一种串联环化反应合成2-氨基-3,3-二氯或二溴代色满酮的方法
本发明涉及化合物合成方法领域,具体涉及一种串联环化反应合成2-氨基-3,3-二氯或二溴代色满酮的方法。
背景技术:
色满酮,也称为苯并吡喃酮,是一类具有明显不同生物活性的化合物,因此被认为是药物化学中的一种基本骨架(eur.j.med.chem.2012,54,914.)。化学上,色满酮表示的是一类含色满酮衍生物的酮,它构成各种天然和合成化合物的基本骨架。其中的色满酮类衍生物可用于治疗血脂异常或2型糖尿病(bioorgan&medicalchem.2019,24,115162.),是设计和鉴定腺苷受体拮抗剂以治疗各种疾病的合适先导(bioorganchem.,2018,77,136–143.)。因此,建立合成具有丰富多样性次级结构的色满酮类化合物,尤其是当前尚未被报道过的色满酮类化合物的方法对于发展和发现色满酮的新应用具有重要意义。本发明报道一种合成2-氨基-3,3-二卤代色满酮类化合物的方法,这一方法所合成的色满酮产物均为新产物,在已有的文献中未见报道,因而对于色满酮在不同领域的应用研究均具有重要指导意义。
技术实现要素:
本发明的目的在于提供一种原料经济、操作简便的串联反应3,3-二卤代色满酮类化合物的方法。
本发明是这样来实现的:
将烯胺酮0.2mmol,n-卤代丁二酰亚胺0.5mmol,0.5mldmf置于装有搅拌磁子的10ml微波反应管中,然后将管密封并用于nova-2s微波反应器。将混合物在微波辐射下于60℃在微波辐射下搅拌8分钟。反应结束后冷却至室温,将所得混合物用乙酸乙酯(3×10ml)萃取。收集有机相,并用少量水洗涤三遍。萃取所得有机相用无水na2so4干燥后,过滤固体,并在减压下除去溶剂。将得到的残余物进行快速硅胶柱色谱处理,通过用混合的石油醚和乙酸乙酯(v/v=50∶1)洗脱得到纯产物。产物结构经过核磁共振、高分辨质谱以及代表性产物的单晶衍射测试等确证无误。
其中,烯胺酮的结构式为
式中r为c4~c5取代氢、氟、氯、溴、碘、硝基或烷基、烷氧基,x为氯或溴。
本发明的技术效果是:本发明所用的原料均简单易得、常态下较稳定,无需任何催化以及无水无氧等保护操作。过程简便,兼容于不同性质的烯胺酮包括二级烯胺酮和三级烯胺酮等进行反应合成不同类型的色满酮产物。可以通过化合物结构上的活性碳-卤键的转化设计多种不同的新有机合成反应。
附图说明
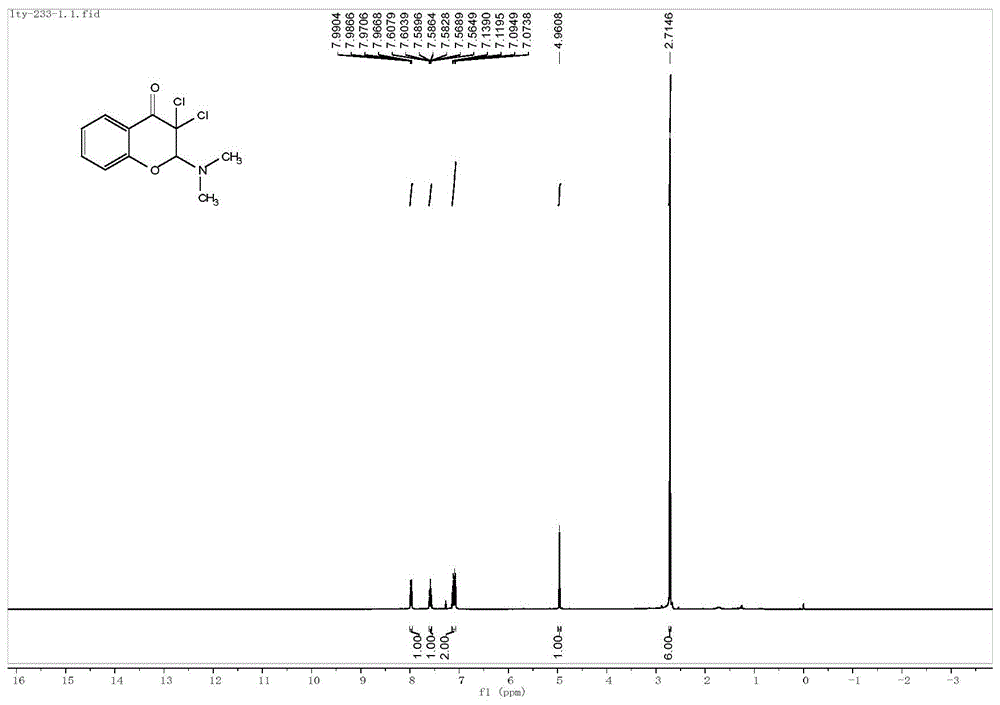
图1为本发明化合物3a的核磁共振氢谱图。
图2为本发明化合物3a的核磁共振碳谱图。
图3为本发明化合物3b的核磁共振氢谱图。
图4为本发明化合物3b的核磁共振碳谱图。
图5为本发明化合物3c的核磁共振氢谱图。
图6为本发明化合物3c的核磁共振碳谱图。
图7为本发明化合物3d的核磁共振氢谱图。
图8为本发明化合物3d的核磁共振碳谱图。
图9为本发明化合物3e的核磁共振氢谱图。
图10为本发明化合物3e的核磁共振碳谱图。
图11为本发明化合物3f的核磁共振氢谱图。
图12为本发明化合物3f的核磁共振碳谱图。
图13为本发明化合物3g的核磁共振氢谱图。
图14为本发明化合物3g的核磁共振碳谱图。
图15为本发明化合物3h的核磁共振氢谱图。
图16为本发明化合物3h的核磁共振碳谱图。
图17为本发明化合物3i的核磁共振氢谱图。
图18为本发明化合物3i的核磁共振碳谱图。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
本发明是这样实现的:将烯胺酮1(0.2mmol),n-卤代丁二酰亚胺2a(0.5mmol)和dmf(0.5ml)装入装有搅拌磁子的10ml微波反应管中。然后将管密封并用于nova-2s微波反应器。将混合物在微波辐射下于60℃在微波辐射下搅拌8分钟。冷却至室温后,将所得混合物用乙酸乙酯(3×10ml)萃取。收集有机相,并用少量水洗涤三遍。用无水na2so4干燥后,过滤固体,并在减压下除去溶剂。将得到的残余物进行快速硅胶柱色谱处理,通过用混合的石油醚和乙酸乙酯(v/v=50:1)洗脱,来得到纯产物。
反应通式如下:
3,3-二卤代色满酮类化合物的结构式为:
实施例1
本实施例中提供了3,3-二氯代色满酮类化合物的制备方法,制备如式3a所示的化合物的方法。
其制备步骤如下:将烯胺酮(0.2mmol),n-氯代丁二酰亚胺(0.5mmol)和dmf(0.5ml)装入装有搅拌磁子的10ml微波反应管中。然后将管密封并用于nova-2s微波反应器。将混合物在微波辐射下于60℃在微波辐射下搅拌8分钟。冷却至室温后,将所得混合物用乙酸乙酯(3×10ml)萃取。收集有机相,并用少量水洗涤三遍。用无水na2so4干燥后,过滤固体,并在减压下除去溶剂。将得到的残余物进行快速硅胶柱色谱处理,通过用混合的石油醚和乙酸乙酯(v/v=50:1)洗脱,来得到纯产物。
产率为74%。式3a所示的化合物结构表征如下:黄色液体;1hnmr(400mhz,cdcl3)δ7.99-7.97(m,1h),7.61–7.56(m,1h),7.14-7.7(m,2h),4.96(s,1h),2.71(s,6h);13cnmr(100mhz,cdcl3)δ180.7,159.6,137.1,129.3,122.5,117.9,116.7,97.7,84.5,41.2.esi-hrmscalcdforc11h12cl2no2+[m+h]+260.0240,found260.0246.
实施例2
本实施例中提供了3,3-二氯代色满酮类化合物的制备方法,制备如式3b所示的化合物的方法。
其制备步骤如下:将烯胺酮(0.2mmol),n-氯代丁二酰亚胺(0.5mmol)和dmf(0.5ml)装入装有搅拌磁子的10ml微波反应管中。然后将管密封并用于nova-2s微波反应器。将混合物在微波辐射下于60℃在微波辐射下搅拌8分钟。冷却至室温后,将所得混合物用乙酸乙酯(3×10ml)萃取。收集有机相,并用少量水洗涤三遍。用无水na2so4干燥后,过滤固体,并在减压下除去溶剂。将得到的残余物进行快速硅胶柱色谱处理,通过用混合的石油醚和乙酸乙酯(v/v=50:1)洗脱,来得到纯产物。
产率为73%。式3b所示的化合物结构表征如下:黄色液体;yellowliquid;1hnmr(400mhz,cdcl3)δ7.90(d,j=8.9hz,1h),6.69-6.66(m,1h),6.51(d,j=1.9hz,1h),4.97(s,1h),3.88(s,3h),2.71(s,6h);13cnmr(100mhz,cdcl3)δ179.6,167.1,161.8,130.9,111.6,110.0,100.5,98.1,84.5,55.8,41.2.esi-hrmscalcdforc12h14cl2no3+[m+h]+290.0345,found290.0353.
实施例3
本实施例中提供了3,3-二氯代色满酮类化合物的制备方法,制备如式3c所示的化合物的方法。
其制备步骤如下:将烯胺酮(0.2mmol),n-氯代丁二酰亚胺(0.5mmol)和dmf(0.5ml)装入装有搅拌磁子的10ml微波反应管中。然后将管密封并用于nova-2s微波反应器。将混合物在微波辐射下于60℃在微波辐射下搅拌8分钟。冷却至室温后,将所得混合物用乙酸乙酯(3×10ml)萃取。收集有机相,并用少量水洗涤三遍。用无水na2so4干燥后,过滤固体,并在减压下除去溶剂。将得到的残余物进行快速硅胶柱色谱处理,通过用混合的石油醚和乙酸乙酯(v/v=50:1)洗脱,来得到纯产物。
产率为75%。式3c所示的化合物结构表征如下:黄色液体;1hnmr(400mhz,cdcl3)δ7.68(s,1h),7.34–7.30(m,1h),6.90(d,j=8.5hz,1h),4.84(s,1h),2.63(s,5h),2.26(s,3h);13cnmr(100mhz,cdcl3)δ180.9,157.7,138.3,132.1,128.6,117.7,116.2,97.6,84.7,41.2,20.4.esi-hrmscalcdforc12h14cl2no2+[m+h]+274.0396,found274.0404.
实施例4
本实施例中提供了3,3-二氯代色满酮类化合物的制备方法,制备如式3d所示的化合物的方法。
其制备步骤如下:将烯胺酮(0.2mmol),n-氯代丁二酰亚胺(0.5mmol)和dmf(0.5ml)装入装有搅拌磁子的10ml微波反应管中。然后将管密封并用于nova-2s微波反应器。将混合物在微波辐射下于60℃在微波辐射下搅拌8分钟。冷却至室温后,将所得混合物用乙酸乙酯(3×10ml)萃取。收集有机相,并用少量水洗涤三遍。用无水na2so4干燥后,过滤固体,并在减压下除去溶剂。将得到的残余物进行快速硅胶柱色谱处理,通过用混合的石油醚和乙酸乙酯(v/v=50:1)洗脱,来得到纯产物。
产率为77%。式3d所示的化合物结构表征如下:黄色液体;1hnmr(400mhz,cdcl3)δ7.35(s,1h),7.19(d,j=9.0hz,1h),7.02(d,j=9.0hz,1h),4.89(s,1h),3.82(s,3h),2.72(s,6h);13cnmr(100mhz,cdcl3)δ180.9,154.849,154.4,126.8,119.3,116.4,109.0,97.6,84.6,55.9,41.3;esi-hrmscalcdforc12h14cl2no3+[m+h]+290.0345,found290.0356.
实施例5
本实施例中提供了3,3-二氯代色满酮类化合物的制备方法,制备如式3e所示的化合物的方法。
其制备步骤如下:将烯胺酮(0.2mmol),n-氯代丁二酰亚胺(0.5mmol)和dmf(0.5ml)装入装有搅拌磁子的10ml微波反应管中。然后将管密封并用于nova-2s微波反应器。将混合物在微波辐射下于60℃在微波辐射下搅拌8分钟。冷却至室温后,将所得混合物用乙酸乙酯(3×10ml)萃取。收集有机相,并用少量水洗涤三遍。用无水na2so4干燥后,过滤固体,并在减压下除去溶剂。将得到的残余物进行快速硅胶柱色谱处理,通过用混合的石油醚和乙酸乙酯(v/v=50:1)洗脱,来得到纯产物。
产率为85%。式3e所示的化合物结构表征如下:黄色液体;1hnmr(400mhz,cdcl3)δ7.35(s,1h),7.19(d,j=9.0hz,1h),7.02(d,j=9.0hz,1h),4.89(s,1h),3.82(s,3h),2.72(s,6h);13cnmr(100mhz,cdcl3)δ180.9,154.849,154.4,126.8,119.3,116.4,109.0,97.6,84.6,55.9,41.3;esi-hrmscalcdforc13h14cl2no2+[m+h]+286.0396,found286.0403.
实施例6
本实施例中提供了3,3-二氯代色满酮类化合物的制备方法,制备如式3f所示的化合物的方法。
其制备步骤如下:将烯胺酮(0.2mmol),n-氯代丁二酰亚胺(0.5mmol)和dmf(0.5ml)装入装有搅拌磁子的10ml微波反应管中。然后将管密封并用于nova-2s微波反应器。将混合物在微波辐射下于60℃在微波辐射下搅拌8分钟。冷却至室温后,将所得混合物用乙酸乙酯(3×10ml)萃取。收集有机相,并用少量水洗涤三遍。用无水na2so4干燥后,过滤固体,并在减压下除去溶剂。将得到的残余物进行快速硅胶柱色谱处理,通过用混合的石油醚和乙酸乙酯(v/v=50:1)洗脱,来得到纯产物。
产率为87%。式3f所示的化合物结构表征如下:黄色液体;1hnmr(400mhz,cdcl3)δ8.01–7.96(m,1h),7.60–7.55(m,1h),7.11(t,j=7.9hz,1h),7.06(d,j=8.4hz,1h),5.19(s,1h),3.28–3.19(m,4h),1.90–1.85(m,4h);13cnmr(100mhz,cdcl3)δ181.2,159.8,137.0,129.3,122.4,118.1,116.6,94.9,84.9,48.7,24.9.esi-hrmscalcdforc17h24cl2no2+[m+h]+344.1179,found344.1187.
实施例7
本实施例中提供了3,3-二氯代色满酮类化合物的制备方法,制备如式3g所示的化合物的方法。
其制备步骤如下:将烯胺酮(0.2mmol),n-氯代丁二酰亚胺(0.5mmol)和dmf(0.5ml)装入装有搅拌磁子的10ml微波反应管中。然后将管密封并用于nova-2s微波反应器。将混合物在微波辐射下于60℃在微波辐射下搅拌8分钟。冷却至室温后,将所得混合物用乙酸乙酯(3×10ml)萃取。收集有机相,并用少量水洗涤三遍。用无水na2so4干燥后,过滤固体,并在减压下除去溶剂。将得到的残余物进行快速硅胶柱色谱处理,通过用混合的石油醚和乙酸乙酯(v/v=50:1)洗脱,来得到纯产物。
产率为71%。式3g所示的化合物结构表征如下:黄色液体;1hnmr(400mhz,cdcl3)δ7.96–7.84(m,1h),7.53–7.46(m,1h),7.06–6.95(m,2h),4.90(s,1h),3.14–3.04(m,2h),3.00–2.90(m,2h),1.08(t,j=7.1hz,6h);13cnmr(100mhz,cdcl3)δ181.5,160.0,136.9,129.4,122.3,118.1,116.5,96.9,85.5,44.60,14.02.esi-hrmscalcdforc13h16cl2no2+[m+h]+288.0553,found288.0559.
实施例8
本实施例中提供了3,3-二氯代色满酮类化合物的制备方法,制备如式3h所示的化合物的方法。
其制备步骤如下:将烯胺酮(0.2mmol),n-溴代丁二酰亚胺(0.5mmol)和dmf(0.5ml)装入装有搅拌磁子的10ml微波反应管中。然后将管密封并用于nova-2s微波反应器。将混合物在微波辐射下于60℃在微波辐射下搅拌8分钟。冷却至室温后,将所得混合物用乙酸乙酯(3×10ml)萃取。收集有机相,并用少量水洗涤三遍。用无水na2so4干燥后,过滤固体,并在减压下除去溶剂。将得到的残余物进行快速硅胶柱色谱处理,通过用混合的石油醚和乙酸乙酯(v/v=50:1)洗脱,来得到纯产物。
产率为57%。式3h所示的化合物结构表征如下:黄色液体;1hnmr(400mhz,cdcl3)δ8.05–8.00(m,1h),6.87–6.82(m,1h),6.80–6.75(m,1h),4.87(s,1h),2.67(s,6h);13cnmr(100mhz,cdcl3)δ179.8,168.1(d,1jc-f=257hz),161.3(d,3jc-f=13hz),131.9(d,3jc-f=12hz),112.9,111.1(d,2jc-f=23hz),104.6(d,2jc-f=24hz),99.3,65.4,41.0.esi-hrmscalcdforc11h11br2fno2+[m+h]+365.9135,found365.9122.
实施例9
本实施例中提供了3,3-二氯代色满酮类化合物的制备方法,制备如式3i所示的化合物的方法。
其制备步骤如下:将烯胺酮(0.2mmol),n-氯代丁二酰亚胺(0.5mmol)和dmf(0.5ml)装入装有搅拌磁子的10ml微波反应管中。然后将管密封并用于nova-2s微波反应器。将混合物在微波辐射下于60℃在微波辐射下搅拌8分钟。冷却至室温后,将所得混合物用乙酸乙酯(3×10ml)萃取。收集有机相,并用少量水洗涤三遍。用无水na2so4干燥后,过滤固体,并在减压下除去溶剂。将得到的残余物进行快速硅胶柱色谱处理,通过用混合的石油醚和乙酸乙酯(v/v=50:1)洗脱,来得到纯产物。
产率为50%。式3i所示的化合物结构表征如下:白色固体;m.p.97–98℃;1hnmr(400mhz,cdcl3)δ8.87(d,j=2.7hz,1h),8.46–8.40(m,1h),7.23(d,j=9.2hz,1h),5.18(s,1h),2.71(s,6h);13cnmr(100mhz,cdcl3)δ178.9,163.6,142.7,131.3,125.7,119.3,116.5,99.4,83.1,41.0.esi-hrmscalcdforc11h11cl2no4+[m+h]+305.0090,found305.0090.
式3i单晶结构:
- 还没有人留言评论。精彩留言会获得点赞!