生与各个候选氨基酸序列对应的候选蛋白质,每个候选基因编码对应的候选氨基酸序列,(ii)通过使用一种或多种测定测量候选蛋白质的值评估候选蛋白质分别表现出所需功能性的程度,和(iii)当迭代循环的一个或多个停止标准未得到满足时,从测量值计算潜在空间中的适合度函数,并且使用适合度函数与机器学习模型的组合来选择用于后续迭代的候选氨基酸序列。
58.根据第四个实施方案的方面,提供了一种设计具有所需功能性的蛋白质的方法。该方法包括以下步骤(i)确定为序列定义的生物分子的候选基因序列,该候选基因序列是使用机器学习模型生成的,该机器学习模型已被训练来学习在序列定义的生物分子的训练数据集中的隐式模式,该机器学习模型表达在潜在空间中学习的隐式模式;和(ii)执行迭代循环。循环的每次迭代包含、包括以下步骤:(i)合成与各个候选基因序列对应的候选基因,每个候选基因编码对应的候选生物分子,(ii)通过使用一种或多种测定测量候选生物分子的值评估候选生物分子分别表现出所需功能性的程度,和(iii)当迭代循环的一个或多个停止标准未得到满足时,从测量值计算潜在空间中的适合度函数,并且使用适合度函数与机器学习模型的组合来选择用于后续迭代的候选基因序列。
59.根据以上内容以及本文的公开内容,本公开包括计算机功能性的改进或至少因为本文所公开的发明叙述了例如通过更新机器学习模型以更准确地预测或鉴定设计或候选蛋白质而不断改进的底层计算机或机器而对其它技术的改进。也就是说,本公开描述了计算机本身的功能的改进或因为随着计算装置的机器学习模型被进一步训练(经由各种迭代循环)以更好地鉴定或预测候选或设计蛋白质由底层计算装置生成的预测或评估随着时间的推移而改进的“任何其它技术或技术领域”的改进。这至少因为现有技术系统在没有人工编码或由人类开发人员进行的开发的情况下无法改进比现有技术有所改进。
60.本公开涉及至少因为本公开描述了应用人工智能(例如,机器学习模型)来设计具有所需功能性的蛋白质的用途而对其它技术或技术领域的改进。
61.本公开包括使用特定机器或通过使用特定机器应用本文的特征,该特定机器例如为用于测量与由本文所述的基于人工智能的系统和方法鉴定的候选或设计蛋白质对应的细胞荧光的微流体设备。
62.本公开包括实现特定物品向不同的状态或事物的转化或还原,例如,将如由本文所述的基于人工智能的系统和方法鉴定的候选或设计蛋白质转化或还原为可以用来生成、生产或以其它方式催化最终产品的细胞。
63.本公开包括除了在该领域中众所周知的、例行的、常规的活动之外的具体特征,或添加将权利要求限制于特定有用的应用(例如,包括设计具有所需功能性的蛋白质的系统和方法,例如用于开发、制造或创建现实世界的产品)的非常规步骤。
64.对于本领域的普通技术人员来说,根据以下对已经通过说明的方式示出和描述的优选实施例的描述,优点将变得更加明显。如将认识到的,本发明实施例可以具有其它和不
同的实施例,并且可以在各个方面对其细节进行修改。因此,附图和说明书将在本质上被视为是说明性的而非限制性的。
具体实施方式
65.本文所述的方法使用数据驱动和基于进化的方法,其利用统计基因组学、机器学习和人工智能技术来学习将氨基酸序列与蛋白质结构、功能和进化性相关联的隐式模式,从而克服以前的蛋白质设计方法的限制和挑战。在一个实施方案中,期望蛋白质通过首先基于训练数据集(例如,同源蛋白质的数据集)中蛋白质的序列信息训练机器学习模型并使用训练过的机器学习模型生成候选氨基酸序列而具有所需的功能性。
66.接下来,执行迭代过程以选择甚至更好的候选氨基酸序列。这个迭代过程包括为候选氨基酸序列产生蛋白质并测定它们以测量它们的功能性。使用来自测定的测量值,生成功能性前景(functionality landscape)(例如,基于功能性的模型)以预测哪些候选序列最有可能表现出所需的功能性。使用基于功能性前景的适合度函数以及机器学习可以确定具有比第一次迭代更好的功能性的新候选序列。然后重复该迭代过程,每次迭代产生比前一次迭代更好的新候选序列,直到达到停止标准并且将最终优化的氨基酸序列输出为设计的蛋白质。例如,图1a中展示了此迭代过程。
67.本文所述的方法使用由序列本身提供的信息(即,基于序列的模型)和通过使用测量蛋白质功能性的测定产生和评估蛋白质而提供的功能性信息(即,基于功能性的模型)。基于序列的模型与基于功能性的模型的组合被称为蛋白质模型。
68.基于序列的模型通常是接收来自蛋白质集合的氨基酸序列作为训练数据的一种无监督机器学习模型。因此,基于序列的模型被称为机器学习模型、无监督模型、基于氨基酸序列的模型或它们的任何组合。
69.通常,基于功能性的模型是监督模型,因为其使用来自另外测量(即监督)的信息生成/训练。基于功能性的模型通常表达为适合度函数和/或功能性前景。例如,当在设计过程中使用多目标多维度优化时,功能性前景是在针对所需功能性进行设计时鉴定哪些氨基酸序列是好的候选者的适合度函数中的成分之一。因此,基于功能性的模型可以被称为监督模型、适合度函数、功能性前景、基于功能性的模型、功能性模型或它们的任何组合。基于功能性的模型是通过机器学习生成的,并且可以是机器学习模型。然而,如本文所用的术语“机器学习模型”通常是指基于序列的模型,除非上下文另有明确说明。
70.如上所论述,蛋白质设计的目标是鉴定具有某些所需特性的新蛋白质。这可以被视为一个优化问题,其中执行搜索以获得最大化给定的定量需要的蛋白质。然而,蛋白质空间中的优化极具挑战性,因为搜索空间很大、是离散且非结构化的(例如,在数据挖掘的上下文中,氨基酸序列被归类为非结构化数据,具有分类所带来的所有挑战)。制备和测试新蛋白质既昂贵又耗时,而且潜在候选物的数量非常庞大。
71.例如,长度为n的可能氨基酸序列的数量为20n,并且该序列空间的大部分被非折叠、非功能的分子占据。定向进化提供了一种通过多轮突变和功能选择在该空间中搜索结构化功能分子的方法,但仅代表对特定自然序列周围的序列空间的非常局部的探索。因此,需要正式的规则来克服定向进化的这些限制,以便指导可以探索可能功能序列的更巨大组合复杂性的搜索。
72.一套规则来自原子间基本力的物理模型-这是基于物理的设计。但是这些方法受到以下限制:(i)力场的不准确性、(ii)施加特定约束来管理搜索的组合复杂性以及(iii)全局优化序列是最佳的原理。因此,即使通过基于物理的设计生成的序列可以形成高度稳定的结构,但迄今为止的证据是在没有定向进化的情况下,此类序列的功能性能很差。
73.与基于物理的设计和定向进化方法相比,本文所述的方法使用进化数据驱动的统计模型。这些类型的模型提供了一种获得搜索序列空间的规则的独特方法,其不依赖于潜在物理力的知识或折叠、功能或进化机制的知识。此外,这些模型不需要三维结构作为起点。相反,这些模型捕获限定蛋白质序列自然集合的统计约束的模式,并且通过这样做间接地捕获蛋白质折叠和功能的物理特性。因此,本文所述的方法不是为了稳定性而全局优化蛋白质,而是优化自然界通过进化历史选择的任何约束,使得能够比以前的方法更巨大的产量和深度对序列空间搜索功能蛋白质。
74.为了解决以前的蛋白质设计方法的上述挑战,本文所述的系统和方法使用数据驱动的基于进化的迭代方法来针对所需蛋白质/酶功能性鉴定/选择最有希望的氨基酸序列。
75.这种方法的数据驱动方面部分源于使用数据来训练模型的组合,该模型包括基于序列的模型和基于功能性的模型。基于序列的模型(也称为无监督模型)可以是被训练以表达从训练数据集(例如,同源蛋白质的多序列比对(msa))中学习到的统计特性和隐式模式的机器学习模型。基于功能性的模型(也称为监督模型)并入了从合成蛋白质的测定中测量的反馈,这些合成蛋白质是基于在先前迭代中被鉴定为有希望的候选者的氨基酸序列。设计这些测定以提供有关所需蛋白质功能性的标记。例如,所需的功能性(例如,结合、变构性或催化)可以与各种特定测定中的生物体生长速率、基因表达或光学特性(诸如吸光度或荧光)的变化定量地相关。
76.这种方法的基于进化的方面部分源于使用迭代反馈回路来鉴定最佳性能氨基酸序列的趋势,以根据趋势(即,计算机模拟诱变)对改进的氨基酸序列进行选择。
77.此外,在某些实施方式中,训练数据集将由同源物组成。因此,通过基于序列的模型学习的隐式模式将包括通过生物进化随时间推移学习的所需功能性的序列模式。
78.因此,本文所述的方法解决了通过使用数据驱动的蛋白质计算模型生成候选氨基酸序列、然后在实验室中使用定量测定物理地产生和评估候选蛋白质、并且最终使用来自这些测定的信息来改进计算模型以生成在下一次迭代中针对所需功能性进一步优化的候选氨基酸序列的迭代循环来快速地鉴定可能表现出所需功能性的合成蛋白质的挑战。因此,建立了一个反馈回路以针对所需的功能性迭代优化氨基酸序列。
79.特别地,蛋白质模型可以分为两个部分:监督模型和无监督模型。在迭代循环的第一次迭代中,可以仅使用无监督模型来确定初始候选氨基酸序列。然后在随后的迭代中,监督模型和无监督模型两者都可以用于在后续迭代中生成候选氨基酸序列。
80.现在参考附图,其中相同的附图标记在几个视图中表示相同或对应的部分,图1a和1b显示了使用序列模型和功能性模型的组合来设计合成蛋白质的方法10的流程图。
81.在方法10的过程22中,使用氨基酸序列的训练数据集来训练基于序列的模型(也称为无监督机器学习模型或更简单地称为无监督模型),以生成具有从训练数据集中学习的相似统计结构和/或模式的新氨基酸序列。一般地说,本文所述的方法适用于任何序列定义的分子,包括生物分子,而不仅仅是蛋白质。例如,可以设计核酸诸如信使rna或微小rna
以具有所需的功能性。在这种情况下,候选序列将是核苷酸序列而不是氨基酸序列;否则方法10将与本领域普通技术人员将理解的相同,仅具有较小的简明的变化。作为另一个实例,可以使用本文所述的方法设计由单体序列定义的聚合物以具有所需的功能性。具体而言,使用蛋白质设计的非限制性实例来展示本文所述的方法和系统,但本文所述的方法和系统是通用的并且包括所有序列定义的分子的设计。
82.在方法10的过程32中,对于在过程22中确定的候选序列,在实验室中物理地产生序列定义的分子(例如,蛋白质)。
83.在方法10的过程42中,测定序列定义的分子以测量它们表现出多少所需的功能性。然后将这些测量值并入到功能性模型中,然后在下一次迭代中的过程22中使用该功能性模型以基于从过程42产生的测量反馈来选择新的、更好的候选者。
84.随着时间的推移,方法10将集中于满足预定义的停止标准(例如,与超过预定义阈值的候选序列对应的所需功能性程度,或者所需功能性从迭代到迭代的增加速率已经减慢到低于另一个预定义阈值)的候选序列。然后,方法10将输出一个或多个优化的候选序列作为设计蛋白质。如本文所用,术语“候选蛋白质”和“设计蛋白质”可以互换使用,其中设计蛋白质指示对于本文所述的系统和/或方法的给定迭代(例如,包括如本文所述的迭代循环和/或机器学习模型的迭代)的候选蛋白质输出。
85.回到过程22,在本文提供的非限制性实例中,将序列模型展示为受限玻尔兹曼机(rbm)、变分自编码器(vae)、生成对抗网络(gan)、统计耦合分析(sca)和直接耦合分析(dca)。
86.前三种类型的序列模型(即rbm、vae和gan)是人工神经网络(ann)的类型,并且通常被称为“生成方法”,因为它们将在ann的可见节点处的输入映射到与可见层相比维度减少的节点隐藏层,从而提供一个其中信息被压缩并且隐式模式被捕获的信息瓶颈。此节点隐藏层定义了一个潜在空间,并且从可见层映射到隐藏空间在本文中被称为“编码”,其中将潜在空间中的点映射回氨基酸序列的原始空间的逆向过程在本文中被称为“解码”或“生成”。由压缩到减小的子空间所产生的学习模式会导致对潜在空间中的点的随机选择,然后解码到点,从而产生具有与训练数据集中那些模式相似的模式的氨基酸序列。
87.例如,使用面孔训练图像训练的vae可以用于生成可识别为面孔的图像,尽管与训练图像中的那些面孔不同的面孔(即,vae学习面孔大体的特征,并且然后可以用于生成具有学习特征的新的面孔图像)。如果希望进一步训练vae不仅识别面孔而且识别具有所需特征的面孔(例如,美丽的面孔),用户可以浏览由vae生成的面孔以根据所需特征标记图形。然后,可以通过使用所需特征的标记来学习美丽面孔或其它一些所需特征的模式来执行监督学习。例如,仅使用美丽面孔训练的vae将学习美丽面孔的模式。类似的原理可以应用于学习特定类型蛋白质(例如,同源蛋白质)的氨基酸序列模式以生成新的候选者,然后通过监督学习技术进一步学习表现出所需功能性的候选氨基酸序列的子集。
88.回到过程22的无监督序列模型,最后两种类型的模型(即sca和dca)通常被称为“统计方法”。这些统计方法也生成候选氨基酸序列,但这样做是通过学习训练数据集中的氨基酸序列的统计特性(例如,一阶和二阶统计量),然后选择符合所学习的统计模型的候选氨基酸序列。也就是说,与生成方法一样,统计方法生成候选氨基酸序列,但统计方法不使用神经网络在序列空间和潜在空间之间进行映射。相反,这些统计方法在序列空间的域
内运行,以学习序列空间(类似于但不同于生成方法中的潜在空间)中可能产生与训练数据集中的那些蛋白质类似的蛋白质的子空间的模式。
89.也就是说,无监督序列模型通过将搜索空间从所有可能的候选氨基酸序列的“绝大多数”数量限制到可能表现出所需功能性/特征的小得多且更易于管理的序列子集来缩小搜索范围。然后,从此子集中选择几个氨基酸序列(例如,大约1,000个)作为候选氨基酸序列以在实验室中产生并进行评估以提供用于监督学习的测量数据/值。应当理解,待产生的所选择的候选氨基酸序列可以包含各种数量和计数,包括作为非限制性实例至少100个氨基酸序列、至少500个氨基酸序列、至少1000个氨基酸序列、至少1500个氨基酸序列等。
90.现在考虑过程42中的监督功能性模型,在某些实施方式中,监督模型可以被认为是潜在空间上的功能性前景,其中峰对应于潜在空间中可能产生表现出更多所需功能性的候选氨基酸序列的区域,并且谷不太可能产生好的候选氨基酸序列。例如,通过对测量值进行回归分析来生成此功能性图景,所述测量值是通过对候选氨基酸序列进行测定以测量所需功能性(例如催化、结合或变构性)而生成的。在本文提供的非限制性实例中,将监督功能性模型展示为通过使用多变量线性回归、支持向量回归(svr)、高斯过程回归(gpr)、随机森林(rf)、决策树(dt)或人工神经网络(ann)将测量值与功能性前景化拟合而生成。例如,高斯过程(gp)在以下文献中进行了论述:c.e.rasmussen和c.k.i.williams,“gaussian processes for machine learning,”the mit press,2006,isbn 026218253x(也可从www.gaussianprocess.org/gpm1获得),所述文献通过引用整体并入。
91.监督模型不仅仅限于考虑功能性,还可以考虑可以增加候选氨基酸序列成功的可能性的其它因素,诸如相似性和稳定性。考虑到基于物理的建模,诸如使用rosseta commons软件套件的数值计算,可以提供稳定性的标记。例如,基于数值计算预测以保留自然折叠的自然结构的序列更有可能具有功能。
92.因此,监督模型可以扩展到考虑其它因素,包括稳定性和相似性。例如,可以在潜在空间中定义稳定性前景,并且可以在潜在空间中定义相似性前景。然后可以使用多维分析确定帕累托前沿面(pareto frontier)(即,(i)功能性、(ii)相似性和(iii)稳定性的多维空间中的凸包),并且可以从位于帕累托前沿面上的潜在空间中的点中选择候选氨基酸序列。
93.力法概述
94.图1b显示了根据一个非限制性实施方案的方法10的更详细流程图。
95.在过程22中,将训练数据集15用于训练氨基酸序列模型。作为非限制性实例,在多区段比对(msa)中n=1388个序列(其中每个序列的长度l为245)的训练数据集可以用作训练数据集15。
96.在过程22的步骤20中,序列模型生成候选氨基酸序列25。如图1c和1d示意性地示出,序列模型具有压缩阶段和生成阶段。模型的压缩阶段也可以被称为编码(例如,编码是从氨基酸序列空间映射到潜在空间),并且模型的生成阶段也可以被称为解码(例如,解码是从潜在空间映射到氨基酸序列空间)。
97.在过程22之后,在过程32的步骤30中,合成基因以编码候选序列25,并且从合成的基因产生候选蛋白质35。在一些实施方式中,可以使用虚拟筛选来加速此搜索。可以使用基于学习代理模型的第一性原理模拟或统计预测来测定包含数十亿到数亿个候选者的虚拟
文库,并且只选择最有希望的线索并进行实验测试。
98.在过程42的步骤40中,进行测定以评估候选蛋白质表现出所需功能性的程度。在步骤50中可以考虑从这些测定获得的指示所需功能性的测量值45以确定是否已经满足停止标准。如果达到停止标准,则将优化的氨基酸序列输出为设计蛋白质55。如果不是,方法10继续到步骤60。
99.在图1d中,将过程22展示为计算建模,并且将步骤32展示为基因合成和蛋白质产生,随后是高通量功能筛选。将高通量功能筛选的测量值反馈到计算模型中,除非高通量功能筛选表明已经满足蛋白质设计目标。在这种情况下,该方法是完整的,并且方法10输出具有所需特征的设计蛋白质。在图1d的示例性实施方式中,使用微流体设备展示了高通量功能筛选,该微流体设备测量含有各个候选蛋白质的细胞的荧光,并且根据所测量的荧光将细胞引导或提供给不同的仓。然而,应当理解的是,替代性方法和系统(例如,替代的或不同于微流体设备)也可以用于执行高通量功能筛选以确定如本文所述的测量值。在此实施方式中测量的荧光被调整为与定义设计目标的蛋白质特性成比例。因此,筛选产生数据用于计算模型的迭代优化。
100.在过程42的步骤60中,基于测量值生成功能性模型。
101.设计或候选蛋白质
102.尽管本公开已经展示了本文所述的方法和系统生成新的酶的用途,但本发明的系统和方法不限于具有特定类型的活性或结构的蛋白质(例如,诸如本文所述的设计或候选蛋白质)。实际上,在各个方面,蛋白质或以其它方式设计的或候选蛋白质可以是抗体、酶、激素、细胞因子、生长因子、凝血因子、抗凝因子、白蛋白、抗原、佐剂、转录因子或细胞受体。实际上,本文所述的系统和方法可用于例如生成或鉴定表现出与抗体、酶、激素、细胞因子、生长因子、凝血因子、抗凝因子、白蛋白、抗原、佐剂或细胞受体一致的生物活性的新蛋白质。此外,本文所述的候选蛋白质可以用于各种应用或功能。举例来说,候选蛋白质可以用于选择性地结合一种或多种其它分子。另外地或可替代地,作为另外的实例,可以提供候选蛋白质以催化一种或多种化学反应。另外地或可替代地,作为另外的实例,可以提供候选蛋白质用于远程信号传导(例如,变构性包括生物大分子(主要是蛋白质)通过其将在一个位点的结合作用传递到另一个位点(通常是远端的功能位点)、从而允许调节活性的过程)。
103.细胞因子包括但不限于趋化因子、干扰素、白细胞介素、淋巴因子和肿瘤坏死因子。还考虑了细胞受体,诸如细胞因子受体。细胞因子和细胞受体的实例包括但不限于肿瘤坏死因子α和β及其受体;脂蛋白;秋水仙碱;促肾上腺皮质激素;加压素;生长抑素;赖氨酸加压素;促胰酶素;亮丙瑞林;α-1-抗胰蛋白酶;心钠素;凝血酶;脑啡肽酶;rantes(受激活调节正常t细胞表达和分泌因子);人巨噬细胞炎症蛋白(mip-1-α);细胞决定蛋白,诸如cd-3、cd-4、cd-8和cd-19;促红细胞生成素;干扰素-α、-β、-γ、-λ;集落刺激因子(csf),例如m-csf、gm-csf和g-csf;il-1至il-10;t细胞受体;和前列腺素。
104.激素的实例包括但不限于抗利尿激素(adh)、催产素、生长激素(gh)、催乳素、生长激素释放激素(ghrh)、促甲状腺激素(tsh)、促甲状腺素释放激素(trh)、促肾上腺皮质激素(acth)、促卵泡激素(fsh)、促黄体生成素(lh)、促黄体生成素释放激素(lhrh)、甲状腺素、降钙素、甲状旁腺激素、醛固酮、皮质醇、肾上腺素、胰高血糖素、胰岛素、雌激素、黄体酮和睾酮。
105.生长因子的实例包括例如血管内皮生长因子(vegf)、神经生长因子(ngf)、血小板衍生生长因子(pdgf)、成纤维细胞生长因子(fgf)、表皮生长因子(egf)、转化生长因子(tgf)、骨形态发生蛋白(bmp)和胰岛素样生长因子i和ii(igf-i和igf-ii)。
106.凝血因子(clotting factor或coagulation factor)的实例包括因子i、因子ii、因子iii、因子v、因子vi、因子vii、因子viii、因子viiic、因子ix、因子x、因子xi、因子xii、因子xiii、血管性血友病因子、前激肽释放酶、肝素辅因子ii、抗凝血酶iii和纤连蛋白。
107.酶的实例包括但不限于血管收缩素转化酶、链激酶、l-天冬酰胺酶等。酶的其它实例包括例如硝酸还原酶(nadh)、过氧化氢酶、过氧化物酶、固氮酶、磷酸酶(例如酸性/碱性磷酸酶)、磷酸二酯酶i、无机二磷酸酶(焦磷酸酶)、脱氢酶、硫酸酯酶、芳基硫酸酯酶、硫代硫酸酯硫转移酶、l-天冬酰胺酶l-谷氨酰胺酶、β-葡糖苷酶、芳基酰基酰胺酶、酰胺酶、转化酶、木聚糖酶、纤维素、脲酶、植酸酶、糖酶、淀粉酶(α-淀粉酶/β-淀粉酶)、阿拉伯木聚糖酶、β-葡聚糖酶、α-半乳糖苷酶、β-甘露聚糖酶、果胶酶、非淀粉多糖降解酶、内切蛋白酶、外切蛋白酶、脂肪酶、纤维素酶、氧化还原酶、连接酶、合成酶(例如氨酰基转移rna合成酶;甘氨酰-trna合成酶)、转移酶、水解酶、裂解酶(例如脱羧酶、脱水酶、脱氨酶、醛缩酶)、异构酶(例如磷酸丙糖异构酶)和胰蛋白酶。酶的另外实例包括过氧化氢酶(例如耐碱过氧化氢酶)、碱性淀粉酶、果胶酶、氧化酶、漆酶、过氧化酶、木聚糖酶、甘露聚糖酶、酰基转移酶、碱性蛋白酶(alcalase)、烷基硫酸酯酶、纤维素分解酶、纤维二糖水解酶、纤维二糖酶、外切-1,4-β-d-葡糖苷酶、氯过氧化物酶、几丁质酶、氰化物酶(cyanidase)、氰化物水解酶、1-半乳糖内酯氧化酶、木质素过氧化物酶、溶菌酶、锰过氧化物酶、胞壁质酶、对硫磷水解酶、果胶酯酶、过氧化物酶和酪氨酸酶。酶的另外实例包括核酸酶(例如,核酸内切酶,诸如锌指核酸酶、转录激活因子样效应物核酸酶、cas核酸酶、工程化大范围核酸酶)。
108.上文的公开内容参考了各种蛋白质类别和亚类中的示例性蛋白质(例如,孕酮作为激素的一个实例)。应当理解,本文所述的系统和方法可以生成在氨基酸序列方面与上文列出的特定蛋白质不同但具有与参考蛋白质相似、等效或改进的活性或其它所需生物学特征的一种蛋白质。
109.蛋白质产生
110.使用多种生物技术工具和方法获得重组蛋白。例如,将包含编码所关注的蛋白质的核酸的表达载体引入宿主细胞中,在允许蛋白质产生的条件下培养该宿主细胞,并且通过例如从培养基中纯化分泌的蛋白质或者裂解细胞以释放细胞内蛋白质并从裂解物中收集所关注的蛋白质来收集蛋白质。
111.将核酸引入宿主细胞中的方法在本领域中是熟知的并且描述于例如cohen等人(1972)proc.natl.acad.sci.usa 69,2110;sambrook等人(2001)molecular cloning,a laboratory manual,第3补充版本cold spring harbor laboratory,cold spring harbor,n.y.;以及sherman等人(1986)methods in yeast genetics,a laboratory manual,cold spring harbor,n.y.
112.通常将编码蛋白质的核酸包装在具有促进蛋白质产生和任选地促进进入宿主细胞中的调控序列的表达载体中。在此上下文中,经常使用质粒或病毒载体。
113.多种宿主细胞适合用于重组蛋白生产。例如,经常将哺乳动物细胞用于生产治疗剂。适合用于重组蛋白生产的哺乳动物宿主细胞的非限制性实例包括但不限于中国仓鼠卵
巢细胞(cho);通过sv40转化的猴肾cv1细胞(cos细胞、cos-7,atcc crl-1651);人胚胎肾细胞(例如,293细胞);小仓鼠肾细胞(bhk,atcc ccl-10);猴肾细胞(cv1、atcc ccl-70);非洲绿猴肾细胞(vero-76,atcc crl-1587;vero,atcc ccl-81);小鼠塞尔托利细胞;人宫颈癌细胞(hela,atcc ccl-2);犬肾细胞(mdck,atcc ccl-34);人肺细胞(w138,atcc ccl-75);人肝癌细胞(hep-g2,hb 8065);和小鼠乳腺肿瘤细胞(mmt 060562,atcc ccl-51)。细菌细胞(革兰氏阳性或革兰氏阴性)是适合用于蛋白质生产的原核宿主细胞。酵母细胞也适合用于重组蛋白生产。
114.最终产品
115.图1e显示了展示由本文所述的系统和/或方法产生的示例性设计或候选蛋白质1e10(例如,设计蛋白质或候选蛋白质1e20、1e22、1e24和/或1e26)的图,其中此类蛋白质可以以一种或多种替代形式提供(例如,作为最终产品)和/或可以应用于各种行业(例如,行业1e30、1e32、1e34、1e36和/或1e38);
116.由本文所述的系统和/或方法产生的蛋白质(例如,候选蛋白质)可以应用于许多行业中的任一个行业,包括生物制药(例如,治疗学和诊断学)1e34、农业(例如,植物和牲畜)1e32、兽医、工业生物技术(例如,生物催化剂)1e30、环境保护和修复1e38和能源1e36。
117.如图1e所示,由本文所述的系统和/或方法产生的蛋白质(例如,设计或候选蛋白质)可以开发或产生用于或以其它方式用于各种行业(例如,1e30、1e32、1e34、1e36和/或1e38)并且以如下数量的一种或多种替代形式提供给最终用户:(1)作为纯化蛋白质1e20,以溶液、作为冻干粉末或其它通常用于传输活性蛋白质分子的形式供应,(2)作为编码在质粒、病毒或粘粒载体中克隆的设计蛋白质的合成基因文库1e22,(3)作为在工程化微生物或其它宿主菌株1e24中表达的基因,和/或(4)作为克隆到基因治疗载体1e26中的基因。作为非限制性实例,如图1e所示,纯化蛋白质1e20可以用于或提供给生物催化1e30、农业1e32和/或生物制药1e34的任何一个或多个行业,用于开发、制造或创造最终产品。作为另外的非限制性实例,合成基因文库1e22可以用于或提供给生物催化1e30、农业1e32、生物制药1e34和/或环境保护与修复1e38的任何一个或多个行业,用于开发、制造或创造最终产品。作为另外的非限制性实例,工程化微生物或其它宿主菌株1e24可以用于或提供给生物催化1e30、农业1e32、生物制药1e34、能源1e36和/或环境保护与修复1e38的任何一个或多个行业,用于开发、制造或创造最终产品。作为另外的非限制性实例,基因疗法载体1e26可以用于或提供给农业1e32和/或生物制药1e34的任何一个或多个行业,用于开发、制造或创造最终产品。本文以下部分提供了关于这些不同方法用于生成、制造、递送、使用和/或以其它方式输出用于开发、制造、创造和/或生成如本文所述的最终产品的工程化蛋白质溶液(例如,设计或候选蛋白质)的另外细节和非限制性实例。
118.纯化的蛋白质
119.本文所述的方法和/或系统的输出可以作为纯化的蛋白质产品(例如,纯化的蛋白质1e20)提供给最终用户。可以以多种方式中的任一种方式来表达和纯化蛋白质。蛋白质表达通常涉及但不限于将编码所关注的蛋白质的基因引入(转化到)宿主微生物中,转化菌株生长到指数期,和使用从许多标准启动子序列之一激活转录的小分子诱导基因表达。然后诱导的培养物生长至饱和期并且收获用于蛋白质纯化。蛋白质纯化技术的实例包括但不限于离心、过滤(例如,切向流微滤)、沉淀/絮凝、色谱法(例如,离子交换色谱、固定化金属螯
合物色谱和/或疏水相互作用色谱)、嗜硫吸附、基于亲和力的纯化方法、结晶等。可以将纯化的蛋白质配制成适合用于在例如治疗、农业和/或工业领域中运输、存储和最终目标用途的液体组合物或冻干(或干燥)组合物。
120.基因文库
121.本文所述的方法和/或系统的输出可以作为基因或基因文库(例如,基因或基因文库1e22)提供。编码设计蛋白质的基因可以通过使用标准密码子表将氨基酸序列反向翻译成dna序列来产生,然后可以将其克隆到各种宿主质粒或病毒载体之一中进行繁殖和扩增。然后,最终用户可以在药品、农产品、生物催化和环境修复中的各种定制制造过程中使用所产生的文库。将基因文库制备为以干燥(或冻干)形式供应的纯dna用于存储和/或运输是标准做法。
122.工程化微生物
123.本文所述的方法和/或系统的输出可以作为通过标准转基因方法整合到宿主生物体的染色体中的基因提供。在这种情况下,可以通过使用标准密码子表将氨基酸序列反向翻译成核酸序列来产生编码设计蛋白质的基因,并与适当的5
′
和3
′
核苷酸序列连接以确保期望的表达、调控和mrna稳定性,并且整合到宿主生物体基因组中。可以此类工程化宿主生物体(例如工程化菌株1e24)供应给最终用户用于许多标准应用之一中。这可能包括在大规模培养和生产设施中的生长、在多步骤工业生物合成途径中使用、或作为工业、农业、制药、环境或能源收获过程的工程化微生物群落的组分。
124.基因疗法载体
125.在各种实施方案中,本文所述的方法或系统的输出是具有所需活性的核酸(例如,其本身具有所需活性或其编码具有所需活性的肽或蛋白质)。在各个方面,将核酸并入表达载体(例如,载体1e26)中。“载体”或“表达载体”是包括用于引入到宿主细胞中的核酸(dna或rna)的任何类型的遗传构建体。在各个实施例中,表达载体是病毒载体,即包括全部或部分病毒基因组的病毒颗粒,其可以用作核酸递送媒剂。包括对所关注的基因产物进行编码的一个或多个外源核酸的病毒载体也被称为重组病毒载体。如本领域将理解的,在一些上下文中,术语“病毒载体”(和类似术语)可以用来指代没有病毒衣壳的载体基因组。在本公开的上下文中使用的病毒载体包含,例如,逆转录病毒载体、基于单纯性疱疹病毒(hsv)的载体、基于细小病毒的载体,例如,基于腺相关病毒(aav)的载体、aav腺病毒嵌合载体和基于腺病毒的载体。这些病毒载体中的任一种可以使用在以下中描述的标准重组dna技术来制备:例如sambrook等人,《分子克隆实验手册(molecular cloning,a laboratory manual)》,第2版,冷泉港出版社(cold spring harbor press),纽约州冷泉港(cold spring harbor,ny.),(1989);ausubel等人,《分子生物学实验指南(current protocols in molecular biology)》,格林出版协会(greene publishing associates)与约翰威立国际出版公司(john wiley&sons),纽约州纽约市(new york,n.y.),(1994);coen d.m,《病毒学中动物病毒的分子遗传学(molecular genetics of animal viruses in virology)》,第2版,b.n.fields(编辑),雷文出版社(ravenpress),纽约(1990)和其中引用的参考文献。
126.表达载体在工业中具有多种用途。例如,可以将表达载体提供给最终用户,例如用于基因疗法方法(即,涉及施用于患者以治疗或预防疾病或病症)中。最终用户可以使用表达载体来产生由核酸编码的所关注的蛋白质。
127.高斯过程回归(gpr)
128.用于生成功能性模型的一个非限制性实例将高斯过程回归(gpr)应用于来自测定的测量值。例如,可以对测量值执行gpr,如在以下文献中所论述:p.a.romero等人,“navigating the protein fitness landscape with gps,”pnas,第110卷,第e193-e201页(2012);c.n.bedbrook等人,“machine learning to design integral membrane channel rhodopsins for efficient eukaryotic expression and plasma membrane localization”plos comput biol.,第13卷,第e1005786页(2017);以及r.g.bombarelli等人,“automatic chemical design using a data-driven continuous representation of molecules,”acs cent.sci,第4卷,第268-276页(2018),所述文献中的每一篇都通过引用整体并入本文。针对与各个候选蛋白质对应的在潜在空间中的位置对测量值执行gpr,导致在潜在空间中的每个位置被分配一个平均值和一个标准变化(即,不确定性)。
129.通过gpr生成的平均值和标准变化前景可以用于根据不同但互补的目标来选择在潜在空间中的候选位置。在一方面,在处于开发模式时的目标是选择最有可能发挥最佳功能的候选位置(例如,在潜在空间上具有最大平均值的区域)。在另一方面,在处于探索模式时的目标是选择最能改善功能性模型的候选位置。这些位置可以是在潜在空间中具有最大不确定性的区域,因为这些区域中的更多样本可以最大程度地缩小不确定性并改善功能性模型的预测能力。探索模式和探索模式的目标是耦合的,因为改进功能性模型可以导致更好地预测潜在空间中的哪些点对应于最佳功能性。此外,在每次迭代期间选择了大量候选者,因此可以通过基于各个目标中的每一个目标或基于它们的组合选择点子集来靶向探索和探索目标。
130.作为位置的函数的平均值可以定义功能性前景。如下文更详细描述的,当处于开发模式时,功能性前景可以用于从前景内与所需功能性中的峰值对应的那些区域中选择那些候选序列。此外,当处于探索模式时,可以通过在这些区域中选择候选蛋白质来鉴定具有较大不确定性的区域进行探索,以更好地估计这些区域中表现出的所需功能性的程度。通过根据开发标准选择一些候选蛋白质并根据探索标准选择其它候选蛋白质,可以同时从事开发和探索两者。
131.此外,在步骤20中,来自先前迭代的候选序列25可以用于更新和精细化序列模型。例如,可以将来自先前迭代的候选序列25添加到训练数据中,并且可以使用扩展/更新的训练数据集进一步训练序列模型。更新序列模型可能会位移或扭曲潜在空间。因此,可以在计算功能性模型和适合度函数65之前更新序列模型。在某些实施方式中,适合度函数65是功能性前景。在其它实施方式中,适合度函数65包括与预测哪些氨基酸序列是有希望的的其它标记(例如,稳定性)组合的功能性前景。
132.在某些实施方式中,构建适合度前景以在蛋白质序列空间上映射适合度的峰和谷。例如,描述符(输入)是蛋白质序列,而适合度的读数(输出)是所需特征(即适合度)的度量。在某些实施方式中,输出可以是所需特征的多维度量。所需特征的这些度量可以包括但不限于活性、与现有自然序列的相似性和蛋白质稳定性。当蛋白质序列的适合度是多维的并且含有多个属性时,可以为适合度的每个成分构建单独的前景。
133.确定适合度的训练数据将来自对通过基因合成和表达产生的序列的实验测定。例如,实验测定可以测量功能活性,包括但不限于结合、催化活性、稳定性或其替代物(例如,
在使其与一种或多种功能活性成比例的环境条件下的生长速率)。此外,在一些实施方式中,计算建模还可以提供用于确定适合度的反馈,例如,序列的计算建模可以用于预测蛋白质稳定性。
134.使用对于适合度函数的输入域的降维潜在空间比使用域的序列空间更加有效。这是因为序列空间的大小和氨基酸相互作用的复杂性使得直接将蛋白质序列作为输入的监督回归模型的构建相当不恰当。因此,优选的是通过将投射到低维潜在空间中的每个序列的表示作为输入来执行回归。
135.可以使用但不限于多变量线性回归、非线性回归、支持向量回归(svr)、高斯过程回归(gpr)、随机森林(rf)和人工神经网络(ann)来拟合监督学习模型。
136.在某些实施方式中,将使用两层法(two tier approach)生成候选序列。在第一层中,基于序列的模型(例如,sca、dca、vae等)选择第一组序列,然后在第二层中,适合度模型基于适合度标准选择第一组序列的子集作为候选序列(即,与适合度前景峰值附近的点对应的序列)。也就是说,第一组序列根据定义适合度每个成分的适合度前景进行计算评估,并且在第一组序列生成的序列中,一个或多个序列被鉴定为向前传递进行实验合成和测定的最佳候选者。
137.潜在空间中的多目标/多维优化
138.在执行多目标优化的实施方式中,最佳序列被鉴定为沿着由上文论述的各种前景定义的适合度的所有成分(例如,相似性和稳定性)表现良好的那些序列。由于适合度的成分可能用作相矛盾的目标(例如,活性可能与稳定性成反比),因此不存在单一的最优解决方案(即单一的最佳序列),因此可以使用帕累托前沿面来鉴定最佳序列以解决多目标优化问题。
139.也就是说,通过将最佳序列的可能选择缩小到多维空间中的低维表面来解决多目标优化问题。换句话说,最佳序列将位于帕累托前沿面(也称为最优前沿面或有效前沿面)。此前沿面和位于其上的序列可以使用如下方法来鉴定,该方法选自但不限于标量化、夹层算法、法线边界相交(nbi)、修订的nbi(nbim)、法线约束(nc)、连续帕累托优化(spo)、定向搜索域(dsd)、非支配排序遗传算法ii(nsga-ii)、强度帕累托进化算法2(spea-2)、颗粒群优化和模拟退火。
140.例如,执行多目标优化问题的一种方式是通过确定帕累托前沿面同时最大化所有竞争的适合度成分。这本质上是一种开发搜索,因为其专注于寻找具有高适合度的候选者。在此实施方式中,适合度模型传递回归模型预测为具有高度适合度的第一序列的那些,并且阻止未被预测为具有高度适合度的第一序列的那些。
141.然而,如果高度的适合度是通过功能性/适合度模型选择的唯一标准,那么搜索空间的欠采样区域可能未被探索,因为这些欠采样区域的高不确定性可能会排除对高度的适合度的预测。因此,功能性/适合度模型还可以根据其中不是对多目标优化解析以最大化由各种回归前景定义的每个适合度成分,而是解析以鉴定在适合度预测模型中具有最高不确定性的那些序列的探索标准来选择候选者。换句话说,探索标准的目标是鉴定回归模型具有最大不确定性的序列。基于探索标准,将形成与具有最大不确定性的区域对应的第一组序列的序列作为候选序列传递给实验合成和测定。这种探索标准是期望的,因为当回归模型对这些序列的特性高度不确定时,收集这些序列的实验数据最能改善模型(即降低它们
的不确定性)。因此,探索对于提供另外的训练数据来再训练模型并增强其预测性能很重要。
142.通常,基于这些开发/探索标准明智地选择序列的方案被称为主动学习。也就是说,机器学习模型正在指导新实验数据的收集,以(i)将实验引导到最有希望的候选者,并且(ii)指导收集对模型再训练最有价值的新数据以提高模型的预测性能。通过这种方式,模型构建和实验合成在正反馈循环中运行。
143.在方法10的某些实施方式中,使用上文所述的多目标优化技术结合贝叶斯(bayesian)优化技术来解决主动学习问题,以利用包括但不限于改善的概率、预期改善、置信区间下/上限的获取函数来控制探索-开发权衡。
144.多次迭代优化
145.图2展示了在方法10的八次迭代中遍历的可能路径。区域210(1)表示长度为l的所有可能序列的集合200内的点的轨迹。实际上,对于候选序列,在方法10的第i次迭代中集合200可以比子集210(i)大许多数量级。因此,图2未按比例绘制。在第二次迭代中生成的候选序列的子集210(2)可以相对于第一次迭代的子集210(1)位移,并且每个后续子集210(i+1)可以相对于之前的子集210(i)位移。因此,随着迭代次数i增加,候选序列的子集210(i)探索更多的空间200并且朝着功能性前景中的峰演进,直到满足所需功能性的探索级别(sought after level)。
146.此外,方法10可以用于在新环境中实现所需的功能性。例如,可能存在在温度x下具有所需催化功能的已知酶,但需要在温度y下具有相同催化功能的新酶。为了设计这种新酶,可以选择一系列的中间温度x<a<b<c<d<y。然后从已知的酶及其同源物开始,可以使用方法10来设计第一组酶,其中在温度a下进行测定以测量所需的催化功能。然后,可以从第一组酶开始重复方法10,但是这次将测定保持在温度b以生成在温度b的环境中时表现出所需催化功能的第二组酶。分别在温度c、d和y下第三次、第四次和第五次重复这一步骤,直到最后一组酶在温度y下时表现出所需的催化功能。因此,方法10可以用于在新环境中(例如,在不同的温度、压力、光照条件下,在不同的ph下,或在溶液/环境中的不同化学/元素浓度下)实现特定的功能性。
147.机器学习的最新进展已经产生了强大的概率生成模型,该概率生成模型在对真实实例进行训练后能够产生真实的合成样本。此类模型通常还产生被建模数据的低维连续表示,从而允许插值或类比推理。如上文所论述,这些生成模型适用于蛋白质设计,例如,使用作为自编码器训练的一对深度网络将表示为氨基酸序列的蛋白质转换为连续向量表示。
148.监督学习和无监督学习
149.如本文的各种实施方案中所述,机器学习模型可以使用监督或无监督机器学习程序或算法来训练。机器学习程序或算法可以采用神经网络,其可以是卷积神经网络、深度学习神经网络或在所关注的特定区域中在两个或更多个特征或特征数据集中学习的组合学习模块或程序。机器学习程序或算法还可以包括自然语言处理、语义分析、自动推理、回归分析、支持向量机(svm)分析、决策树分析、随机森林分析、k-最近邻分析、朴素贝叶斯分析、聚类分析、强化学习和/或其它机器学习算法和/或技术。机器学习可能涉及鉴定和识别现有数据中的模式(诸如蛋白质氨基酸序列训练数据集中的候选蛋白质),以便促进对后续数据进行预测、分类或输出(例如,合成候选基因和产生与各个候选氨基酸序列对应的候选蛋
白质)。
150.可以基于示例性(例如,“训练数据”)输入或数据(其可以被称为“特征”和“标签”)创建和训练机器学习模型(诸如本文所述的那些模型),以便对新输入(诸如测试级别或生产级别的数据或输入)进行有效和可靠的预测。在监督机器学习中,可以向在服务器、计算装置或其它处理器上运行的机器学习程序提供示例性输入(例如,“特征”)及其相关的或观察到的输出(例如,“标签”),以便机器学习程序或算法确定或发现将此类输入(例如,“特征”)映射到输出(例如,标签)上的规则、关系、模式或其它机器学习“模型”,例如,通过确定和/或分配权重或其它度量给模型的各种特征类别。然后可以向此类规则、关系或其它模型提供后续输入,以便在服务器、计算装置或其它处理器上执行的模型基于发现的规则、关系或模型来预测、分类或输出预期的输出。
151.在无监督机器学习中,可能需要服务器、计算装置或其它处理器在未标记的示例性输入中找到其自身的结构,其中例如由服务器、计算装置或其它处理器执行多次训练迭代以训练多代模型直到生成令人满意的模型,例如,当给出测试级别或生产级别数据或输入时提供足够预测准确度的模型。本文的公开内容可以使用此类监督或无监督机器学习技术中的一种或两种。
152.图3a、图3b和图3c展示了方法10的另一个非限制性实施方式。例如,图3a、图3b和图3c使用作为vae或rbm的序列模型(即,无监督模型)的非限制性实例来展示。可以将方法10细分为两个部分:(i)无监督学习过程102和(ii)监督学习过程138。无监督学习过程102从训练数据集105开始,并且从其生成可能表现出给定的定量需要(例如,具有所需的功能性)的候选序列135。在无监督学习过程102中,机器学习模型115被训练以在可见变量(即蛋白质的氨基酸序列)和定义潜在空间的隐藏变量之间来回映射。机器学习模型115可以是生成模型。相对于氨基酸序列的维度,潜在空间具有减小的维度(例如,长度为n的氨基酸序列可以具有20n的维度以解释在n个残基的每个残基处的20个可能的氨基酸)。为了从较高维度空间映射到较低维度空间,机器学习模型115学习训练数据集中的各个氨基酸序列的残基之间的隐式模式和相关性,以更紧凑地编码信息。也就是说,从可见变量到隐藏变量相关性的映射压缩了信息,以使用降低维度更紧凑地表示该信息。因此,训练数据集中的氨基酸序列之间的模式和相关性在机器学习模型115中被隐式地学习和表达。
153.此外,可以将机器学习模型115用于生成新的氨基酸序列。当映射到潜在空间时,具有相似功能性的蛋白质集合可以定义点的聚簇。例如,此聚簇的平均值和方差可以用于定义多变量高斯分布,以表示该聚簇的概率密度函数(pdf)。通过随机选择这其中的点并且然后使用机器学习模型115将这些点映射回氨基酸序列,机器学习模型115可以为可能与用于定义该聚簇的原始蛋白质具有相似结构并因此具有相似功能性的新合成蛋白质生成候选序列135。
154.监督学习过程138从候选序列135开始,并且合成基因序列以产生候选蛋白质,该候选蛋白质具有候选序列135。然后评估这些候选蛋白质的特性。例如,可以测定候选蛋白质以测量它们表现出所需功能性(例如,需要)的程度。然后根据测量值142确定适合度函数145,并且开始迭代循环以通过以迭代的方式更新机器学习模型115、候选序列135、测量值142和适合度函数145直到达到一些预定义的停止标准来搜索更好的蛋白质。
155.关于所需的功能性,蛋白质表现出以下以各种组合可能有助于实现所需的设计目
标的特性:(i)折叠速率和产率(折叠速度和折叠概率),(ii)热力学稳定性(未折叠状态与折叠状态之间的自由能差异),(iii)结合亲和力(分离未结合状态和结合状态的自由能),(iv)结合特异性(所需底物和非所需底物之间的结合差异),(v)催化能力(分离酶-底物复合物和反应过渡态的自由能),(vi)变构性(蛋白质中氨基酸的远程通讯)和(vii)进化性(产生遗传变异的能力)。
156.如上文所论述,可以定制高通量测定以生成用于评估所需功能性的测量值。这里提供了与蛋白质的各个特性对应的各种测定类型实例。可以例如通过针对通过凝胶过滤色谱法随后的紧密度或环境敏感荧光团的荧光的测定来评估折叠速率和产率。可以例如使用差示扫描量热法或1h-15n hsqc nmr来测量热力学稳定性。可以通过荧光或滴定量热法(例如,使用液滴微流体)或细胞中的双杂合基因表达方法来测量结合。可以通过吸光度/荧光法(例如,使用液滴微流体)或通过存活率或生长速率测量来测量催化能力。变构性可以通过多种方法测量,其中包括通过细胞中的调节来测量变构性。在当前和新的功能两者的上下文中,可以通过比较氨基酸对深度突变扫描的敏感性来测量进化性。
157.在某些实施方式中,用于方法10的方法可以分解为两个部分:(i)蛋白质序列的低维潜在空间嵌入的无监督学习,和(ii)潜在空间内的功能性前景的监督学习。也就是说,方法10执行搜索以找到保留关于其序列和功能的关键信息的蛋白质序列的低维表示,并且从这些表示中预测具有甚至更好性能的新序列的可能功能性。
158.在方法10的步骤110中,使用训练数据集105训练机器学习模型115。在一个非限制性实例中,机器学习模型115是变分自编码器(vae)。在另一个非限制性实例中,机器学习模型115是受限玻尔兹曼机(rbm)。vae和rbm两者都是生成模型,并且其它生成模型可以用于机器学习模型115,如本领域普通技术人员将理解的。如上文所论述,除了vae和rbm之外,可以使用生成对抗网络(gan)、统计耦合分析(sca)或直接耦合分析(dca)来执行无监督学习。使用哪种无监督学习方法的决定可以基于对哪种方法在提供稳健的潜在空间表示和准确的生成性能方面提供最佳性能的经验评估。
159.统计耦合分析(sca)模型
160.在一种非限制性实施方式中,统计耦合分析(sca)模型可以用作基于序列的模型,训练数据用于计算由守恒加权相关矩阵(conservation-weighted correlation matrix)(例如,所有氨基酸对之间共同进化的sca矩阵)定义的sca模型,如在k.a.reynolds等人,“evolution-based design ofproteins,”methods in enzymology,第523卷,第213-235页(2013)中所描述和在o.rivoire等人,“evolution-based functional decomposition of proteins,”plos comput biol,第12卷,第e1004817页(2016)中所论述,所述文献中的每一篇都通过引用整体并入本文。也就是说,训练数据集的信息被sca模型压缩成单个成对相关矩阵。此外,sca矩阵的奇异值分解或特征值分解表明,大多数模式与采样噪声无法区分,而前几个模式(对应于潜在空间)捕获了统计学上显著的相关性。这些前几个模式定义了扇区,其包含一组或多组共同进化的氨基酸。基于sca的蛋白质设计可以通过从随机序列开始并进化(计算机模拟)合成序列的计算模拟来执行,所述合成序列受在sca共同进化矩阵中捕获的观察到的进化统计量的约束。
161.例如,基于sca的蛋白质设计使用metropolis monte carlo模拟退火(mcsa)算法来探索与氨基酸之间的应用约束集合一致的序列空间。mcsa算法是一种用于从任意状态开
validation partition)上的损失函数不再减小时通过提前停止来终止训练,以防止过度拟合。vae的数量在不同的训练/测试拆分上进行了优化。此外,潜在空间的维度被优化以选择潜在空间的大小,在该大小处验证损失不再随着维度的增加而减小。最佳vae通过其在训练中未使用的测试分区上的重建精度进行验证。
170.经训练的模型可以用于通过使用高斯随机数对潜在空间进行采样并将它们传递给解码器以将它们转化为蛋白质序列,从而有效地生成数百万个新序列。由于实验表明潜在空间编码自然界对可行蛋白质的规则,因此预期来自该潜在空间的样本也产生可行的蛋白质,其中一些不是通过自然选择产生的。
171.图4和图5显示了用作生成模型的vae的示意图。在图4中,vae包括编码器部分、解码器部分和自回归部分,如在以下文献中所论述:costello,z.和garcia martin,h,“how tohallucinate functional proteins,”预出版于https://arxiv.org/abs/1903(2019),该文献通过引用整体并入本文。在每个模块内部,阴影立方体各自代表一种层类型。阴影层表示具有呈残差网络(resnet)风格的跳跃连接的一维(1d)扩张卷积层。阴影层的暗度表示扩张的幅度。逐渐变深的阴影或其它交叉影线表示更大的扩张。这是在图4中使用的模式中完成的。末端层(例如,末端层400re1和400re2)表示1d卷积,其中输入的长度减半,跨步为2,并且通道加倍。转置的末端层(例如,末端层400ge1和400ge2)通过转置的一维(1d)跨步卷积指示末端层(例如,末端层400re1和400re2)的反向操作。
172.生成模型(诸如vae)产生具有与训练它们时相同的统计属性的数据。也就是说,当vae在其天然宿主中折叠的功能性蛋白质序列上进行训练时,vae应该产生可能与训练数据集中的那些蛋白质类似地折叠和发挥功能的蛋白质。因此,可以验证模型的行为方式与此假设一致。
173.vae被训练来重建它自己的输入。例如,vae首先将蛋白质序列编码为特征向量(即,潜在空间中的向量)。特征向量可以被认为是蛋白质序列中的重要信息的汇总。这些特征向量所在的向量空间通常被称为潜在空间。然后变分自编码器从该特征向量重建原始蛋白质序列。损失函数可以表示来自网络的输出与输入匹配的程度。如果vae是无损的,则输出将始终与输入匹配。然而,总的来说,vae有一些损失。
174.完全训练的vae可以在两种模式下使用:作为编码器或作为解码器。编码器可以用于获取蛋白质序列并找到其相关的特征向量。然后,此特征向量可以用于下游分类或回归任务。例如,可以确定给定蛋白质在给定其序列的情况下可能定位的位置。解码器可以用于通过从潜在空间中采样来生成可能折叠和发挥功能的任意序列。此外,可以选择潜在空间样本,使得序列也可能具有所需的表型。在本节的其余部分中,将详细描述模型设计及其用途。
175.如图5中所示,不同的蛋白质组(例如,“a”、“b”、“c”、“d”、“e”、“f”和“g”)定位并聚集到潜在空间内的不同区域。与氨基酸序列的离散空间相比,潜在空间还具有连续空间的优点。连续和数据驱动的蛋白质表示方法具有几个优点,如以下文献中所论述的:r.g
ó
mez-bombarelli等人,“automatic chemical design using a data-driven continuous representation of molecules,”acs cent.sci.第4卷,第268-276页(2018)以及s.sinai等人,“variational auto-encoding of protein sequences,”预出版于https://arxiv.org/abs/1712.03346(2018),所述两篇文献均整体并入本文,所述文献通过引用整
体并入本文。首先,手动指定的变异规则是不必要的,因为可以通过修改向量表示然后解码来自动地生成新的化合物。其次,使用从蛋白质表示映射到所需属性的可微模型,有可能使用基于梯度的优化在搜索蛋白质空间时进行更大的步骤。基于梯度的优化可以与贝叶斯推理方法相结合,以选择可能为全局最优提供信息的氨基酸序列。第三,数据驱动的表示可以利用大的蛋白质集合(例如,包括表现出所需功能性的蛋白质以及不表现出所需功能性的蛋白质)来自动地构建更大的隐式文库,然后使用表现出所需功能性的较小的蛋白质集合来建立从连续表示到所需特性的回归模型,该回归模型被并入适合度函数145)。因此,即使许多蛋白质具有未知特性,也可以使用大型蛋白质数据库来训练vae。
176.受限玻尔兹曼机(rbm)模型
177.执行无监督学习的机器学习模型的一个非限制性实例是rbm(在图3c中示意性地示出)。在编码方向,氨基酸序列作为输入应用到神经元节点的可见层,并且通过取输入到可见层的值的加权和的偏置s形函数来计算隐藏层。rbm是玻尔兹曼机的一种变体,其限制是它们的神经元必须形成一个二分图:来自两组单元(通常分别称为可见单元和隐藏单元)中的每一组的一对节点可能在它们之间具有对称连接;并且组内的节点之间没有连接。这种限制允许比可用于一般类别的玻尔兹曼机的更有效的训练算法,特别是基于梯度的对比散度算法。受限玻尔兹曼机也可以通过堆叠rbm在深度学习网络中用作深度信念网络,并且任选地使用梯度下降和反向传播对所产生的深度网络进行微调。
178.受限玻尔兹曼机被训练以最大化分配给某些训练集v(一个矩阵,其中的每一行都被视为一个可见向量v)的概率乘积,如以下公式所提供:
[0179][0180]
可替代地并且等效地,以最大化训练样本v的预期对数概率,可以使用以下公式,例如:
[0181][0182]
在上述公式中,p(v)=∑hp(v,h)=z-1
∑hexp(-e(v,h))是可见的布尔值向量在所有可能的隐藏层配置上求和的边际概率,其中z是一个配分函数,其对概率分布p(v,h)进行归一化,并且能量函数为e(v,h)=-a
tv‑‑bt
h-v
t
wh,其中w是与隐藏单元h和可见单元v的值之间的连接相关的权重矩阵,并且a足可见单元的偏置权重(偏移量),b足隐藏单元的偏置权重(偏移量)。
[0183]
可以训练rbm以使用对比散度(cd)优化节点之间的权重w。也就是说,在梯度下降过程中使用吉布斯采样(类似于在训练前馈神经网络时在这种过程中使用反向传播的方式)来计算权重更新。在单步对比散度过程中,执行以下步骤:(i)获取训练样本v,计算隐藏单元的概率并从该概率分布中采样隐藏激活向量h;(ii)计算v和h的外积并将其称为正梯度;(iii)从h中,对可见单元的重建v
′
进行采样,然后从中重新采样隐藏激活h
′
(吉布斯采样步骤);(iv)计算v
′
和h
′
的外积并将其称为负梯度;(v)使权重矩阵w的更新为正梯度减去负梯度,乘以某个学习率,例如,由以下公式提供:
[0184]
δw=∈(vh
t-v
′h′
t
)
[0185]
此外,步骤(vi)可以包括以类似方式更新偏置a和b,例如,由以下公式提供:
[0186]
δa=∈(v-v
′
)andδb=∈(h-h
′
)。
[0187]
隐藏层定义了潜在空间,并且通过选择隐藏层节点的值并应用rbm将隐藏层值解码为氨基酸序列来生成候选氨基酸序列作为候选序列。也可以使用这种方法的变化形式,如以下文献中所论述的:tubiana等人,“learning protein constitutive motifs from sequence data,”elife,第8卷,第e39397页(2019),所述文献通过引用整体并入本文。
[0188]
方法概述
[0189]
返回图3a,在方法10的步骤120中,在潜在空间内选择候选点。例如,k均值聚类可以用于鉴定潜在空间内可能与具有所需功能性/属性的蛋白质对应的区域/邻域。可替代地,可以执行统计分析以确定所需功能性/属性的概率密度函数(pdf)。然后可以使用随机数生成器在潜在空间内选择具有统计学上代表性的点样本。也可以使用其它方法来确定潜在空间内可能与具有所需功能性的蛋白质对应的点样本。
[0190]
在方法10的步骤130中,使用机器学习模型115来选择候选氨基酸序列。这些候选序列是基于它们与训练数据集中的那些序列的相似性来选择的。例如,训练数据集可以具有一个子集,该子集特别表现出所需的功能性,并且可以对该子集进行聚类以鉴定潜在空间的特定邻域。然后,可以基于所鉴定的邻域选择候选序列。例如,因为机器学习模型115是生成模型,所鉴定的邻域内或紧邻所鉴定的邻域的点可以映射到用作候选序列的氨基酸序列上。
[0191]
搜索性能更好的氨基酸序列可以具有探索和开发的竞争/互补目标。鉴于此,选择与那些被鉴定为高性能者的氨基酸序列有多少偏差/差异可能取决于在搜索的给定阶段是否更需要探索或开发。
[0192]
在方法10的步骤140中,合成基因序列,然后将其用于生成具有来自步骤130的候选氨基酸序列的蛋白质。然后测定/评估所生成的蛋白质以确定它们的功能性/属性。然后将表示所生成的蛋白质的功能性/属性的值传递到步骤150,在该步骤中生成适合度函数以指导对未来候选序列的选择。
[0193]
本文更详细地描述了合成和测定方法。
[0194]
在方法10的步骤150中,根据在步骤140中测量的值确定适合度函数。例如,可以通过在潜在空间中对测量值进行回归分析来确定适合度函数。在某些实施方式中,使用高斯过程回归从测量值生成适合度函数。本文讨论了用于确定适合度函数的方法的其它非限制性实例。
[0195]
在方法10的过程160中,候选序列的优化搜索使用迭代循环继续,如图2和图3所示。
[0196]
在过程160的步骤162中,使用在步骤130中或在过程160的循环的先前迭代中生成的候选序列来更新机器学习模型。通过扩展训练数据集中的氨基酸序列的数量,可以进一步训练机器学习模型115以精细化和改进机器学习模型115的性能。
[0197]
在过程160的步骤164中,基于机器学习模型115和适合度函数来选择新的候选序列。
[0198]
在过程160的步骤166中,合成基因序列,然后将其用于生成具有来自步骤164的新候选序列的蛋白质。然后评估所生成的蛋白质以测量代表其所需功能性的新值。例如,可以测定所生成的蛋白质以测量与功能性对应的需要。
[0199]
在过程160的步骤168中,可以评估各种停止标准以确定是否满足停止标准。例如,停止标准可以包括迭代次数是否超过或等于预定的最大迭代次数。另外地/可替代地,停止标准可以包括预定数量的候选序列的功能性是否超过预定的功能性阈值。另外地/可替代地,停止标准可以包括,从迭代到迭代,候选序列的功能性的改进率是否已经减慢或收敛,使得改进率已经下降到预定义的改进阈值以下。
[0200]
如果满足停止标准,则过程160进行到步骤172,其中最高功能蛋白质候选者的序列被储存和/或呈递给用户。否则,过程160进行到步骤170。
[0201]
在过程160的步骤170中,使用来自步骤170的新测量值来更新适合度函数。例如,可以使用对新测量值的回归分析来更新适合度函数。所执行的回归分析可以使用当前迭代和所有先前迭代的所有候选序列的所有测量值。
[0202]
可以改变步骤162、164、166、168和170的顺序而不偏离过程160的精神。例如,对是否满足停止标准的询问可以在除了步骤166和170之外的不同对的步骤之间执行。此外,在某些迭代中可以省略步骤162。例如,可以针对过程160中的循环的各个迭代更新适合度函数,并且机器学习方法115可以保持不变。通过从迭代到迭代精细化适合度函数,可以学习和改进作为潜在空间函数的所需功能性的前景,以更好地选择具有所需功能性的候选者。
[0203]
此外,用于选择候选者的参数可以在迭代之间变化。例如,在搜索的早期,相对于开发而言,探索可能更有利。然后,可以从更广泛的分布中选择候选者。此策略可能有助于避免陷入功能性前景的局部最大值。也就是说,功能性前景具有峰值和值,并且鼓励探索的全局优化方法可以避免迭代到作为局部最大值但不是全局最大值的峰值中的氨基酸序列。全局优化方法的一个实例是模拟退火。
[0204]
除了功能性前景之外的其它因素对于选择氨基酸序列的最佳候选者可能很重要。例如,这些因素可以包括相似性和稳定性。从潜在空间采样生成的数百万个可能序列中,最优选择方法将集中在最有可能具有所需特性的那些。实际上,迭代过程160执行计算机模拟诱变。也就是说,过程160执行对无监督模型生成的那些序列进行计算自然选择,以挑选预测稳定且功能强大的那些序列。这是通过向潜在空间解码生成的每个序列分配与其合意性相关联的分数来实现的。
[0205]
在某些实施方式中,候选序列的选择是使用包括三个成分的评分向量来执行的:(i)依据与已知自然序列的接近程度的相似性-与自然序列接近的新序列预期具有更高的机会保留功能,(ii)使用预测蛋白质结构的软件工具通过计算建模预测的稳定性-预测保留自然折叠的自然结构的序列可能更具功能性,以及(iii)通过实验测定测量的功能性(例如,来自步骤166的测量值)。可以对于每个预测序列以高通量的方式计算确定前两个分数(即相似性和稳定性)。在某些实施方式中,通过将监督回归模型拟合到先前已经合成和实验测定(例如,在步骤140或步骤166中)的所有蛋白质来近似确定第三个分数。监督回归模型可以被认为是潜在空间中的前景。可以使用但不限于多变量线性回归、支持向量回归(svr)、高斯过程回归(gpr)、随机森林(rf)和人工神经网络(ann)来拟合监督学习模型。通过沿着这三个度量对每个预测序列进行评分,可以对那些序列进行排序,并且可以选择预计最有希望的那些序列进行实验合成。
[0206]
例如,可以通过鉴定在该3d多维优化空间中的帕累托前沿面并且选择在该前沿面上的那些序列作为建议用于合成的序列来执行候选序列选择。在某些实施方式中,位于前
沿面上的序列子集可以通过将可调整的权重分配给三个优化标准来进一步精细化。例如,与已知自然序列的稳定性和同源性相关的高权重可以呈现更保守的候选集合,然而低权重允许离自然序列更远的更雄心勃勃的候选集合。也就是说,相似性和稳定性的这些低权重可能有利于探索。随着模型通过多次迭代变得更加准确,模型往往会更可靠,从而使得能够选择离自然序列更远的更雄心勃勃的序列。
[0207]
例如,蛋白质的目标可能是在非自然环境(例如,高温、高压)中表现出所需的功能性。在这种情况下,可以合理地假设蛋白质结构会偏离自然界在室温和压力下选择为最佳的结构。也就是说,更雄心勃勃的序列可能是有利的以便于使序列“以计算的方式进化”向非自然环境(例如,高温、高压)中的功能性,该功能性在工业应用中有价值但在自然界中将永远不会被选择,其在很大程度上在室温和压力下运行。
[0208]
此外,回归模型可以解释功能性预测中的不确定性。这些不确定性可以在开发-探索模式中使用,以便于平衡在合成由模型鉴定的最有希望的序列中的竞争利益与在合成序列以探索模型具有高不确定性和新实验数据将对改进模型贡献最大的搜索空间中的利益。
[0209]
过程160有利地使用反馈,以与机器学习模型115一起精细化和改进适合度函数的预测能力。例如,在步骤140和166中产生和测定的序列将被反馈到监督和无监督学习模型(例如,适合度函数和机器学习模型115)中。机器学习模型115可以学习蛋白质的更大多样性的更好的潜在空间表示。此外,适合度函数将成为更好的功能预测器。这样,计算模型在序列制造和测试的每一轮迭代中变得越来越强大。此外,可以在并入新的候选蛋白质集合的迭代过程中使用数据驱动的表示,以自动构建更大的隐式文库,然后使用较小的标记实例集合来构建从连续表示到所需属性的回归模型。
[0210]
基因合成
[0211]
关于步骤140和166中的基因合成,可以采用使用管合成的(高纯度)寡核苷酸和自动化细菌克隆技术合成候选氨基酸序列的基因序列的过程。然而,这个过程通常很昂贵。因此,需要改进的、更便宜的基因合成过程。
[0212]
一种较便宜的方法(例如,大规模生产约500bp长的基因,即数干个序列,摊销成本约为2美元/基因)使用带寡核苷酸条形码功能化的珠粒来将给定基因所需的所有寡核苷酸的拷贝分离到油包水乳液的单个液滴中,随后将重叠的寡核苷酸聚合酶循环组装(pca)成全长基因,如以下文献中所述:w.stemmer等人,“single-step assembly ofa gene and entire plasmid from large numbers of oligodeoxyribonucleotides,”gene.164(1995)49-53以及c.plesa等人,“multiplexed gene synthesis in emulsions for exploring protein functional landscapes,”science.359(2018)343-347,所述两篇文献均通过引用整体并入本文。尽管错误率很大(例如,约5%的正确氨基酸序列),但是这种方法可以以约2美元/基因的摊销成本大规模生产例如约500bp长的基因(即数千个序列)。此方法在本文中被称为珠粒和条形码方法(bead-and-barcode method)。
[0213]
另一种甚至更便宜的方法从较长的寡核苷酸开始并且使用单独的微型池(minipool),从而省略了珠粒和条形码方法的珠粒杂交的需要。此方法在本文中被称为微型池方法(minipool method)。在微型池方法中,预商业化的寡核苷酸以阵列形式合成,与先前可用的产物相比有两个显著的改进。首先,合成长度多达300nt的预商业化寡核苷酸。其次,合成预商业化寡核苷酸的错误率为约1∶1300。长度的这种增加(与以前可用的200nt
的长度相比)降低了组装反应的复杂性,因为每个寡核苷酸的60-80nt被用于对最终基因产物没有贡献的
‘
开销(overhead)’序列。因此,对于300-nt寡核苷酸,每个寡核苷酸可用的有效序列增加了75%(从约130nt到约230nt),从而允许将1kb基因跟与先前可用的预商业化寡核苷酸使用的相似数量的寡核苷酸组装制成500bp的基因。
[0214]
较低的错误率导致组装基因中的序列错误较少,因为寡核苷酸中的单碱基插入和缺失是序列错误的主要来源。此外,通过将寡核苷酸作为仅含有每个基因所需的寡核苷酸的单独
‘
微型池’提供,可以省略珠粒和条形码方法的珠粒杂交步骤,同时降低成本和复杂性。
[0215]
寡核苷酸合成中的错误不是随机分布的,而是与其序列相关的。例如,嘌呤碱基在合成期间比嘧啶更容易降解,并且由生长的寡核苷酸链形成紧凑的折叠结构会阻碍后续核苷酸的添加。遗传密码的灵活性(多个密码子编码相同的氨基酸,如图7中所示)应允许设计具有更高合成准确性的寡核苷酸。可以通过使用高通量(ht)测序测量递送的寡核苷酸的准确性,鉴定与性能不佳相关的序列模式并且更新寡核苷酸设计算法避免这些情况来指导选择使用哪个密码子来编码特定的氨基酸。
[0216]
关于聚合酶循环组装(pca)的优化,pca通过退火重叠序列然后聚合酶延伸将寡核苷酸组装成更大的基因(类似于聚合酶链反应(pcr)技术,但具有扮演引物的重叠序列)来生成/合成表达候选氨基酸序列的基因序列,如图6a和6b中所示的示意图所示。因此,重叠序列的设计对于成功组装非常重要,就像成功的pcr扩增需要好的引物一样。重叠序列与基因中的其它重叠序列正交,但也在相似的温度下彼此退火。基因的氨基酸序列限制了选择重叠序列的自由度,但确实提供了有限的自由度,如前一节中所述。此外,可以在寡核苷酸之间选择断点以便优化这两个参数(即退火温度和重叠序列之间的正交性)中的任一者以实现高效组装。
[0217]
遗传序列合成方法的变化在本文公开的方法的精神内。例如,基因合成可以使用连接酶链反应方法、热力学平衡内外合成、和通过连接的基因合成以及各种错误校正方法(例如,回嚼(chew back)、退火和修复)来进行。
[0218]
直接耦合分析(dca)模型
[0219]
如上所论述,可以使用不同类型的机器学习模型115(在某些实施方案中也称为无监督学习模型115)来执行方法10。现在,为无监督学习模型115提供了使用dca的非限制性实施方案。
[0220]
基于直接耦合分析(dca),图8a显示了一种受进化启发的蛋白质设计方法,该方法是一种最初设想用于预测蛋白质三维结构中氨基酸之间的接触的方法。通常,该算法从天然同源物的多序列比对开始,从中计算经验一阶和二阶统计量。这些量用于学习由对氨基酸(hi)和成对相互作用(j
ij
)的内在约束组成的最小统计模型。接下来,统计模型可以用于生成多得多的概括了自然统计量的人工序列,并且可以筛选出所需的活性。
[0221]
图12显示了细菌和真菌中莽草酸途径的一部分,导致芳香族氨基酸酪氨酸和苯丙氨酸的生物合成;分支酸变位酶(cm)的aroq家族在一个分支点运行。
[0222]
图8c显示了具有两个功能活性位点的二聚体(例如,条目800c1、800c2和800c3)大肠杆菌cm的原子结构;每个活性位点由两种原体(protomer)贡献的氨基酸构成。结合的底物类似物以洋红色棍键(stick bond)显示。
[0223]
起点是一个蛋白质家族的大而多样的多序列比对(msa),从中估计了观察到的所有氨基酸频率(f
ia
)和成对相关性-一阶统计量和二阶统计量。从这些数量中,推断出一个模型,其包含最佳地解释观察到的统计量的一组固有氨基酸倾向(字段hi)和最小的一组成对交互作用(耦合j
ij
)。该模型被定义为:
[0224]
p(σ1,
…
,σ
l
)~exp[-h(σ1,
…
,σ
l
)],
[0225]
在上述模型中,p是氨基酸序列(σ1,
…
,σ
l
)发生的概率,l是蛋白质的长度并且h(σ1,
…
,σ
l
)=∑
ihi
σi+∑
i<jjij
σiσj是为每个序列的可能性提供定量分数的统计能量(或哈密顿量)。较低的能量与较高的概率相关,允许我们通过蒙特卡洛采样来生成非自然序列库,然后可以筛选出所需的功能活性。如果成对相关性通常足以捕获蛋白质序列的信息内容,并且如果模型推理足够准确,则合成序列应概括自然蛋白质的功能多样性和特性。
[0226]
现在通过使用分支酸变位酶(cm)同源物的多序列比对(msa)来训练dca无监督学习模型115的非限制性实例来展示方法10的这种dca实施方式。
[0227]
为了展示使用dca作为序列模型的方法10,使用分支酸变位酶(cm)的aroq家族(一种理解催化和酶设计原理的经典模型)执行该方法。这些酶可以存在于细菌、植物和真菌中,并且在莽草酸途径的分支点运作,导致酪氨酸和苯丙氨酸的生物合成(如图12中所展示)。cm通过克莱森(claisen)重排催化中间代谢物分支酸向预苯酸(prephenate)的转化,显示该反应加速超过一百万倍速率,并且是细菌细胞生长所必需的。例如,缺乏cm的大肠杆菌菌株对酪氨酸和苯丙氨酸是营养缺陷型的,这些氨基酸的补充程度和cm的表达水平两者都定量地决定了生长速率。在结构上,aroq cm形成了相对较小原体(约100个氨基酸,图1e)的结构域交换二聚体,其连同对细菌生长的要求和良好生物化学测定的存在使它们成为测试从msa推断的统计模型的能力的优秀设计靶标。
[0228]
首先,创建包含大量序列的msa。在一种实施方式中,使用大肠杆菌p蛋白质的残基1-95作为3轮psi-blast(参考)的起始询问和1e-4的e得分截止值来获取序列。生成初始比对,从pdb条目1ecm、2d8e、3nvt和1ybz的结构比对开始并且迭代生成比对谱,并使用肌肉(参考)将来自psi-blast结果的最近邻序列与该谱进行比对。将产生的比对调整并修剪到1ecm中看到的区域,以去除短序列(少于82个残基),去除添加了不良表示的缺口(<30%被占用)的序列,并减少冗余(>90%最高命中同一性)。以这种方式创建了1,259个序列的msa,其用作序列设计和测试在功能序列上的实验程序的输入。如本领域普通技术人员将理解的,可以通过与对程序的变化类似的方式生成其它同源物的其它msa。
[0229]
接下来,使用msa执行dca分析。例如,msa可以用于推断potts模型,从而分配概率,例如:
[0230][0231]
这种概率可以分配给l=95氨基酸或比对缺口的每个比对序列统计能量(或哈密顿量)可以由以下公式提供:
[0232][0233]
potts模型的统计能量(或哈密顿量)是根据在位置i和j中的氨基酸a和b之间的直
接协同进化耦合j
ij
(a,b)、以及在位置i中的氨基酸a的使用的偏置(或场)hi(a)给出的。这些参数是使用bmdca(重新加权阈值为0.8,正则化强度为10-2
和10-3
)推断的。正式的逆温度(例如,由提供)在推理期间被设置为1。
[0234]
所产生的模型的目的是准确再现在位置i中具有氨基酸a的自然序列的经验分数fi(a)、和在位置i和j中同时具有氨基酸a和b的序列的分数f
ij
(a,b),从而将残差守恒和协同变异结合在一起,例如:
[0235][0236][0237]
为了检查推断模型的准确性,将自然序列的序列统计量与从以下公式中提取的mcmc(马尔可夫链蒙特卡洛)样本进行比较:这可以使用连接的二残差相关性和三残差相关性来执行,例如,分别由以下公式[1]和[2]提供:
[0238]cij
(a,b)=f
ij
(a,b)-fi(a)fj(b)
ꢀꢀ
[1]
[0239]cijk
(a,b,c)=f
ijk
(a,b,c)-f
ij
(a,b)fk(c)-f
ik
(a,c)fj(b)-f
jk
(b,c)fi(a)+2fi(a)fj(b)fk(c)
ꢀꢀ
[2]
[0240]
如上文所引用,公式[1]和[2]分别描述了无法通过低阶统计量来解释的经验二残基和三残基分数的一部分。因此,它们本质上比f
ij
(a,b)和f
ijk
(a,b,c)更难以再现并且对模型的准确性构成更严格的检验。
[0241]
关于正则化的使用,统计模型取决于许多参数{j,h},它们是从有限数据中推断山来的。为了避免强烈的过拟合效应,dca可以使用l2正则化,即罚分,其可以由例如以下公式表示:
[0242][0243]
这样,罚分值(例如,如由上述公式所输出)可以添加到数据的可能性中。这种罚分系统地降低了bmdca推理中的参数值,从而避免了由于欠采样的罕见事件而导致的极大参数值。这在以下公式中修改了模型之间的一致性方程和经验频率计数:
[0244][0245][0246]
如果将自然序列(nat-频率计数f)的统计能量与在(mcmc-由p给出的频率计数)从推断模型中采样的序列进行比较,观察到平均能量被系统地位移,例如,如由以下公式提供:
[0247][0248]
也就是说,在模型中,自然序列比采样序列具有系统更低的能量,因此具有更高的概率,例如,由以下公式表示:
[0249][0250]
为了克服这一差距,较低温度t<1被引入,迫使mcmc以与自然序列相容的较低统计能量进行采样。
[0251]
一旦dca模型被训练,dca模型就可以用于生成新的候选序列。mcmc采样用于从(mcmc)生成序列。pott模型中的温度t(即,β=1/t)控制着采样的宽度。例如,可以增加温度以扩大采样能量的范围特别是至较低能量因此更高的概率作为说明性实例,图10c-e显示了分别在温度下获取的样本的实验结果。
[0252]
dca用于为广泛涵盖细菌和真菌谱系的多样性的1259种自然aroq cm酶的比对制作统计模型。技术上,对于任何蛋白质来说从在msa中观察到的统计量(fi,f
ij
)推导参数(hi,j
ij
)通过直接手段在计算上都是棘手的,但是许多近似算法是可能的。这里,使用了bmdca近似算法,这是一种基于玻尔兹曼机学习的计算量大但精度高的方法。在其它实施方式中,可以使用平均场解法、蒙特卡洛梯度下降法或伪似然最大化法来获得dca模型的参数(hi,j
ij
)。
[0253]
图9a和图9b分别显示了从bmdca模型中采样的序列概括了经验一阶和二阶msa统计量。从这些结果可以观察到,模型拟合良好。图9c显示了bmdca模型还概括了msa中未用于训练模型的三阶相关性,表明该模型在统计学上是完整的。
[0254]
图9d显示了在msa中所有自然cm序列之间的距离矩阵的前两个主成分(例如条目900d1的阴影圆形条目或点)。这里,cm家族跨越的序列空间的结构被可视化。从bmdca模型推导出的序列(例如条目900d2的阴影圆形条目或点)也以与自然cm序列一致的方式填充序列空间。大肠杆菌cm序列的位置由点900ds指示。
[0255]
因此,很明显,通过从模型中蒙特卡洛采样生成的序列再现了用于拟合的自然序列的经验一阶和二阶统计量。统计量显示在图9a、图9b和图9c中,分别显示了一阶、二阶和三阶统计量。此外,可以观察到该模型概括了msa中从未用于推断模型的高阶统计特征。这包括三向残差相关性(参见图9c)和序列空间中蛋白质家族的不均匀聚集的系统发育组织(参见图9d),表明统计模型捕获了通过进化控制自然cm序列分歧的基本规则。相比之下,只保留在位点(hi)处的氨基酸的内在倾向并且遗漏成对耦合的一个更简单的模型甚至无法再现msa的二阶统计量,并且无法解释自然cm蛋白中序列分歧的模式。
[0256]
这个实例说明bmdca提供了用于生成新的候选序列的统计模型,这意味着自然序列和从概率分布p(σ)中采样的序列尽管有相当大的序列差异,但在功能上是等效的。为了说明这一点,使用大肠杆菌中的cm的高通量定量体内互补测定来评估使用bmdca模型生成的候选蛋白质的所需功能性。这里,催化能力用作所需功能性来说明该方法。高通量定量体内互补测定适合用于在单个内部控制实验中研究大量的自然和设计的cm。
[0257]
图9e显示了cm的定量高通量功能测定,其中cm变体文库在缺乏分支酸变位酶的大肠杆菌菌株中表达,在选择性条件下作为混合群体生长,然后进行下一代测序以计数输入和选择群体中每个cm等位基因的频率。
[0258]
图9f显示了可以从定量高通量功能测定的测量中计算相对富集(r.e.)。r.e.提供了催化能力(例如,所需功能性)的标记,因为r.e在大致5个对数级范围内与催化能力(ln(kc/km))接近线性。该“标准曲线”是使用一组跨越宽广的特定活性范围的大肠杆菌cm点突变体制作的。
[0259]
现在提供高通量测定的简要描述,并且下面提供更详细的描述。cm变体(自然的和/或合成的,例如,在冷启动中)的文库是使用能够快速且相对便宜地大规模组装新的dna序列的定制的从头基因合成方案制备的。例如,制备了包含msa中每个自然cm同源物(总共1,259个)的文库,并且制备了超过1,900个合成变体来探索bmdca模型的各种设计参数。这些文库在缺乏cm的细菌菌株(ka12)中表达,并在缺乏苯丙氨酸和酪氨酸的选择性培养基中作为单一种群一起生长,以选择分支酸变位酶活性(如图9e中所示)。选择前后对群体的深度测序允许我们计数每个等位基因相对于野生型的对数频率(一种称为“相对富集”(r.e.)的量),其在诱导、生长时间和温度的特定条件下定量且可再现地报告分支酸变位酶的催化活性(如图9f中所示)。这种“select-seq”测定在宽广的催化能力范围内接近线性,并且用作在单个内部控制实验中严格比较大量的自然和合成变体的体内功能活性的一种有效工具。
[0260]
第一项研究用于检查自然cm同源物在select-seq测定中的性能,这是bmdca设计序列的阳性对照。自然序列显示中心在大肠杆菌cm的值(定义为零,参见图10a)附近的bmdca统计能量单峰分布,但是它们在测定中使用的特定大肠杆菌菌株和实验条件下将如何发挥作用尚不清楚。例如,cm家族的成员在任何特定环境中的活性可能以未知的方式变化,并且msa包括一些部分的执行相关但不同的化学反应的旁系同源酶。select-seq测定表明,msa中的1,259个自然cm同源物在测定中表现出互补的双峰分布,其中一种模式包含约31%的以野生型大肠杆菌cm水平为中心的序列,其余的包含以无效等位基因的水平为中心的模式(参见图10b)。标记有绿色荧光蛋白(gfp)的文库版本表明,与大肠杆菌变体相比,互补的双峰性与表达水平的差异没有明显关系;相反,双峰性推测起源于氨基酸在指定分支酸催化能力方面的协同性、以及旁系同源序列的可能性。出于本研究的目的,双峰性允许将完整分布的减少以简化序列在测定中互补功能的概率。重要的是,标准曲线显示该量是对高分支酸变位酶活性的严格测试(参见图9f)。
[0261]
为了评估bmdca模型的生成潜力,将蒙特卡洛采样用于从模型中随机抽取相对于自然msa跨越了一系列统计能量的序列。例如,图10c-e展示了具有低能量的序列将是功能性分支酸变位酶。
[0262]
关于采样过程,应注意序列数据本质上是有限的(例如,并非所有氨基酸都出现在每个位置),并且相对于msa中的所有对位点处的所有氨基酸的组合数量即使大的序列家族也存在欠采样。鉴于这些限制,为了避免过度拟合,在制作bmdca模型时使用了正则化推理。正则化的使用导致从模型中采样的序列平均具有比自然序列更小的概率(即更高的统计能量)。为了对具有低能量的序列进行采样,在模型中引入正式的计算“温度”t<1,例如,由以下公式提供:
[0263][0264]
这种模型补偿了正则化的影响。例如,在t∈{0.33,0.66}处采样产生出具有更密切影响自然分布并且表现出几乎不依赖正则化参数的统计能量的序列,例如,如图10c和图10d中所示。相比之下,在t=1处采样的序列显示出显著偏离自然分布并且更强烈地依赖于正则化强度的广泛的统计能量分布,例如,如图10e所示。
[0265]
制作并测试了350个合成序列的文库。这些文库在t∈{0.33,0.66,1.0}处采样,每个都来自bmdca模型。图10f-h显示,总体而言,这些序列还显示出互补的双峰分布,具有接近野生型大肠杆菌序列水平的许多互补功能。与假设一致,bmdca统计能量很好地预测了互补的概率,从t∈{0.33,0.66}中提取的低能量序列基本上概括或甚至在某种程度上超过了自然序列的性能(图10f-g)。相比之下,从t=1提取的序列表现出较差的性能,与bmdca模型的偏离一致(参见图10h)。整个地,总共1,050个合成序列中有521个合成序列(约50%)挽救了测定中的生长,包括44-92%的任何自然分支酸变位酶的一系列最高命中同一性。这些包括与msa中的蛋白质的同一性低于65%的48个序列,对应于与最接近的自然对应物相距至少33个突变。与大肠杆菌cm的序列差异的范围为19%至42%。蛋白质中对bmdca统计能量贡献最大的位置表示突出显示分布在活性位点内并延伸通过cm三级结构以包括二聚体界面的残基(参见图11f)。
[0266]
图10a-10i显示了自然和合成cm序列的功能分析。图10a显示了msa中的自然cm序列集合包含中心在大肠杆菌cm的值(定义为零)附近的bmdca序列单峰分布。图10b表明自然cm的相对能量(r.e)分数显示双峰分布,其中一个模型包含约31%的在大肠杆菌cm水平(定义为零r.e.或根据归一化r.e.的一个序列,虚线1000b1)附近的序列、以及在cm无效等位基因水平附近的其余序列(红色虚线)。
[0267]
图10c-10d分别显示了在三个不同计算温度(0.33、0.66、1)下采样的序列的bmdca统计能量。在033和066温度下采样的序列非常接近地再现自然序列的能量,但在t=1时提取的序列不会再现自然序列的能量。
[0268]
在图10f-10h中,功能分析表明,在温度t为0.33和0.66时采样的序列概括或甚至超过了自然序列的性能,但在t=1时采样的序列大多都是无功能的。
[0269]
图10i和10j表明通过保留一阶统计量但忽略相关性产生的序列显示出很大的统计能量并且根本没有显示任何功能。因此,bmdca模型在合成cm序列中编码类似自然的功能。
[0270]
图11a显示了所有合成cm序列的散点图,显示了bmdca统计能量和催化功能之间的关系。显示了功能序列(例如条目1100a1的阴影条)和非功能序列(例如条目1100a2的阴影条)。数据显示该功能是由低bmdca能量预测的,基本上没有e dca<40的序列补充cm无效表型。
[0271]
图11b显示了由图9d中的自然cm序列定义的序列变异的前两个主要成分,如图11a中所指示或以其它方式加阴影。大肠杆菌序列由点1100bs标记。这些数据表明,在大肠杆菌中归类为有功能的自然cm序列定位于序列变异总体模式内的特定区域。
[0272]
图11c显示了合成cm在同一空间上的投影,表明功能序列定位于相同的簇。关于功能序列定位/聚集在pca定义域的那些区域的信息可以用于定义分数(例如,功能性前景),
其然后用于从与功能序列簇对应的区域中选择候选序列。例如,玻尔兹曼分布可以定义一个概率密度函数,根据该概率密度函数随机抽取候选序列。这个概率密度函数可以被偏置以增加抽取的与功能序列簇对应的序列的密度。在图11a-c中,功能性与非功能性的确定是二元的,但在其它实施方式中,可以使用连续规模的功能性,并且可以使用gpr例如生成功能性前景。
[0273]
图11d和图11e显示了在没有(图11d)或有(图11e)从自然cm序列的功能互补模式衍生的额外统计条件(p(x=1|σ))的情况下edca<40的合成序列的互补模式。数据表明,使用在自然cm中发挥功能的先验知识可以显著增强对特定背景中互补功能的合成序列的预测。
[0274]
图11f显示了大肠杆菌cm的结构,其中对低统计能量贡献最大的位置显示在球体(例如条目1100f2的球体或圆圈阴影)中,以及对大肠杆菌特定功能有贡献的位置(例如条目1100f1的球体或圆圈阴影)。数据表明,一般设计约束集中在活性位点和延伸到二聚体界面的残基的物理连续模式上,并且对大肠杆菌特异性功能的额外约束位于更外围。
[0275]
作为另一个对照,产生了326个序列,其具有与在t=0.66时的bmdca设计序列相同的序列同一性分布,但仅保留一阶统计量并忽略相关性。这些序列预计显示出高bmdca能量并且根本没有显示互补(参见图10i和图10j),表明酶功能从根本上取决于通过在j
ij
中的耦合施加的相关性模式而不仅仅是序列变化的幅度。
[0276]
将所有数据放在一起,可以观察到bmdca统计能量与cm活性之间的显著陡峭关系-当统计能量低于由自然序列的能量分布宽度设定的阈值(edca<50)时,近50%的设计序列挽救了cm无效表型,并且基本上没有序列在高于该值下发挥功能(参见图11a)。因此,bmdca是一种有效的生成模型,如果统计能量在自然同源物的范围内,则能够在相当大序列多样性的情况下设计出类似自然的酶促活性。
[0277]
bmdca模型捕获蛋白质家族的总体统计量,而不关注该家族单个成员的特定功能活性。因此,就像自然cm同源物一样,大多数bmdca设计的序列在测定的特定条件下不互补功能(参见图11a)。可以改进生成模型以推断使蛋白质序列对特定表型最优的额外信息。例如,测定中挽救功能的序列占据由自然cm序列跨越的序列空间。在大肠杆菌中互补功能的自然cm分布在几个不同的簇中(参见图11b),但有趣的是,功能性合成序列也遵循相同的模式(参见图11c)。这表明在大肠杆菌的特定环境和测定条件下关于cm功能的信息存在于自然序列的统计量中,并且可能被学习。在此类实施方案中,在一项实验性试验中获得的知识可以用于正式训练计算模型以预测编码特定蛋白质表型的合成序列和有机体环境。
[0278]
作为对上述实施方案的测试,从自然msa中的序列训练/生成dca模型,但现在用二进制值x注释表明它们在测定中发挥功能的能力(对于功能性,x=1;如果不是,则为零)。从这个模型中,可以计算出任何合成序列σ将在大肠杆菌select-seq测定中互补功能的概率;也就是说,p(x=1|σ)。图11d和图11e显示,对于来自原初bmdca模型的低能量类似cm的合成序列(参见图11d),额外的条件是p(x=1|σ)现在有效地预测在测定的背景下互补的子集(83%,参见图11e)。绘制对大肠杆菌特异性cm序列有显著贡献的顶部位置显示在活性位点外围的氨基酸的聚集排列(参见图11f)。因此,这些位置变构地起作用控制催化活性,这是一种提供对反应参数的背景依赖性调整的机制。这些结果支持针对特定蛋白质表型的迭代设计策略,其中bmdca模型随着每一轮选择而更新以最佳地靶向所需表型。
[0279]
这里描述的结果验证并扩展了蛋白质家族的实际可用序列比对中的成对氨基酸相关性足以规定蛋白质折叠和功能的概念。bmdca模型是捕获这些相关性的一种方法。
[0280]
现在提供对用于上文论述的图10a-j和图11a-e的非限制性高通量基因构建和测定的更详细描述。使用在微阵列芯片上合成的寡核苷酸的pcr重叠延伸构建cm基因。为每个基因设计了两个寡核苷酸(230聚体),具有用于“基因特异性引物”(gsp)的一对独特侧翼正交引物退火位点和一个在扩增后去除侧翼区域的btsαi限制位点。重叠设计为使用最近邻法计算的至少16个碱基长,具有3
′
g或c碱基,并且具有至少59℃的解链温度,如在以下文献中所论述:breslauer等人“predicting dna duplex stability from the base sequence,”proc natl acad sci,.第83卷,第3746-3750页。(1986),所述文献通过引用整体并入本文。在10u1总体积中使用q5聚合酶和1x q5缓冲液、0.2μm dntp和一对0.5μm gsp在384孔板中进行pcr。通过分别在98℃、61℃和72℃下进行35个循环的每次解链10秒、退火和延伸对与单个基因对应的寡核苷酸进行扩增。为了去除gsp退火位点并扩增全长基因,将扩增产物500倍稀释到含有0.1u/μl btsαi和侧翼引物5
’‑
agcgatctcggtgacgatgg-3’和5
’‑
cattaacgatgcaagtctcgtgg-3’的pcr反应中并且在55℃下孵育60分钟,然后在61℃退火温度下扩增10个循环和在65℃退火温度下扩增35个循环。
[0281]
克隆:将基因合并,用ndei和xhoi消化,连接到质粒pktctet,进行柱纯化并转化到足够的电感受态neb 10β细胞中,每个被克隆的基因产生>1000x的转化子。整个转化在含有100ug/ml amp的500ml lb中培养过夜,其后将质粒纯化,稀释至1ng/ul以最小化用多个质粒转化单个细胞,并且转化到含有质粒pkimp/uauc的cm缺陷型菌株ka12中,每个基因产生>1000x的转化子。整个转化在含有100ug/ml amp和30ug/ml cam的500ml lb中培养,补充16%甘油并在-80℃下冷冻。
[0282]
分支酸变位酶选择测定:将ka12甘油储液在lb培养基中于30℃下培养过夜,在非选择性m9cfy中稀释至od600为0.045,并且在30℃下生长至od600为约0.2,并用m9c(无fy)洗涤。将该预选择培养物接种到含有100ug/ml amp的lb中,生长过夜并收获用于质粒纯化以产生“输入”样本。对于选择,将培养物以1e-4的计算起始od600稀释到含有3ng/ml多西环素的500ml m9c中,并在30℃下生长24小时。通过离心收获50ml培养物,重悬于含有100ng/ml amp的2ml lb中,生长过夜并收获用于质粒纯化。
[0283]
测序:从输入和选定培养物中纯化的质粒使用kod聚合酶进行两轮pcr扩增,以添加用于illumina测序的连接器和指示物。在第一轮中,使用与质粒退火并添加6至9个n以帮助初始聚焦的引物以及i5或i7连接器的一部分对dna进行扩增。示例性引物是5
’‑
tgactggagttcagacgtgtgctcttccgatctnnnnnnacgactcactatagggagac-3’和5
’‑
cactctttccctacacgacgctcttccgatctnnnnnntgactagtcattattagtgg-3’。在第二轮pcr中,添加了剩余的连接器和truseq指示物。示例性引物是5
’‑
caagcagaagacggcatacgagatcgagtaatgtgactggagttcagacgtg-3’和5
’‑
aatgatacggcgaccaccgagatctacactatagcctacactctttccctacacgac-3’。对于两轮pcr,低循环(16轮)和高初始模板浓度用于最小化扩增诱导的偏差。最终产物通过凝胶纯化,使用qubit进行定量,并在miseq中以2x 250个循环进行测序。
[0284]
使用flash(参考)连接配对末端读数,修剪到ndei和xhoi克隆位点并翻译。仅计数与设计基因的完全匹配。最后,计算相对富集值(r.e)。
[0285]
在图12中,还显示了用于从蛋白质测定设备810、基因合成设备820(也称为基因合
成设备/系统820)和基因表达设备830获取、储存、处理和分发数据的电路系统和硬件。该电路系统和硬件包括:处理器870、网络控制器874、存储器878和数据采集系统(das)876。蛋白质优化系统800可以包括数据通道(未示出),其将来自各个设备(例如,蛋白质测定设备810、基因合成设备820(也称为基因合成设备/系统820)和基因表达设备830)检测测量结果递送到das 876、处理器870、存储器878和网络控制器874。数据采集系统876可以控制来自各种传感器和检测器的检测数据的采集、数字化和递送。如本文所论述,处理器870执行包括训练机器学习模型115、拟合功能性前景和控制各个设备的功能。
[0286]
处理器870可以被配置为执行本文所描述的方法和过程的各个步骤。处理器870可以包括可以作为离散逻辑门、专用集成电路(asic)、现场可编程门阵列(fpga)或其它复杂可编程逻辑装置(cpld)实施的cpu。fpga或cpld实施方式可以用vhdl、verilog或任何其它硬件描述语言编码,并且代码可以直接储存在fpga或cpld内的电子存储器中,或作为单独的电子存储器储存。此外,存储器可以是非易失性的,诸如rom、eprom、eeprom或flash存储器。存储器也可以是易失性的,诸如静态或动态ram,并且可以提供诸如微控制器或微处理器之类的处理器来管理电子存储器以及在fpga或cpld与存储器之间的交互作用。
[0287]
可替代地,处理器870中的cpu可以执行包括执行方法10和/或方法10
′
的各个步骤的一组计算机可读指令的计算机程序,该程序被储存在任何上述非临时电子存储器和/或硬盘驱动器、cd、dvd、flash驱动器或任何其它已知的存储介质上。此外,计算机可读指令可以作为实用应用程序、后台守护程序或操作系统的组件或它们的组合提供,与处理器(诸如来自美国英特尔(intel)的xenon处理器或来自美国amd的opteron处理器)和操作系统(诸如microsoft vista、unix、solaris、linux、apple、mac-os和本领域技术人员已知的其它操作系统)结合执行。此外,cpu可以作为并行协作的多个处理器实施以执行指令。
[0288]
存储器878可以是硬盘驱动器、cd-rom驱动器、dvd驱动器、flash驱动器、ram、rom或本领域已知的任何其它电子存储器。
[0289]
网络控制器874(诸如来自美国英特尔公司的英特尔以太网pro网络接口卡)可以在蛋白质优化系统800的各个部分之间进行接口。此外,网络控制器874还可以与外部网络接口。可以理解,外部网络可以是公共网络(诸如互联网)或专用网络(诸如lan或wan网络)或它们的任何组合,并且还可以包括pstn或isdn子网络。外部网络也可以是有线的,诸如以太网,或者可以是无线的,诸如包括edge、3g和4g无线蜂窝系统的蜂窝网络。无线网络也可以是wifi、蓝牙或任何其它已知的无线通信形式。
[0290]
现在提供训练人工神经网络(例如,vae)的更详细描述(例如,过程310)。这里,目标数据是例如输出氨基酸序列,并且输入数据是相同的输出氨基酸序列,如上文所述。
[0291]
图13显示了训练过程310的一种实施方式的流程图。在过程310中,输入数据和目标数据被用作训练数据来训练人工神经网络,导致训练的人工神经网络370从过程310的步骤319输出。离线dl训练过程310使用输入数据的大量氨基酸序列来训练人工神经网络,以训练人工神经网络。
[0292]
在过程310中,获得一组训练数据,并且迭代地更新网络以减少误差(例如,由损失函数产生的值)。人工神经网络推断训练数据所隐含的映射,并且损失函数产生与目标数据和通过将人工神经网络的当前化身应用于输入数据所产生的结果之间的不匹配相关的误差值。例如,在某些实施方式中,损失函数可以使用均方误差来最小化均方误差。在多层感
知器(mlp)神经网络的情况下,反向传播算法可以用于通过使用(随机)梯度下降法最小化基于均方误差的损失函数来训练网络。
[0293]
在过程310的步骤316中,为人工神经网络的系数生成初始猜测。例如,初始猜测可以基于lecun初始化、xavier初始化和kaiming初始化之一。
[0294]
过程310的步骤316至319提供了用于训练人工神经网络的优化方法的非限制性实例。
[0295]
在应用当前版本的网络之后,计算(例如,使用损失函数或损失函数)误差以表示在目标数据(即基本实况)与输入数据之间的差异的度量(例如,距离度量)。可以使用任何已知的损失函数或距离度量(包括上文所述的那些损失函数)来计算误差。此外,在某些实施方式中,可以使用合页损失(hinge loss)和交叉熵损失(cross-entropy loss)中的一种或多种来计算误差/损失函数。在某些实施方式中,损失函数可以是在目标数据与将输入数据应用于人工神经网络的结果之间的差异的l
p
范数。l
p
范数中的不同的“p”值可以用于强调噪声的不同方面。在某些实施方式中,损失函数可以表示相似性(例如,使用峰值信噪比(psnr)或结构相似性(ssim)指数),而不是最小化在目标数据与来自输入数据的结果之间的差异的l
p
范数。
[0296]
在某些实施方式中,使用反向传播训练网络。反向传播可以用于训练神经网络,并且与梯度下降优化方法结合使用。在前向传递(forward pass)期间,算法基于当前参数(θ)计算网络的预测。然后将这些预测输入损失函数中,通过该损失函数将它们与对应的基本实况标签(即高质量目标数据)进行比较。在后向传递(backward pass)期间,模型计算损失函数相对于当前参数的梯度,其后通过在最小化损失的方向上采取预定义大小的步长来更新参数(例如,在加速方法(诸如nesterov动量法和各种自适应方法)中,可以选择步长以更快收敛来优化损失函数)。
[0297]
执行反投影的优化方法可以使用梯度下降、批量梯度下降、随机梯度下降和小批量随机梯度下降中的一种或多种。可以通过网络的各个层逐步执行前向传递和后向传递。在前向传递中,执行通过经由第一层输送输入开始,从而为后续层创建输出激活。重复这个过程,直到达到最后一层的损失函数。在后向传递期间,最后一层根据其自身的可学习参数(如果有的话)以及关于其自身的输入(其用作前一层的上游导数)计算梯度。重复这个过程,直到到达输入层。
[0298]
返回图13,过程310的步骤317确定可以计算作为网络变化函数的误差变化(例如,误差梯度),并且该误差变化可以用于选择方向和用于随后更改人工神经网络的权重/系数的步长。以这种方式计算误差梯度与梯度下降优化方法的某些实施方式是一致的。在某些其它实施方式中,该步骤可以被省略和/或替换为根据另一种优化算法(例如,非梯度下降优化算法,如模拟退火或遗传算法)的另一步骤,如本领域普通人员所理解的那样。
[0299]
在过程310的步骤317中,为人工神经网络确定一组新的系数。例如,可以使用在步骤317中计算的变化来更新权重/系数,如在梯度下降优化方法或过松弛加速方法中。
[0300]
在过程310的步骤318中,使用人工神经网络的更新的权重/系数来计算新的误差值。
[0301]
在步骤319中,使用预定义的停止标准来确定网络的训练是否完成。例如,预定义的停止标准可以评估新的误差和/或所执行的迭代总数是否超过预定义值。例如,如果新的
误差低于预定义的阈值或者如果达到最大迭代次数,则可以满足停止标准。当不满足停止标准时,在过程310中执行的训练过程将通过返回并使用新的权重和系数重复步骤317而继续回到迭代循环的开始(迭代循环包括步骤317、318和319)。当满足停止标准时,完成在过程310中执行的训练过程。
[0302]
图14显示了人工神经网络中各层之间的互连的实例。人工神经网络可以包括全连接层、卷积层和池化层,所有这些都在下文进行解释。在人工神经网络的某些优选实施方式中,卷积层靠近输入层放置,然而执行高级推理的全连接层则放置在进一步朝向损失函数的体系结构的下方。池化层可以在卷积之后插入,并且证明减少了过滤器的空间范围,从而降低了可学习参数的量。激活函数也被并入到各个层中,以引入非线性并使得网络能够学习复杂的预测关系。激活函数可以是饱和激活函数(例如,s形或双曲线正切激活函数)或整流激活函数(例如,在上文论述的第一和第二实例中应用的整流线性单元(relu))。人工神经网络的层也可以结合批量归一化,如上文所论述的第一和第二实例中所例示的。
[0303]
图14显示了具有n个输入、k个隐藏层和三个输出的通用人工神经网络(ann)的实例。每层由节点(也称为神经元)组成,并且每个节点执行输入的加权求和并将加权求和的结果与阈值进行比较以生成输出。ann构成一类函数,其中该类的成员通过改变阈值、连接权重或体系结构的细节(诸如节点数量和/或它们的连接性)来获得。ann中的节点可以被称为神经元(或神经元节点),并且神经元可以在ann系统的不同层之间进行互连。突触(即神经元之间的连接)储存在计算中操纵数据的称为“权重”(也可互换地称为“系数”或“加权系数”)的值。ann的输出取决于三种类型的参数:(i)神经元的不同层之间的互连模式,(ii)更新互连权重的学习过程,以及(iii)将神经元加权输入转化为其输出激活的激活函数。
[0304]
数学上,神经元的网络函数m(x)被定义为其它函数ni(x)的成分,其可以进一步被定义为其它函数的成分。这可以方便地表示为一个网络结构,箭头描绘变量之间的依赖关系,如图14中所示。例如,ann可以使用非线性加权和,其中m(x)=k(∑iwini(x)),其中k(通常称为激活函数)是一些预定义函数(诸如双曲线正切)。
[0305]
在图14中,神经元(即节点)由阈值函数周围的圆圈描绘。对于图14中所示的非限制性实例,输入被描绘为在线性函数周围的圆圈,并且箭头指示神经元之间的定向连接。
[0306]
尽管已经描述了某些实施方式,但这些实施方式仅以实例的方式呈现并且不旨在限制本公开的教导。实际上,本文所述的新颖方法、设备和系统可以以多种其它形式体现;此外,在不背离本公开的精神的情况下,可以对本文所述的方法、设备和系统的形式进行各种省略、替换和改变。
[0307]
本公开的方面
[0308]
本公开的以下方面仅是示例性的并且不旨在限制本公开的范围。
[0309]
1.一种设计具有所需功能性的蛋白质的方法,所述方法包括:使用机器学习模型确定合成蛋白质的候选氨基酸序列,所述机器学习模型已被训练来学习在蛋白质的训练数据集氨基酸序列中的隐式模式,所述机器学习模型在经训练的模型中表达学习到的隐式模式;执行迭代循环,其中所述循环的每次迭代包括:合成候选基因并且产生与各个候选氨基酸序列对应的候选蛋白质,每个所述候选基因编码所述对应的候选氨基酸序列;通过使用一种或多种测定测量指示所述候选蛋白质的特性的值来评估所述候选蛋白质分别表现出所需功能性的程度;以及,当所述迭代循环的一个或多个停止标准未得到满足时,从所测量
的值计算分配给每个序列的适合度函数,并且使用所述适合度函数连同所述机器学习模型的组合来选择用于后续迭代的新的候选氨基酸序列。
[0310]
2.如方面1所述的方法,其中所述隐式模式是在潜在空间中学习的,并且其中确定所述候选氨基酸序列还包括确定所述潜在空间相对于所述训练数据集的氨基酸序列的特征维度具有减小的维度。
[0311]
3.如方面1-2中任一项所述的方法,其中所述训练数据集包含进化相关蛋白的多序列比对,所述多序列比对中的氨基酸序列具有序列长度l,并且所述训练数据集的特征维度大到足以容纳与所述序列长度l对应的20
l
氨基酸组合。
[0312]
4.如方面1-3中任一项所述的方法,其中所述训练数据集包含进化相关蛋白的多序列比对,并且所述训练数据集的氨基酸序列的特征维度为乘积l
×
k,其中l是所述训练数据集的一个氨基酸序列的长度次数,并且k是可能的氨基酸类型的数量。
[0313]
5.如方面4所述的方法,其中所述氨基酸是天然氨基酸并且k等于或小于20。
[0314]
6.如方面4所述的方法,其中所述可能的氨基酸类型中的至少一种是非天然氨基酸。
[0315]
7.如方面1-6中任一项所述的方法,其中所述训练数据集包含与共同功能相关的蛋白质,所述共同功能是以下中的至少一种:(i)共同结合功能、(ii)共同变构功能和(iii)共同催化功能。
[0316]
8.如方面1-7中任一项所述的方法,其中用于训练所述机器学习模型的所述训练数据集包括与以下中的至少一种相关的蛋白质:(i)共同祖先、(ii)共同三维结构、(iii)共同功能、(iv)共同结构域结构和(v)共同进化选择压力。
[0317]
9.如方面1-8中任一项所述的方法,其中执行所述迭代循环的步骤还包括:当一个或多个停止标准未得到满足时,基于包括所述候选蛋白质的氨基酸序列的更新的蛋白质训练数据集更新所述机器学习模型,并且在基于所述更新的训练数据集进行更新之后使用所述适合度函数与所述机器学习模型的组合来选择用于所述后续迭代的所述新的候选氨基酸序列。
[0318]
10.如方面1-x中任一项所述的方法,其中所述机器学习模型是以下中的一种:(i)变分自编码器(vae)网络、(ii)受限玻尔兹曼机(rbm)网络、(iii)直接耦合分析(dca)模型、(iv)统计耦合分析(sca)模型和(v)生成对抗网络(gan)。
[0319]
11.如方面2所述的方法,其中所述机器学习模型是执行编码和解码/生成的网络模型,所述编码通过将输入氨基酸序列映射到所述潜在空间中的点来执行,并且所述解码/生成通过将所述潜在空间中的点映射到输出氨基酸序列来执行,并且所述机器学习模型被训练以优化目标函数,所述目标函数的一个成分表示所述输入氨基酸序列和所述输出氨基酸序列匹配的程度,使得当使用所述训练数据集进行训练时,所述机器学习模型生成与用作所述机器学习模型的输入的所述训练数据集的氨基酸序列大致匹配的输出氨基酸序列。
[0320]
12.如方面1-x中任一项所述的方法,其中所述机器学习模型是基于所述训练数据集的氨基酸序列的一阶统计量和二阶统计量来学习设计规则的基于无监督统计的模型,并且所述机器学习模型是通过机器学习方法训练以生成与所述学习的设计规则一致的输出氨基酸序列的生成模型。
[0321]
13.如方面1-12中任一项所述的方法,所述方法还包括使用所述训练数据集训练
所述机器学习模型以学习potts模型的外部场和残基-残基耦合以生成所述训练数据集的dca模型,所述dca模型被用作所述机器学习模型。
[0322]
14.如方面13所述的方法,其中所述dca模型使用玻尔兹曼机学习方法、平均场解法、蒙特卡洛梯度下降法和伪似然最大化法中的一种来训练。
[0323]
15.如方面13所述的方法,其中确定所述候选氨基酸序列的步骤还包括基于在一个或多个一个或多个预定义温度下训练的potts模型的哈密顿量从玻尔兹曼统计分布中选择所述候选氨基酸序列,所述候选氨基酸序列使用马尔可夫链蒙特卡洛(mcmc)法、模拟退火法、模拟加热法、遗传算法、跳盆法(basin hopping method)、采样法和优化法中的至少一种从所述玻尔兹曼统计分布中抽取样本来选择。
[0324]
16.如方面15所述的方法,其中选择用于所述后续迭代的所述新的候选氨基酸序列的步骤还包括基于在一个或多个预定义温度下训练的potts模型的哈密顿量从玻尔兹曼统计分布中对氨基酸序列选择进行偏置,其中对所述氨基酸序列选择的偏置基于所述适合度函数来增加与所述测量值表明所需功能性大于所述测量值的平均值、中值或众数的所测量候选蛋白质的氨基酸序列更紧密地匹配的被选择氨基酸序列的数量。
[0325]
17.如方面15所述的方法,其中选择用于所述后续迭代的所述新的候选氨基酸序列的步骤还包括从统计分布中随机抽取氨基酸序列,其中基于所训练的potts模型的哈密顿量的玻尔兹曼统计分布通过适合度函数被加权以增加所述样本从所述潜在空间内的如下区域中抽取的可能性,所述区域更能代表比与所述潜在空间的其它区域对应的候选氨基酸序列表现出更多所需功能性的候选氨基酸序列。
[0326]
18.如方面1-17中任一项所述的方法,所述方法还包括使用所述训练数据集训练所述机器学习模型来学习位置协同进化矩阵以生成所述训练数据集的sca模型,所述sca模型被用作所述机器学习模型。
[0327]
19.如方面18所述的方法,所述方法还包括:通过使用sca模型执行模拟退火或模拟加热来生成氨基酸序列样本集,所述氨基酸序列样本集表达所述训练数据集的学习隐式模式,并且从所生成的氨基酸序列样本集中选择所述候选氨基酸序列。
[0328]
20.如方面1-19中任一项所述的方法,其中选择用于所述后续迭代的所述新的候选氨基酸序列的步骤还包括对所述候选蛋白质的所述候选氨基酸序列进行线性或非线性降维以对低维模型的成分进行排序,并且对所述氨基酸序列的选择进行偏置以增加在所述低维模型的前导成分空间内的一个或多个邻域中选择的氨基酸序列的数量,其中与测量值对应的氨基酸序列表明高度的所需功能性聚簇。
[0329]
21.如方面20所述的方法,其中所述非线性降维是主成分分析,并且所述低维模型的前导成分是由与相关矩阵的一组最大特征值对应的一组特征向量表示的主成分分析的主要成分。
[0330]
22.如方面20所述的方法,其中所述非线性降维是独立成分分析,其中所述特征向量经受旋转和缩放操作以鉴定序列变化的功能独立模式。
[0331]
23.如方面11所述的方法,其中确定所述候选氨基酸序列的步骤还包括:鉴定在所述潜在空间内与被选择为可能表现出所需功能性的蛋白质的氨基酸序列对应的邻域,选择在所述潜在空间内所鉴定的邻域内的点,以及使用由所述机器学习模型执行的所述解码/生成将所选定的点映射到各个候选氨基酸序列,然后将其用作所述候选氨基酸序列。
[0332]
24.如方面11所述的方法,其中选择用于所述后续迭代的所述新的候选氨基酸序列的步骤还包括:基于所述适合度函数鉴定在所述潜在空间内表现出所需功能性或比其它区域更可能表现出所需功能性或采样太稀疏而无法关于所述所需功能性进行统计学上显著的估计的区域,选择在所述潜在空间内所鉴定的区域内的点,以及使用由所述机器学习模型执行的所述解码/生成将所选定的点映射到各个候选氨基酸序列,然后将其用作所述后续迭代的所述新的候选氨基酸序列还包括。
[0333]
25.如方面24所述的方法,其中:鉴定所述潜在空间内的所述区域的步骤还包括基于所述适合度函数在所述潜在空间内生成密度函数,并且选择在所述潜在空间内所鉴定的区域内的点的步骤还包括选择在统计学上代表所述密度函数的点。
[0334]
26.如方面1-25中任一项所述的方法,其中计算所述适合度函数的步骤还包括执行功能性前景的监督学习,所述功能性前景将所述候选蛋白质的测量值近似为所述潜在空间内的对应位置的函数,其中所述适合度函数至少部分基于所述功能性前景。
[0335]
27.如方面26所述的方法,其中对于在所述潜在空间中的给定点,所述功能性前景为所述给定点的对应氨基酸序列提供功能性估计值,并且所述功能性的估计值是以下中的至少一个:(i)所述对应氨基酸序列的基于所述机器学习模型的统计概率,(ii)折叠所述对应氨基酸序列的统计能量或物理能量,所述统计能量基于统计评分函数通过计算预测,和(iii)所述统计能量在执行特定结构或功能角色方面的活性,所述活性通过计算预测或通过实验测量。
[0336]
28.如方面26所述的方法,其中所述适合度函数是功能性前景。
[0337]
29.如方面26所述的方法,其中所述适合度函数基于功能性前景和至少一个选自序列相似性前景和稳定性前景的其它参数,所述序列相似性前景估计与所述潜在空间中的点对应的蛋白质跟预定义的蛋白质集合相似的程度,并且所述稳定性前景估计与所述潜在空间中的点对应的蛋白质稳定的程度。
[0338]
30.如方面29所述的方法,其中所述稳定性前景基于与所述潜在空间中的点对应的稳定的蛋白质的蛋白质折叠的数值模拟。
[0339]
31.如方面29所述的方法,其中所述功能性前景和所述至少一个其它参数定义多目标优化空间,并且用于所述后续迭代的所述候选氨基酸序列通过以下方式来选择:确定在所述多目标优化空间内的凸包作为帕累托前沿面,选择在所述潜在空间内位于所述帕累托前沿面上的点,并且使用所述机器学习模型将所选定的点映射到氨基酸序列,然后将其用作用于所述后续迭代的所述候选氨基酸序列。
[0340]
32.如方面29所述的方法,其中通过使用监督分类或回归分析执行监督学习来生成功能性前景,所述监督学习是以下中的一种:(i)多变量线性、多项式、步进、套索、岭回归、核心回归或非线性回归方法,(ii)支持向量回归(svr)方法,(iii)高斯过程回归(gpr)方法,(iv)决策树(dt)方法,(v)随机森林(rf)方法,和(vi)人工神经网络(ann)。
[0341]
33.如方面30所述的方法,其中所述功能性前景还包括作为所述潜在空间内的位置的函数的不确定性值,所述不确定性值表示已经针对所述功能性前景与所述测量值的近似程度而估计的不确定性。
[0342]
34.如方面33所述的方法,所述方法还包括选择用于所述后续迭代的所述候选氨基酸序列中的一些以对应于所述潜在空间中具有比其它区域更大的不确定性值的区域,使
得在所述后续迭代中,与所述候选氨基酸序列中的一些对应的测量值由于在所述较大不确定值的区域中采样的增加将使所述较大不确定值减小。
[0343]
35.如方面1-34中任一项所述的方法,其中测量所述候选蛋白质的值的步骤包括使用以下中的至少一种来测量所述值:(i)测量生长速率作为所需功能性的标记的测定,(ii)测量基因表达作为所需功能性的标记的测定,和(iii)使用微流体和荧光来测量基因表达或活性作为所需功能性的标记的测定。
[0344]
36.如方面1-34中任一项所述的方法,其中合成所述候选基因的步骤还包括使用其中在溶液中提供具有重叠延伸部分的寡核苷酸(oligo)的聚合酶循环/链组装(pca),其中所述寡核苷酸被循环通过一系列温度,由此通过以下步骤将寡核苷酸组合成更大的寡核苷酸:(i)使寡核苷酸变性,(ii)使所述重叠延伸部分退火,和(iii)延伸非重叠延伸部分。
[0345]
37.如方面1-34中任一项所述的方法,其中执行所述迭代循环的步骤还包括演化从起始值演化为最终值的一个或多个测定的参数,使得在第一次迭代期间当以所述起始值测量时,所述候选基因表现出所述所需功能性,但当以所述最终值测量时不表现出所述所需功能性,并且在最后一次迭代期间,所述候选基因当以所述最终值测量时表现出所述所需功能性。
[0346]
38.如方面37所述的方法,其中所述参数是以下中的一种:(i)温度、(ii)压力、(iii)光照条件、(iv)ph值和(v)介质中用于一种或多种测定的物质的浓度。
[0347]
39.如方面37所述的方法,其中选择所述一种或多种测定的参数以关于内部表型和外部环境条件的组合评估所述候选氨基酸序列。
[0348]
40.如方面1-39中任一项所述的方法,其中执行所述迭代循环的步骤还包括:当满足所述迭代循环的所述一个或多个停止标准时,停止所述迭代循环并且输出与一个或多个最表现出所需功能性的候选基因对应的一个或多个遗传密码的信息。
[0349]
41.一种用于设计具有所需功能性的蛋白质的系统,所述系统包括:基因合成系统,所述基因合成系统被配置成基于编码各个氨基酸序列的输入基因序列合成基因,并且从所合成的基因中生成蛋白质;测定系统,所述测定系统被配置成测量从所述基因合成系统接收的蛋白质的值,所测量的值提供所需功能性的标记;和处理电路系统,所述处理电路系统被配置成:使用机器学习模型确定合成蛋白质的候选氨基酸序列,所述机器学习模型已被训练来学习蛋白质氨基酸序列训练数据集中的隐式模式,所述机器学习模型在训练模型中表达所学习的隐式模式并且执行迭代循环,其中所述循环的每次迭代包括向所述基因合成系统发送所述候选氨基酸序列以基于所述候选氨基酸序列生成候选蛋白质,从所述测定系统接收基于所述候选氨基酸序列与候选蛋白质对应的测量值,并且当所述迭代循环的一个或多个停止标准未得到满足时,从所述测量值计算分配给每个氨基酸序列的适合度函数,并且使用所述适合度函数与所述机器学习模型的组合来选择用于后续迭代的新的候选氨基酸序列。
[0350]
42.如方面41所述的系统,其中所述机器学习模型表达在潜在空间中所学习的隐式模式,以及所述处理电路系统还被配置成确定所述候选氨基酸序列,所述潜在空间相对于所述训练数据集的氨基酸序列的特征维度具有减小的维度。
[0351]
43.如方面41-42中任一项所述的系统,其中所述训练数据集包含同源蛋白质的多序列比对,所述多序列比对中的氨基酸序列具有序列长度l,并且所述训练数据集的特征维
度大到足以容纳与所述序列长度l对应的20
l
氨基酸组合。
[0352]
44.如方面42所述的系统,其中所述训练数据集包含进化相关蛋白的多序列比对,并且所述训练数据集的氨基酸序列的特征维度为乘积l
×
k,其中l是所述训练数据集的一个氨基酸序列的长度,并且k是可能的氨基酸类型的数量。
[0353]
45.如方面44所述的系统,其中所述氨基酸是天然氨基酸并且k等于或小于20。
[0354]
46.如方面44所述的系统,其中所述氨基类型中的至少一种是非天然氨基酸。
[0355]
47如方面41-46中任一项所述的系统,其中所述训练数据集包含与共同功能相关的蛋白质,所述共同功能是以下中的至少一种:(i)共同结合功能、(ii)共同变构功能和(iii)共同催化功能。
[0356]
48.如方面41-47中任一项所述的系统,其中用于训练所述机器学习模型的所述训练数据集包括与以下中的至少一种相关的蛋白质:(i)共同祖先、(ii)共同三维结构、(iii)共同功能、(iv)共同结构域结构和(v)共同进化选择压力。
[0357]
49.如方面41-48中任一项所述的系统,其中所述处理电路系统还被配置成执行所述迭代循环,当一个或多个停止标准未得到满足时,基于包括所述候选蛋白质的氨基酸序列的更新的蛋白质训练数据集更新所述机器学习模型,并且在基于所述更新的训练数据集进行更新之后使用所述适合度函数与所述机器学习模型的组合来选择用于所述后续迭代的所述新的候选氨基酸序列。
[0358]
50.如方面41-49中任一项所述的系统,其中所述机器学习模型是以下中的一种:(i)变分自编码器(vae)网络、(ii)受限玻尔兹曼机(rbm)网络、(iii)直接耦合分析(dca)模型、(iv)统计耦合分析(sca)模型和(v)生成对抗网络(gan)。
[0359]
51.如方面42所述的系统,其中所述机器学习模型是执行编码和解码/生成的网络模型,所述编码通过将输入氨基酸序列映射到所述潜在空间中的点来执行,并且所述解码/生成通过将所述潜在空间中的点映射到输出氨基酸序列来执行,并且所述机器学习模型被训练以优化目标函数,所述目标函数的一个成分表示所述输入氨基酸序列和所述输出氨基酸序列匹配的程度,使得当使用所述训练数据集进行训练时,所述机器学习模型生成与用作所述机器学习模型的输入的所述训练数据集的氨基酸序列大致匹配的输出氨基酸序列。
[0360]
52.如方面41-51中任一项所述的系统,其中所述机器学习模型是基于所述训练数据集的氨基酸序列的一阶统计量和二阶统计量来学习设计规则的基于无监督统计的模型,并且所述机器学习模型是被训练以生成与所学习的设计规则一致的输出氨基酸序列的生成模型。
[0361]
53.如方面41-52中任一项所述的系统,其中所述处理电路系统还被配置成使用所述训练数据集训练所述机器学习模型以学习potts模型的外部场和残基-残基耦合以生成所述训练数据集的dca模型,所述dca模型被用作所述机器学习模型。
[0362]
54.如方面53所述的系统,其中所述dca模型使用玻尔兹曼机学习方法、平均场解法、蒙特卡洛梯度下降法和伪似然最大化法中的一种来训练。
[0363]
55.如方面53所述的系统,其中所述处理电路系统还被配置成通过基于在一个或多个预定义温度下训练的potts模型的哈密顿量从玻尔兹曼统计分布中选择所述候选氨基酸序列来确定所述候选氨基酸序列,所述候选氨基酸序列使用马尔可夫链蒙特卡洛(mcmc)法、模拟退火法、模拟加热法、遗传算法、跳盆法、采样法和优化法中的至少一种从所述玻尔
兹曼统计分布中抽取样本来选择。
[0364]
56.如方面55所述的系统,其中所述处理电路系统还被配置成通过基于在一个或多个预定义温度下训练的potts模型的哈密顿量从玻尔兹曼统计分布中对氨基酸序列选择进行偏置来选择用于所述后续迭代的所述新的候选氨基酸序列,其中对所述氨基酸序列选择的偏置基于所述适合度函数来增加与所述测量值表明所需功能性大于所述测量值的平均值、中值或众数的所测量候选蛋白质的氨基酸序列更紧密地匹配的被选择氨基酸序列的数量。
[0365]
57.如方面53所述的系统,其中所述处理电路系统还被配置成通过从统计分布中随机抽取氨基酸序列选择用于所述后续迭代的所述新的候选氨基酸序列,其中基于所训练的potts模型的哈密顿量的玻尔兹曼统计分布通过所述适合度函数被加权以增加所述样本从所述潜在空间内的如下区域中抽取的可能性,所述区域更能代表比与所述潜在空间的其它区域对应的候选氨基酸序列表现出更多所需功能性的候选氨基酸序列。
[0366]
58.如方面41-57中任一项所述的系统,其中所述处理电路系统还被配置成使用所述训练数据集训练所述机器学习模型来学习位置协同进化矩阵以生成所述训练数据集的sca模型,所述sca模型被用作所述机器学习模型。
[0367]
59.如方面58所述的系统,其中所述处理电路系统还被配置成通过使用所述sca模型执行模拟退火或模拟加热来生成氨基酸序列样本集,所述氨基酸序列样本集表达所述训练数据集的所学习的隐式模式,以及其中所述处理电路系统还被配置成从所述氨基酸序列样本集中选择所述候选氨基酸序列。
[0368]
60.如方面41-59中任一项所述的系统,其中所述处理电路系统还被配置成通过以下方式来选择用于所述后续迭代的所述新的候选氨基酸序列:对所测量的候选蛋白质的所述候选氨基酸序列进行线性或非线性降维以对低维模型的成分进行排序,并且对所述氨基酸序列的选择进行偏置以增加在所述低维模型的前导成分空间内的一个或多个邻域中选择的氨基酸序列的数量,其中与测量值对应的氨基酸序列表明高度的所需功能性聚簇。
[0369]
61.如方面60所述的系统,其中所述非线性降维是主成分分析或独立成分分析,并且所述低维模型的前导成分是所述主成分分析的主要成分或所述独立成分分析的独立成分。
[0370]
62.如方面51所述的系统,其中所述处理电路系统还被配置成通过以下方式来确定所述候选氨基酸序列:鉴定在所述潜在空间内与被选择为可能表现出所需功能性的蛋白质的氨基酸序列对应的邻域,选择在所述潜在空间内所鉴定的邻域内的点,以及使用由所述机器学习模型执行的所述解码/生成将所选定的点映射到各个候选氨基酸序列,然后将其用作所述候选氨基酸序列。
[0371]
63.如方面51所述的系统,其中所述处理电路系统还被配置成通过以下方式来选择用于所述后续迭代的所述新的候选氨基酸序列:基于所述适合度函数鉴定在所述潜在空间内表现出所需功能性或比其它区域更可能表现出所需功能性或采样太稀疏而无法关于所述所需功能性进行统计学上显著的估计的区域,选择在所述潜在空间内所鉴定的区域内的点,以及使用由所述机器学习模型执行的所述解码/生成将所选定的点映射到各个候选氨基酸序列,然后将其用作所述后续迭代的所述新的候选氨基酸序列。
[0372]
64.如方面63所述的系统,其中所述处理电路系统还被配置成:通过基于所述适合
度函数在所述潜在空间内生成密度函数来鉴定所述潜在空间内的区域,并且通过选择在统计学上代表所述密度函数的点来选择所述潜在空间内的所鉴定区域内的点。
[0373]
65.如方面41-64中任一项所述的系统,其中所述处理电路系统还被配置成在计算所述适合度函数时,执行功能性前景的监督学习,所述功能性前景将所述候选蛋白质的测量值近似为所述潜在空间内的对应位置的函数,其中所述适合度函数至少部分基于所述功能性前景。
[0374]
66.如方面65所述的系统,其中对于在所述潜在空间上的给定点,所述功能性前景为所述给定点的对应氨基酸序列提供功能性估计值,并且所述功能性的估计值是以下中的至少一个:(i)所述对应氨基酸序列的基于所述机器学习模型的统计概率,(ii)折叠所述对应氨基酸序列的统计能量或物理能量,所述统计能量基于统计评分函数通过计算预测,和(iii)所述统计能量在执行特定结构或功能角色方面的活性,所述活性通过计算预测或通过实验测量。
[0375]
67.如方面65所述的系统,其中所述适合度函数是功能性前景。
[0376]
68.如方面65所述的系统,其中所述适合度函数基于功能性前景和至少一个选自序列相似性前景和稳定性前景的其它参数,所述序列相似性前景估计与所述潜在空间中的点对应的蛋白质跟预定义的蛋白质集合相似的程度,并且所述稳定性前景估计与所述潜在空间中的点对应的蛋白质稳定的程度。
[0377]
69.如方面68所述的系统,其中所述稳定性前景基于与所述潜在空间中的点对应的稳定的蛋白质的蛋白质折叠的数值模拟。
[0378]
70.如方面69所述的系统,其中所述功能性前景和所述至少一个其它参数定义多目标优化空间,并且用于所述后续迭代的所述新的候选氨基酸序列通过以下方式来选择:确定在所述多目标优化空间内的凸包作为帕累托前沿面,选择在所述潜在空间内位于所述帕累托前沿面上的点,并且使用所述机器学习模型将所选定的点映射到氨基酸序列,然后将其用作用于所述后续迭代的所述新的候选氨基酸序列。
[0379]
71.如方面65所述的系统,其中通过使用监督分类或回归分析执行监督学习来生成功能性前景,所述监督学习是以下中的一种:(i)多变量线性、多项式、步进、套索、岭回归、核心回归或非线性回归方法,(ii)支持向量回归(svr)方法,(iii)高斯过程回归(gpr)方法,(iv)决策树(dt)方法,(v)随机森林(rf)方法,和(vi)人工神经网络(ann)。
[0380]
72.如方面65所述的系统,其中所述功能性前景还包括作为所述潜在空间内的位置的函数的不确定性值,所述不确定性值表示已经针对所述功能性前景与所述测量值的近似程度而估计的不确定性。
[0381]
73.如方面41-72中任一项所述的系统,其中所述处理电路系统还被配置成选择用于所述后续迭代的所述新的候选氨基酸序列中的一些以对应于所述潜在空间中具有比其它区域更大的不确定性值的区域,使得在所述后续迭代中,与所述候选氨基酸序列中的一些对应的测量值由于在所述较大不确定值的区域中采样的增加将使所述较大不确定值减小。
[0382]
74.如方面41-73中任一项所述的系统,其中所述测定系统还被配置成使用以下中的至少一种来测量所述候选蛋白质的值:(i)测量生长速率作为所需功能性的标记的测定,(ii)测量基因表达作为所需功能性的标记的测定,和(iii)使用微流体和荧光来测量基因
表达或活性作为所需功能性的标记的测定。
[0383]
75.如方面41-74中任一项所述的系统,其中所述基因合成系统还被配置成使用其中在溶液中提供具有重叠延伸部分的寡核苷酸(oligo)的聚合酶循环/链组装(pca)合成所述候选基因,其中所述寡核苷酸被循环通过一系列温度,由此通过以下步骤将寡核苷酸组合成更大的寡核苷酸:(i)使寡核苷酸变性,(ii)使所述重叠延伸部分退火,和(iii)延伸非重叠延伸部分。
[0384]
76.如方面41-x中任一项所述的系统,其中所述处理电路系统还被配置成执行所述迭代循环,使得所述一个或多个测定的参数从起始值演化为最终值,使得在第一次迭代期间当以所述起始值测量时,所述候选基因表现出所述所需功能性,但当以所述最终值测量时不表现出所述所需功能性,并且在最后一次迭代期间,所述候选基因当以所述最终值测量时表现出所述所需功能性。
[0385]
77.如方面76所述的系统,其中所述一个或多个测定的参数是以下中的一种:(i)温度、(ii)压力、(iii)光照条件、(iv)ph值和(v)介质中用于一种或多种测定的物质的浓度。
[0386]
78.如方面76所述的系统,其中将所述一种或多种测定的参数用于关于内部表型和外部环境条件的组合评估所述候选氨基酸序列。
[0387]
79.一种包括可执行指令的非暂时性计算机可读存储介质,其中所述指令在由电路系统执行时使所述电路系统执行包括以下步骤的方法:使用已训练以学习蛋白质训练数据集中的隐式模式的机器学习模型确定合成蛋白质的候选氨基酸序列,所述机器学习模型表达所学习的隐式模式,以及执行迭代循环,其中所述循环的每次迭代包括:基于所述候选氨基酸序列确定候选基因序列,向基因合成系统发送待合成为候选基因的所述候选基因序列以产生候选蛋白质,从测定系统接收通过使用一种或多种测定测量所述候选蛋白质而产生的测量值,并且当所述迭代循环的一个或多个停止标准未得到满足时,从所述测量值计算适合度函数并使用所述适合度函数与所述机器学习模型的组合来选择用于后续迭代的另外的候选氨基酸序列。
[0388]
80.一种设计具有所需功能性的序列定义的分子的方法,所述方法包括:确定序列定义的分子的候选序列,所述候选序列是使用机器学习模型生成的,所述机器学习模型已被训练来学习在序列定义的分子的训练数据集中的隐式模式,所述机器学习模型表达所学习的隐式模式;以及执行迭代循环,其中所述循环的每次迭代包括:合成与所述候选分子对应的候选序列,通过使用一种或多种测定测量所述候选分子的值来评估所述候选分子分别表现出所需功能性的程度,并且当所述迭代循环的一个或多个停止标准未得到满足时,从所述测量值计算适合度函数,并且使用所述适合度函数与所述机器学习模型的组合来选择用于后续迭代的另外的候选序列。
[0389]
81.如方面80所述的方法,其中执行所述迭代循环的步骤还包括:当一个或多个停止标准未得到满足时,基于包括所述候选分子的序列的更新的分子训练数据集来更新所述机器学习模型,并且在基于所述更新的训练数据集被更新之后,使用所述适合度函数与所述机器学习模型的组合来选择用于后续迭代的另外的候选序列。
[0390]
82.如方面80所述的方法,其中所述分子是dna分子并且所述序列是核苷酸序列。
[0391]
83.如方面80所述的方法,其中所述分子是rna分子并且所述序列是核苷酸序列。
[0392]
84.如方面80所述的方法,其中所述分子是聚合物并且所述序列是化学单体的序列。
[0393]
85.如方面1-40中任一项所述的方法,其中所述候选蛋白质包括以下中的一种或多种:抗体、酶、激素、细胞因子、生长因子、凝血因子、抗凝因子、白蛋白、抗原、佐剂、转录因子或细胞受体。
[0394]
86.如方面1-40中任一项所述的方法,其中提供所述候选蛋白质用于选择性地结合一种或多种其它分子。
[0395]
87.如方面1-40中任一项所述的方法,其中提供所述候选蛋白质以催化一种或多种化学反应。
[0396]
88.如方面1-40中任一项所述的方法,其中提供所述候选蛋白质用于长程信号传导。
[0397]
89.如方面1-40中任一项所述的方法,所述方法还包括基于所述候选蛋白质生成或制造最终产品。
[0398]
90如方面1-40中任一项所述的方法,其中一种或多种细胞由所述候选蛋白质产生。
[0399]
91.如方面90所述的方法,其中将由所述候选蛋白质产生的细胞导向或置于一个或多个仓中。
[0400]
92.如方面1-40中任一项所述的方法,其中所述候选蛋白质通过高通量功能筛选来确定。
[0401]
93.如方面92所述的方法,其中所述高通量功能筛选通过测量与所述候选蛋白质对应的细胞的荧光的微流体设备来实施。
[0402]
附加考虑因素
[0403]
尽管本文的公开阐述了许多不同实施例的详细描述,但是应当理解的是,这一描述的法律范围由在本专利和等效物的结尾处阐述的权利要求书的文字来定义。具体实施方式仅被解释为示例性的,并未描述每个可能的实施例,因为描述每个可能的实施例将是不切实际的。可以使用当前技术或在本专利申请日之后开发的技术来实施许多替代性实施例,所述实施例将仍落入权利要求书的范围内。
[0404]
以下附加考虑适用于上述讨论。在整个说明书中,多个实例可以实现被描述为单个实例的组件、操作或结构。尽管一种或多种方法的单独操作示出并被描述为单独的操作,但是单独操作中的一个或多个可以同时地执行,并且不需要按照所示顺序执行操作。在实例配置中呈现为独立部件的结构和功能可以实现为组合结构或部件。类似地,作为单个部件呈现的结构和功能可以作为单独的部件实施。这些和其它变化、修改、添加和改进都落入本文主题的范围内。
[0405]
另外,某些实施例在本文中被描述为包含逻辑或多个例程、子例程、应用或指令。其可以构成软件(例如,在机器可读介质上或在传输信号中具体化的代码)或硬件。在硬件中,例程等是能够执行某些操作的有形单元并且可以按照某种方式进行配置或布置。在实例实施例中,一个或多个计算机系统(例如,独立的客户端或服务器计算机系统)或者计算机系统的一个或多个硬件模块(例如,处理器或处理器组)可以通过软件(例如,应用或应用部分)被配置成操作以执行如本文所描述的某些操作的硬件模块。
[0406]
在各个实施例中,硬件模块可以机械地或电子地实施。例如,硬件模块可以包括被永久地配置成执行某些操作的专用电路系统或逻辑(例如,专用处理器,如场可编程门阵列(fpga)或专用集成电路(asic))。硬件模块还可以包括通过软件被临时地配置成执行某些操作的可编程逻辑或电路系统(例如,如专用处理器或其它可编程处理器中所包含的)。应了解到,在专用且永久配置的电路系统中或在临时配置的电路系统中(例如,通过软件进行配置)机械地实施硬件模块的决策可能受成本和时间考虑驱使。
[0407]
因此,术语“硬件模块”应被理解为涵盖有形实体,是指被物理地构造、永久地配置(例如,硬连线)或临时地配置(例如,编程)为按照一定方式操作或者执行本文所述的某些操作的实体。考虑到硬件模块被临时配置(例如,编程)的实施例,无需在任何一个时刻配置或实例化每个硬件模块。例如,在硬件模块包括使用软件来配置的通用处理器的情况下,通用处理器在不同时间可以被配置成对应的不同硬件模块。因此,软件可以配置处理器例如以在一个时刻构成特定的硬件模块并且在不同时刻构成不同的硬件模块。
[0408]
硬件模块可以向其它硬件模块提供信息,并且从其它硬件模块接收信息。因此,所述硬件模块可以被认为是通信地耦接的。在同时存在多个此类硬件模块的情况下,可以通过连接硬件模块的信号传输(例如,通过适当的电路系统和总线)来实现通信。在其中在不同时间配置或实例化多个硬件模块的实施例中,可以例如通过在多个硬件模块能够访问的存储器结构中存储和检索信息来实现此类硬件模块之间的通信。例如,一个硬件模块可以执行操作并将这种操作的输出存储在其所通信耦合的存储器装置中。然后,另一个硬件模块可以在以后的时间访问这一存储器装置以检索和处理所存储的输出。硬件模块还可以启动与输入或输出装置的通信,并且可以对资源(例如,信息的集合)进行操作。
[0409]
本文所述的示例方法的各种操作可以至少部分地由被临时配置(例如,通过软件)或永久配置为执行相关操作的一个或多个处理器执行。无论是临时配置还是永久配置,此类处理器都可以构成处理器实现的模块,这些模块运行以执行一个或多个操作或功能。在一些示例性实施方案中,本文所指的模块可以包括处理器实现的模块。
[0410]
类似地,本文所述的方法或例程可以至少部分地由处理器实现。例如,一种方法的至少一些操作可以由一个或多个处理器或处理器实现的硬件模块执行。某些操作的性能可以分布在一个或多个处理器之间,不仅驻留在单个机器内,而且可以跨多个机器部署。在一些示例实施例中,一个或多个处理器可以位于单个位置,而在其它实施例中,处理器可以跨多个位置分布。
[0411]
某些操作的性能可以分布在一个或多个处理器之间,不仅驻留在单个机器内,而且可以跨多个机器部署。在一些示例性实施例中,一个或多个处理器或处理器实施的模块可以位于单个地理位置(例如,在家庭环境、办公室环境或服务器场内)。在其它实施例中,一个或多个处理器或处理器实施的模块可以跨多个地理位置分布。
[0412]
此详细描述应被解释为仅示范性并且未描述每个可能的实施例,因为描述每个可能的实施例将是不切实际的,即使不是不可能的。本领域的普通技术人员可以使用当前技术或在本技术的提交日期之后开发的技术来实施很多替代实施例。
[0413]
本领域的普通技术人员将认识到,在不脱离本发明的范围的情况下,可以关于上述实施例做出各种各样的修改、变更和组合,并且此些修改、变更和组合将被视为处于本发明概念的范围内。
[0414]
在本专利申请的末尾的专利权利要求并不旨在根据35u.s.c.
§
112(f)进行解释,除非明确地叙述了传统的手段加功能(means-plus-function)语言,例如在权利要求中明确叙述的“用于......的构件”或“用于......的步骤”。本文中所描述的系统和方法涉及对计算机功能的改进,并改进传统计算机的功能。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1