一种基于铃木反应的联苯吡菌胺的合成方法与流程
本发明属于农药分子有机化学合成领域,具体涉及一种使用自主涉及的、创新的合成方法,制备和产业化联苯吡菌胺这一农药分子。
背景技术:
联苯吡菌胺(bixafen)是一种由拜耳作物科学公司开发的吡唑酰胺雷杀菌剂,也是目前增长最快的琥珀酸脱氢酶抑制剂(sdhi)雷杀菌剂中的重要成员。其结构于2006年公开并于2010年完成首次登记。自2011年上市以来,已有富美实及住友化工等公司参与其相关研发,拜耳作物科学公司预估其年销售额将突破3亿欧元(折合约20亿人民币)。
联苯吡菌胺为内吸性杀菌剂,具有广泛的杀菌谱,一般用于叶面喷雾。联苯吡菌胺通过干扰病原菌线粒体呼吸电子传递链中复合体ii上的琥珀酸脱氢酶,抑制线粒体的相关功能,继而阻止其通过呼吸作用产生能量,抑制病原菌生长并最终导致细胞死亡。据报道,联苯吡菌胺队大麦网斑病、苹果白粉病等相关病原菌具有极好的治疗和保护效果,而其于丙硫菌唑的复配产品对壳针孢菌引起的病害具有非常好的抑制效果。
联苯吡菌胺自2011年上市后,发展迅速,尤其是随着联苯吡菌胺与丙硫菌唑复配产品xpro系列的成功上市,其市场份额增长更为显著。仅2011年上半年,其销售额已突破1亿欧元(折合约7亿人民币)大关。直到2020年,其市场份额仍处于快速增长中,未来市场仍乐观可期。
以下为联苯吡菌胺的分子结构。
目前,联苯吡菌胺的合成主要为以下两种方式:
方法一使用3-二氟甲基-1-甲基-1-哌唑-4-酰氯为原料,与2-(3,4-二氯苯基)-4-氟苯胺发生缩合反应得到产物;
该方法步骤简便,但所用原料价格较高,且酰氯化合物性质不稳定,极易降解产生杂质,导致反应所得产物纯度不高,于纯化生产不利;
方法二使用3-二氟甲基-4-溴-1-甲基吡唑作为原料,与2-(3,4-二氯苯基)-4-氟苯胺在钯金属催化剂和一氧化碳氛围下发生羰化反应,得到产物;
该方法所得产物纯度较高,但过程使用的贵金属催化剂制备困难,且反应涉及一氧化碳气体,毒性较高并需特种气体反应设备,生产难度较大。
综上,针对联苯吡菌胺的合成工艺改良非常有必要。
技术实现要素:
针对现有技术的不足,为解决联苯吡菌胺合成中安全风险大等问题,本发明提供了一种经过自主设计的、创新的合成方法。该方法的创新点在于,采用逆合成分析方式,通过使用廉价的3-二氟甲基-1-甲基-1-哌唑-4-羧酸为原料进行两步反应,得到产物。
本发明所提供的联苯吡菌胺的合成方法包括:以3-二氟甲基-1-甲基-1-哌唑-4-羧酸为原料,在偶联试剂的作用下,与2-碘-4-氟苯胺(式ii)发生缩合反应,后在钯金属催化剂的作用下与3,4-二氯苯基硼酸(式iv)发生铃木反应,生成联苯吡菌胺产物。反应过程如下式所示:
根据本发明的一种实施方式,所述合成方法包括以下步骤:
s1.3-二氟甲基-1-甲基-1-哌唑-4-羧酸在偶联试剂和4-二甲基氨基吡啶的作用下,在有机溶剂中与2-碘-4-氟苯胺发生缩合反应,生成3-二氟甲基-n-(2-碘-4-氟苯基)-1-甲基-1-哌唑-4-酰胺;
s2.3-二氟甲基-n-(2-碘-4-氟苯基)-1-甲基-1-哌唑-4-酰胺在钯金属催化剂和碱性试剂存在的条件下,于有机溶剂中发生铃木反应,生成3-二氟甲基-1-甲基-n-[4-氟-3’-4’-二氯-(1,1’-联苯基)-2-基]-1-哌唑-4-酰胺,即联苯吡菌胺产品。
优选地,在步骤s1中,所述偶联试剂为1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,所述有机溶剂为二氯甲烷;在步骤s2中,所述钯金属催化剂为四(三苯基膦)钯,所述碱性试剂为碳酸钾,所述有机溶剂为四氢呋喃。
步骤s1中使用偶联试剂及有机溶剂:所述偶联试剂为n,n’-羰基二咪唑、二环己基碳二亚胺、二异丙基碳二亚胺、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、羟基苯并三唑;所述有机溶剂为二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺、甲醇、乙醇、异丙醇中的至少一种或其组合;优选地,所述偶联试剂为1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,所述有机溶剂为二氯甲烷。步骤s2中使用钯金属催化剂、碱性试剂及有机溶剂:所述钯金属催化剂为四(三苯基膦)钯、二氯化钯/三苯基膦、醋酸钯/三苯基膦、三氟乙酸钯/三苯基膦中的至少一种或其组合;碱性试剂为氢氧化锂、碳酸锂、氢氧化钠、碳酸钠、氢氧化钾、碳酸钾中的至少一种或其组合;有机溶剂为二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺、甲醇、乙醇、异丙醇中的至少一种或其组合;优选地,所述把金属催化剂为四(三苯基膦)钯,所述碱性试剂为碳酸钾,所述有机溶剂为四氢呋喃。
与现有合成方式相比,本发明所提供的合成路线具有如下特点:
1.本发明所提供的合成路线不涉及高活性原料或化合物,不产生降解,所得产物纯度较高;
2.本发明所提供的合成路线不涉及有毒、易燃易爆气体,无需特种气体反应设备,制备工艺简单,安全性高;
3.本发明涉及的贵金属催化剂简单易得,催化剂制备成本较低。
本发明所提的合成路线反应条件相对温和,一并解决了已有路线的若干缺点,具有重大的商业价值及潜力。
附图说明
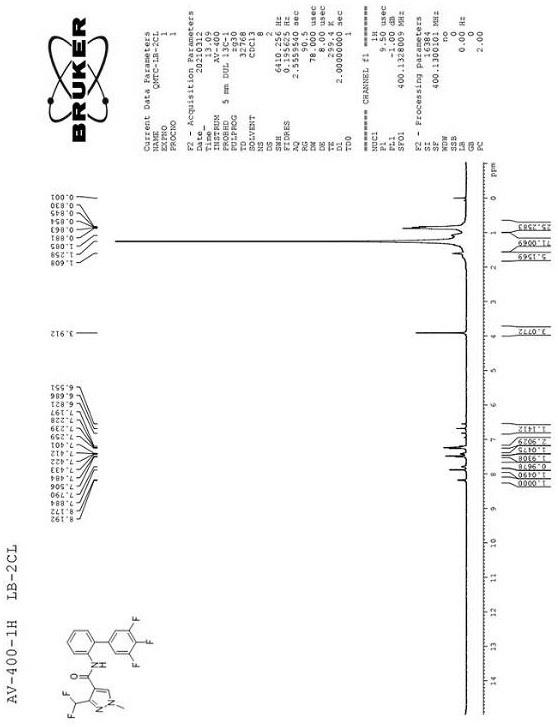
图1是化合物联苯吡菌胺核磁共振氢谱图。该核磁共振氢谱计算纯度为95%。
实施例一
取一圆底烧瓶,内置一搅拌子。向其中加入3-二氟甲基-1-甲基-1-哌唑-4-羧酸(5.00g,28.5mmol)、2-碘-4-氟苯胺(7.50g,34.2mmol)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(6.0g,31.35mmol)、4-二甲基氨基吡啶(4.18g,34.2mmol)及二氯甲烷(50ml)。所得混合物在室温下搅拌隔夜,薄层色谱监测反应完成后,向反应体系内加入蒸馏水(50ml)。分出有机相,水相以二氯甲烷(50ml)洗涤3次。合并有机相并将有机相蒸发至干,所得粗产品经柱层析纯化得到白色固体状3-二氟甲基-n-(2-碘-4-氟苯基)-1-甲基-1-哌唑-4-酰胺纯品,产率82%,核磁共振氢谱计算纯度为92%。
实施例二
取一圆底烧瓶,内置一搅拌子。向其中加入3-二氟甲基-n-(2-碘-4-氟苯基)-1-甲基-1-哌唑-4-酰胺纯品(1g,2.65mmol)、3,4-二氯苯基硼酸(0.61g,3.18mmol)、四(三苯基膦)钯(500mg)、碳酸钾(1.46g,10.6mmol)及四氢呋喃(9ml)蒸馏水(3ml)。所得混合物至于氮气氛围下,加热至回流,反应隔夜。薄层色谱检测反应完成后,向其中加入水(20ml)及二氯甲烷(50ml),搅拌30分钟。分出有机相,有机相以饱和氯化铵溶液(20ml)洗涤并干燥,浓缩至干。所得粗品以柱层析进行纯化,得到联苯吡菌胺纯品,为淡黄色固体,产率91%,核磁共振氢谱计算纯度为95%。
- 还没有人留言评论。精彩留言会获得点赞!