一种低毒性的抗心律失常的化合物及其制药用途
本发明属于制药领域,具体涉及一种低毒性的抗心律失常的化合物及其制药用途。
背景技术:
毛茛科ranunculaceae乌头属aconituml.植物全世界约有300余种,广泛分布于北温带地区,具有重要的药用价值,我国有乌头属植物200余种,各省区均有分布,主要分布在四川西部、云南西北部和西藏东部的高山地区,可以入药的有76种,在中医和少数民族医临床中应用广泛。例如,川乌、草乌和附子是中医常用品种,铁棒锤和黄草乌是藏医、羌医和彝医常用品种,具有祛风除湿,温经止痛和解热的功效,用于风寒湿痹,关节疼痛等症。
二萜类生物碱是乌头属植物发挥镇痛、抗炎作用的活性成分,但同时也具有心脏毒性和神经毒性。例如乌头碱引起毒性的机制是其能够与心肌细胞、神经细胞细胞膜上的电压敏感型钠通道特异性结合,使钠通道持续激活,钠离子持续内流,破坏内环境稳态,从而影响细胞功能,出现四肢麻木、呼吸抑制和心律失常,严重者可导致多器官功能损害甚至死亡。
草乌甲素是滇西乌头(aconitumbulleyanumdiels)、粗茎乌头(a.crassicaulew.t.wang)中的主要成分,现已经被开发为注射剂,片剂等多种剂型的药物,用于治疗风湿性和类风湿性关节炎、肩周炎等。有学者对来源于我国国家药品不良反应监测中心2009年-2017年的草乌甲素片不良反应报告进行了统计分析,发现在403例单用草乌甲素片患者中,有28.78%的服用者出现心悸的现象,17.62%的服用者出现味觉减退的情况,说明草乌甲素仍存在较强的毒性。心律失常是指心脏冲动的频率、节律、起源部位、传导速度或激动次序的异常。心律失常是心血管疾病中重要的一组疾病,它可单独发病,亦可与其他心血管病伴发。其预后与心律失常的病因、诱因、演变趋势、是否导致严重血流动力障碍有关,可突然发作而致猝死,亦可持续累及心脏而致其衰竭。
目前市场上的抗心律失常常用药物多数伴有不可忽视的不良反应,如利多卡因常表现为眩晕、感觉异常、意识模糊等神经系统不良反应,胺碘酮则有甲减或甲亢、肺损害等不良反应,维拉帕米若剂量调整不当则也会出现心血管、神经、内分泌等方面的不良反应,难以寻找到各方面均理想的抗心律失常药物。如何克服现有技术中抗心律失常药物的不良反应的问题,是目前亟需解决的问题。
实际上,在中医和民族医药临床中,大多数乌头类药材不是直接使用,而是要经过炮制后再使用。但是,在炮制过程中,乌头类药材中的成分转化规律还不明确。如果能够研究出不仅具有抗心律失常的作用,还能够降低毒性的炮制转化产物,对开发新的抗心律失常药物具有重要意义。
技术实现要素:
本发明的一个目的在于提供一种低毒性的抗心律失常的化合物及其制药用途。
本发明的另一个目的在于提供前述化合物在制备砂炒滇西乌头或砂炒粗茎乌头标志性成分中的用途。
本发明提供了一种式i所示化合物、或其药学上可接受的盐、或其立体异构体:
式i中,r1、r2、r3、r4各自独立的选自c1~4烷基。
进一步地,所述化合物的结构如式ii所示:
进一步地,所述化合物的结构如下所示:
本发明还提供了前述化合物1、或其药学上可接受的盐、或其立体异构体的制备方法,其特征在于:所述方法包括以下步骤:
(1)制备草乌甲素炮制产物:取草乌甲素,加有机溶剂溶解,蒸干有机溶剂,油浴,得到草乌甲素炮制产物;
(2)分离纯化:将草乌甲素炮制产物用有机溶剂溶解后进行硅胶柱层析分离纯化,梯度洗脱后收集包含权利要求3所述化合物的流分,然后分离出rf值为0.41的洗脱物,采用制备液相色谱纯化,得到权利要求3所述化合物。
进一步地,步骤(1)中,所述有机溶剂为二氯甲烷,所述油浴的温度为150~170℃,时间为20~40min。
进一步地,步骤(1)中,所述有机溶剂为二氯甲烷,所述油浴的温度为160℃,时间为30min。
进一步地,步骤(2)中,所述硅胶柱层析分离纯化时,梯度洗脱的洗脱剂为石油醚-丙酮-三乙胺混合溶液,石油醚-丙酮-三乙胺的体积比依次为8:1:0.01,6:1:0.01,3:1:0.01;
所述rf值为0.41的洗脱物是在以下薄层层析色谱条件下检测的:展开剂为石油醚:丙酮:三乙胺体积比=3:3:0.01的混合溶液;所述制备液相色谱纯化时的洗脱剂为:体积比为42:58的乙腈-0.03mol/l碳酸氢铵溶液,ph为9.5。
本发明还提供了一种低毒性的抗心律失常药,它是以上述化合物、或其药学上可接受的盐、或其立体异构体为活性成分,加上药学上可接受的辅料制得的制剂。
进一步地,所述制剂为口服制剂、注射制剂、栓剂或透皮制剂。
本发明还提供了上述化合物、或其药学上可接受的盐、或其立体异构体在制备低毒性的抗心律失常药中的用途。
进一步地,所述抗心律失常药引起的心脏毒性低;和/或,所述抗心律失常药引起的急性毒性低。
本发明还提供了一种砂炒滇西乌头或砂炒粗茎乌头,其特征在于:它含有上述的化合物。
本发明还提供了上述的化合物在制备砂炒滇西乌头或砂炒粗茎乌头标志性成分中的用途。
滇西乌头、粗茎乌头为毛茛科植物滇西乌头(aconitumbulleyanumdiels)、粗茎乌头(a.crassicaulew.t.wang)的干燥块根。
实验结果表明,本发明提供的草乌甲素炮制产物化合物1具有优异的抗心律失常活性,能延缓乌头碱诱发心律失常大鼠的室性早搏(vpb)潜伏期,有效阻止vpb的进一步发展,并且在0.60mg/kg剂量下,化合物1的抗心律失常作用甚至优于阳性药物利多卡因。
更重要的是,本发明化合物1给药30min内未引起vpb、室性心动过速(vt)和室颤(vf)等心律失常现象,与草乌甲素相比,化合物1的心脏毒性明显降低;与已知的印乌碱砂炒产物δ15,16-16-demethoxyindaconitine相比,本发明化合物1的急性毒性明显更低,安全性更高,并且具有剂量范围更广的优势。本发明为制备低毒性的抗心律失常药物提供了一种新的选择。
草乌甲素是滇西乌头、粗茎乌头药材中的主要成分。以本发明化合物1为标志性成分,能够在砂炒滇西乌头、粗茎乌头时将本发明化合物1用作控制砂炒滇西乌头或砂炒粗茎乌头质量的标准品。本发明为砂炒滇西乌头或砂炒粗茎乌头的质量控制提供了新的方法。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
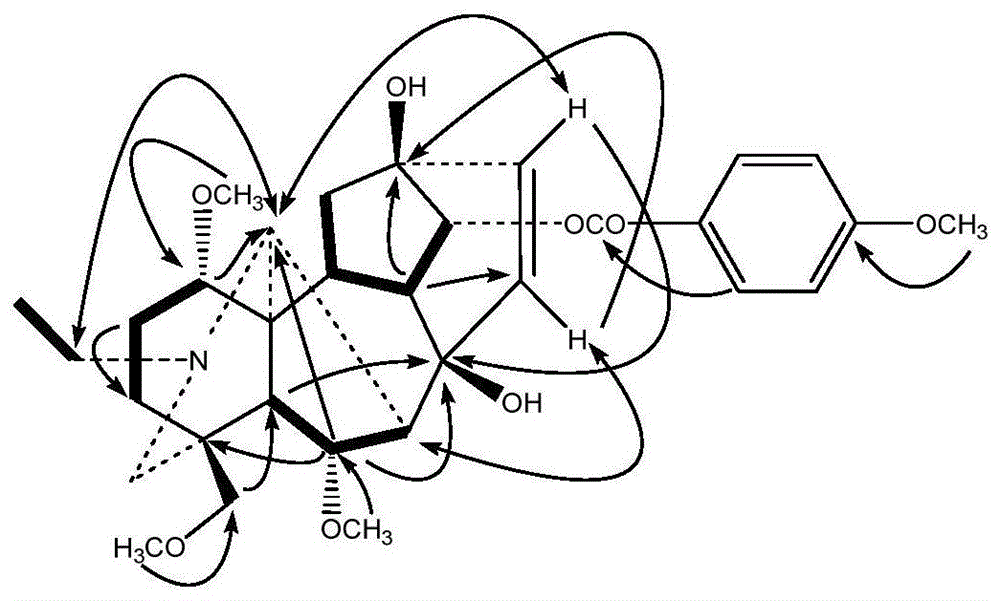
图1为化合物1的1h-1hcosy(-),hmbc(h→c)和noesy
图2为sd大鼠的特征心电图;图中,a:正常心电图;b:室性早搏;c:室性心动过速;d:室颤。
图3为化合物1组(16-demethoxy-δ15(16)-8-deacetylcrassicaulinea)、模型对照组(ctrl)、阳性对照组利多卡因(lid)、阳性对照组普罗帕酮(pro)对sd大鼠vpb潜伏期的影响;图中,与模型对照组比较:*p<0.05;与阳性对照组利多卡因比较:+p<0.05;与阳性对照组普罗帕酮比较:#p<0.05。
图4为不同剂量化合物1对sd大鼠vt发生率的影响。
图5为化合物1(16-demethoxy-δ15(16)-8-deacetylcrassicaulinea)与利多卡因(lid)心律失常完全抑制率结果。
图6为化合物δ15,16-16-demethoxyindaconitine的结构。
图7为化合物1的高分辨质谱图。
图8为化合物1的1h-nmr图谱。
图9为化合物1的13c-nmr图谱。
图10为化合物1的hmbc代表性图谱。
图11为化合物1的1h-1hcosy代表性图谱。
图12为化合物1的noesy代表性图谱。
具体实施方式
本发明所用原料与设备均为已知产品,通过购买市售产品所得。
实施例1本发明化合物1的制备
1.柱色谱分离样品的制备
取草乌甲素1.83g,置250ml圆底烧瓶中,加二氯甲烷25ml溶解,用旋转蒸发仪在40℃条件下蒸干溶剂,使草乌甲素均匀地附着在圆底烧瓶内壁,并于160℃下油浴30min,取出,放凉,得到草乌甲素炮制产物(1.65g),供上柱用。
2.分离纯化
将草乌甲素炮制产物用二氯甲烷溶解后,进行硅胶柱层析(200-300目,120g)分离纯化,分别用石油醚-丙酮-三乙胺8:1:0.01(0.4l),6:1:0.01(3l),3:1:0.01(1.2l)梯度洗脱,薄层板检视,合并相近流分后,得到a、b、c三个组分。组分b(1g)进一步采用制备薄层层析,石油醚:丙酮:三乙胺(3:3:0.01)展开,室温下晾干后,碘蒸汽在边缘显色后,发现有三个斑点,rf值分别为0.41、0.51和0.77,分别刮下相应条带上的硅胶,用甲醇洗脱,回收溶剂后,得到三个洗脱物。rf值为0.41的洗脱物(200mg)采用制备液相进一步纯化,色谱条件为乙腈-0.03mol/l碳酸氢铵溶液42:58(浓氨水调ph9.5,检测波长为260nm),收集的流分减压回收至无乙腈味,水相用二氯甲烷萃取两次,合并二氯甲烷萃取液,加无水硫酸钠脱水后过滤,滤液浓缩,得到化合物16-demethoxy-δ15(16)-8-deacetylcrassicaulinea(即化合物1,110mg)。
3.化合物1结构表征
化合物1的结构表征图谱如图7~图12所示。
化合物1:白色粉末,
草乌甲素的c-15、c-16的化学位移分别为39.3和83.7,从13c-nmr中可以看出:化合物1缺少c-16位的甲氧基信号,并且c-15、c-16的化学位移分别向低场迁移到130.1和131.8。因此,在1h-nmr中,δh5.61(1h,d,j=9.5hz)和δh5.91(1h,d,j=9.5hz)分别归属为h-15和h-16,表明c-15和c-16之间形成了双键,hmbc谱中,h-15与c-13,h-16与c-8远程相关,也证实c-15与c-16之间存在双键结构(图1),根据1h-nmr,13c-nmr及2d-nmr(hmbc,1h-1hcosy,noesy)数据(表1),可以确定化合物1的结构为16-demethoxy-δ15(16)-8-deacetylcrassicaulinea(n-乙基-δ15(16)-14α-[(4′-甲氧基)苯甲酰氧基]-1α,6α,18-三甲氧基-乌头烷-8β,13β-二醇)。
表1化合物1的nmr数据
以下通过实验例证明本发明的有益效果。
本发明实验例采用的试药、试剂与动物:
草乌甲素(dst190704-057,纯度≥98%,成都德思特生物技术有限公司);乌头碱(wtz10013,纯度99%,陕西昊辰生物科技有限公司);盐酸普罗帕酮(101190-201702,纯度99.8%)和盐酸利多卡因(100341-201403,纯度93.4%)购自中国食品药品检定研究院;乌拉坦(2017090501,成都市科龙化工试剂厂)。
spf级昆明小鼠,体重(18±2)g,雌雄兼用;spf级sd大鼠,雌雄兼用,体重(200±20)g,均由成都达硕实验动物有限公司提供,生产许可证号:scxk(川)-2020-030。
实验例1草乌甲素及其砂炒产物化合物1的心脏毒性试验1.试验方法
取spf级sd大鼠20只,雌雄各半,随机分为草乌甲素组和化合物1组(n=10,实验前12小时禁食不禁水)。腹腔注射20%乌拉坦(剂量1.2g/kg)麻醉,仰位固定,将针形电极插入四肢皮下,使用bl-420f多功能生理记录仪观察大鼠ⅱ导联心电图20min后给药,观察给药后30min内心电图变化。通过前期预实验,发现草乌甲素0.10mg/kg可引起大鼠出现室性早搏(ventricularprematurebeat;vpb)、室性心动过速(ventriculartachycardia;vt)和室颤(ventricularfibrillation;vf)等心律失常现象,通过观察相同剂量下化合物1是否引起上述心律失常现象,可直观反映经过炮制后,草乌甲素结构上的改变是否引起其毒性的降低。
2.心脏毒性试验结果
结果见表2和图2。0.10mg/kg草乌甲素可引起正常大鼠出现vpb、vt及vf等心律失常现象,心律失常潜伏期为(182.8±84.6)s,同时伴随胸部鼓起、抽搐,喉部发生不适声等反应;但是相同剂量下化合物1未出现心律失常现象和行为学反应。所以,与草乌甲素相比,化合物1心脏毒性明显降低。
表2草乌甲素及其砂炒产物的心脏毒性比较(
“—”表示在给药30min内,未出现任何心律失常现象。
上述实验结果表明,与草乌甲素相比,本发明砂炒产物化合物1的心脏毒性明显降低。
实验例2化合物1抗心律失常活性研究
1.试验方法
取111只sd大鼠,雌雄兼用,随机分为8组,即空白溶剂组(配制方法:取4ml1%盐酸乙醇,加生理盐水定容至100ml)、模型对照组(乌头碱;剂量0.03mg/kg)、阳性对照组(利多卡因组,剂量5mg/kg;普罗帕酮组,剂量3.2mg/kg)、化合物1不同剂量组(0.05mg/kg、0.20mg/kg、0.40mg/kg和0.60mg/kg)。乌头碱、阳性药和各受试药物均采用空白溶剂的配制方法进行配制。空白溶剂组只给等体积的空白溶剂,观察溶剂是否对sd大鼠心电图有影响。
实验开始前,sd大鼠腹腔注射20%乌拉坦(剂量1.2g/kg)麻醉,仰位固定,将针形电极插入四肢皮下,使用bl-420f多功能生理记录仪记录大鼠正常ⅱ导联心电图20min后,暴露股静脉,从股静脉分别注射相应剂量的受试药物、阳性药和等体积的生理盐水,10min后再从股静脉注射乌头碱(0.03mg/kg)建立心律失常模型,记录30min内各组大鼠心律失常的发生率、第一次出现vpb的时间以及是否出现心动过速。
2.数据处理
实验数据采用spss20.0软件进行统计学分析,数据用
3.实验结果
3.1化合物1对乌头碱诱发大鼠vpb潜伏期的影响
vpb是心律失常模型大鼠的初期表现,其心电图表现为qrs波群提前出现,形态宽大畸形,t波方向与qrs主波方向相反,并且p波消失。vpb潜伏期是指注射完乌头碱后到第一次出现vpb的时间,vpb潜伏期的长短可以衡量受试药物对抗乌头碱导致的心律失常作用的大小,潜伏期越长,表明受试药物的抗心律失常作用越强。
模型对照组大鼠经股静脉注射乌头碱后出现典型的vpb、vt,并有一部分大鼠可出现vf,并且持续30min以上,表明心律失常模型复制成功。
乌头碱致心律失常模型对照组中,sd大鼠vpb潜伏期为(116.5±36.4)s;阳性药利多卡因组(lid)为(280.3±128.7)s,普罗帕酮组(pro)为(193.3±39.9)s;与模型对照组相比,两个阳性药均有显著性差异(p<0.05)。
与模型对照组相比,除了0.05mg/kg组外,化合物1其他剂量组(0.20mg/kg、0.40mg/kg和0.60mg/kg)均有显著性差异(p<0.05);0.40mg/kg和0.60mg/kg剂量组的vpb潜伏期分别为(493.7±148.7)s和(547.3±241.8)s;与利多卡因组相比,0.40mg/kg剂量组有显著性差异(p<0.05);与普罗帕酮组相比,0.40mg/kg和0.60mg/kg剂量组有显著性差异(p<0.05),如图3所示。
上述结果表明化合物1能够剂量依赖性地延缓乌头碱诱发心律失常出现的时间(vpb潜伏期),并且0.40mg/kg和0.60mg/kg剂量组的抗心律失常作用明显优于阳性药。
3.2化合物1对乌头碱诱发大鼠vt发生率的影响
vt是vpb进一步发展的结果,vt的心电图特点为:连续出现3次或3次以上的室性期前收缩,qrs波群宽大畸形,无恒定的p波。vt发生率可以评价受试药物是否能够阻止vpb的进一步发展,vt发生率越低,表明受试药物越能阻止vpb的进一步发展,药效越好。
按照卡方检验的数据要求,需要将vt发生率相近的剂量组进行合并,因此,化合物1由原来的4个组变为3个组:0.05-0.20mg/kg、0.40mg/kg和0.60mg/kg剂量组。卡方检验结果显示:3个剂量组之间的vt发生率有差异(χ2=14.704,p=0.001),如表3所示;两两比较结果显示:与0.05-0.20mg/kg剂量组相比,0.60mg/kg剂量组的vt发生率显著降低(χ2=11.313,p=0.001),仅为18.2%,如图4所示。
上述结果表明,化合物1能够剂量依赖性地降低vt发生率,有效阻止vpb的进一步发展。
表3化合物1不同剂量组组间vt发生率总卡方分析
3.3化合物1对乌头碱诱发大鼠心律失常完全抑制率的影响
心律失常发生率是指大鼠预先静脉推注受试药物,再用乌头碱建立心律失常模型后,30min内出现心律失常的比例(出现vpb、vt或vf中的任何一种心电图变化,均算作出现心律失常)。心律失常完全抑制率(%)=100%-心律失常发生率(%),即30min内未出现心律失常的比例,可以评价受试药物是否能够完全抑制住乌头碱的致心律失常作用。心律失常完全抑制率是最能直观反映受试药物药效强弱的指标,心律失常完全抑制率越高,说明受试药物越能够抵抗乌头碱的致心律失常作用,药效越好。
由于0.05mg/kg和0.20mg/kg剂量组不出现心律失常的大鼠只数偏少,按照原来的分组无法满足卡方检验的要求,因此,将0.05mg/kg和0.20mg/kg剂量组合并为0.05-0.20mg/kg剂量组进行卡方分析。
卡方检验结果显示:化合物1不同剂量组(0.05-0.20mg/kg、0.40mg/kg和0.60mg/kg)及利多卡因(5.0mg/kg)组的心律失常完全抑制率有差异(χ2=15.457,p=0.001),并且,在0.05mg/kg-0.60mg/kg剂量范围内,随着剂量增大,化合物1的心律失常完全抑制率呈现逐渐上升的趋势,如表4,图5所示。两两比较结果显示,与0.05-0.20mg/kg剂量组相比,0.60mg/kg剂量组和利多卡因的心律失常完全抑制率显著升高(χ2=14.258,p=0.0002;χ2=6.820,p=0.009)。并且0.60mg/kg剂量组的心律失常完全抑制率为56%,利多卡因为42.9%,在该剂量下,化合物1的抗心律失常作用更好。
表4化合物1和利多卡因心律失常完全抑制率总卡方分析
上述实验结果表明,化合物1有优异的抗心律失常活性,能延缓乌头碱诱发心律失常大鼠的vpb潜伏期,有效阻止vpb的进一步发展,并且在0.60mg/kg剂量下,化合物1的抗心律失常作用优于阳性药物利多卡因。
实验例3化合物1与δ15,16-16-demethoxyindaconitine急性毒性的比较
1.实验方法
急性毒性是药物的毒理学研究和安全性评价的重要组成部分,是指机体(人或者动物)24h内一次或者多次给予受试药物后,在短期时间内(一般不超过14d)产生的毒性反应。急性毒性是认识和研究药物对机体毒效应的第一步,同时,也可以通过观察机体(人或动物)出现的中毒症状初步判断其毒性作用靶器官。急性毒性所获得的信息对于某些药物ⅰ期临床试验起始剂量的选择具有重要参考价值,并可以提供一些与人类药物过量急性中毒的信息。
急性毒性试验有很多测定方法,其中半数致死剂量法(ld50)是比较经典的方法,本实验采用bliss法计算受试药物的ld50,bliss法是目前公认最准确的测定方法,也是目前我国卫生部规定作为新药ld50测定评定必须采用的方法。ld50是指引起一组受试实验动物半数死亡的剂量,ld50数值越小表示受试药物的毒性越强;ld50数值越大,则毒性越低。
1.1实验药物配制
δ15,16-16-demethoxyindaconitine是在印乌碱的砂炒产物中分离得到的一种二萜生物碱,结构如图6所示。取δ15,16-16-demethoxyindaconitine(参照申请专利号为cn201911107946.7的中国专利申请记载的方法制得)、化合物1(按照实施例1的方法制得,纯度>95%),用少量1%盐酸乙醇溶解后用生理盐水配制成不同浓度,充分混匀,放入冰箱中冷藏保存备用。
1.2预实验
选取小鼠32只,雌雄各半,按体重分层后随机分组,每组4只。每个样品按照不同浓度各选择一组小鼠进行预实验,每只小鼠均按0.15ml/10g尾静脉给药一次(实验前12h均禁食不禁水),分别密切观察小鼠给药后4h内出现的毒性反应及死亡的情况,最后计算出每个药物的最高致死剂量(dm,此处指刚好导致实验动物全部死亡的剂量)以及最低致死剂量(dn,此处指刚好不引起实验动物死亡的剂量)供正式实验参考。
根据预实验结果推测δ15,16-16-demethoxyindaconitine和化合物1的最高致死剂量(dm)和最低致死剂量(dn)约分别为20mg/kg,26.78mg/kg和8.192mg/kg,10.04mg/kg。
1.3正式实验
选用spf级昆明小鼠110只,体质量(18±2)g,雌雄各半,按体重分层后随机分为11组,给药前12h禁食不禁水。根据预实验结果,选择组间等比0.8设计各剂量组,分别为δ15,16-16-demethoxyindaconitine6.55、8.19、10.24、12.8、16mg/kg组,化合物110.31、12.89、16.12、20.15、25.18mg/kg组,同时设置溶剂对照组。每只小鼠均按照0.15ml/10g尾静脉给药1次,分别观察给药后7d内出现的中毒症状,死亡情况及死亡时间,对观察期满或毒死的小鼠进行解剖,肉眼观察主要脏器的改变。按bliss法计算动物的半数致死剂量(ld50)及其95%的可信限。
2.实验结果
表5不同化合物死亡情况、中毒症状及尸检结果
表6δ15,16-16-demethoxyindaconitine及化合物1急性毒性实验结果
小鼠中毒症状见表5,存活小鼠在24h内逐渐恢复正常,此后未出现异常行为,观察7天后全部处死,解剖毒死和观察期满处死的动物,均未见内脏明显异常。结果表明,δ15,16-16-demethoxyindaconitine和化合物1的毒性靶器官均可能为中枢神经系统。
采用bliss法计算ld50,结果见表6。化合物δ15,16-16-demethoxyindaconitine的ld50为9.017mg/kg,95%可信区间为7.990~10.031mg/kg;化合物1的ld50为12.108mg/kg,95%可信区间为10.646~13.358mg/kg。与δ15,16-16-demethoxyindaconitine相比,化合物1的毒性明显更低(ld50=12.108mg/kg>9.017mg/kg),即安全性更高,并且具有剂量范围更广的优势(0.05mg/kg~0.60mg/kg)。
上述实验结果表明,与δ15,16-16-demethoxyindaconitine相比,本发明化合物1的急性毒性明显更低,安全性更高,并且具有剂量范围更广的优势。
综上,本发明提供了式i所示低毒性的抗心律失常的化合物及其制药用途。实验结果表明,本发明提供的化合物具有优异的抗心律失常活性,能延缓乌头碱诱发心律失常大鼠的vpb潜伏期,有效阻止vpb的进一步发展,并且在0.60mg/kg剂量下,化合物的抗心律失常作用甚至优于阳性药物利多卡因。更重要的是,与草乌甲素相比,本发明化合物的心脏毒性明显降低;与已知的印乌碱砂炒产物δ15,16-16-demethoxyindaconitine相比,本发明化合物的急性毒性明显更低,安全性更高,并且具有剂量范围更广的优势。本发明为制备低毒性的抗心律失常药物提供了一种新的选择,为砂炒滇西乌头或砂炒粗茎乌头的质量控制提供了一种新的方法。
- 还没有人留言评论。精彩留言会获得点赞!