一种半日花烷型二萜类化合物、制备方法及应用与流程
本发明涉及天然药物
技术领域:
,具体而言,涉及一种半日花烷型二萜类化合物、制备方法及应用。
背景技术:
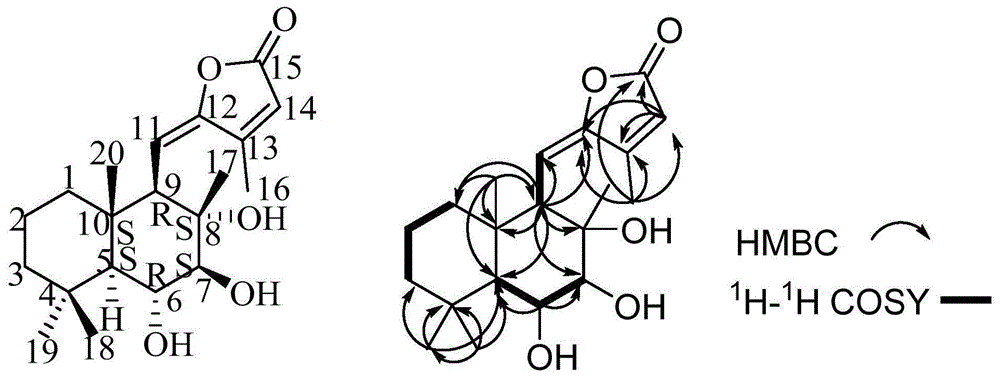
:裸花紫珠(callicarpanudiflorahook.etarm)为马鞭草科(verbenacea)紫珠属(callicarpal.)植物,用药部位为干燥叶,主要生长在海南、江西、广东、广西等国内地区,新加坡、印度、越南、马来西亚等国外地区。裸花紫珠首次记载于唐代陈藏器《本草拾遗》中的“紫荆”一品;后受到海南黎族土医的广泛使用,在上世纪50年代得以“裸花紫珠”独立命名;2020年新增录入《中国药典》(2020版)一部。裸花紫珠味苦、微辛,性平,具有止血,抗炎,消肿,清热解毒,化湿去浊等功效,临床上主要用于治疗多种内外伤出血、细菌性感染引起炎症肿毒,急性传染性肝炎等疾病。目前,裸花紫珠的化学成分以及其活性成分尚不明晰。鉴于此,特提出本发明。技术实现要素:本发明的目的在于提供一种半日花烷型二萜类化合物、制备方法及应用以解决上述技术问题。本发明是这样实现的:本发明提供了一种半日花烷型二萜类化合物,半日花烷型二萜类化合物具有如下的结构式:一种半日花烷型二萜类化合物的制备方法,其包括如下步骤:以裸花紫珠叶为原料,用乙醇回流提取,浓缩制得总提取物;将获得的总提取物经吸附树脂,分别以水、25%-100%的乙醇进行梯度洗脱,收集乙醇洗脱液,经过除醇除水处理获得粗提取物;将粗提取物经硅胶柱色谱分离,以洗脱液梯度洗脱得到fr.1~fr.6共6个组份,将fr.3组份经硅胶柱色谱,经梯度洗脱得到fr.3-1~fr.3-8共8个组份,将其中的fr.3-2组份经硅胶柱,梯度洗脱后得到fr.3-2-1~fr.3-2-7共7个组份,将fr.3-2-4组份经ods柱色谱,依次用20%、40%、60%乙腈水溶液梯度洗脱,并将乙腈水溶液梯度洗脱后的洗脱液经液相柱色谱分离得到半日花烷型二萜类化合物。在本发明应用较佳的实施方式中,上述吸附树脂为da-201大孔吸附树脂,分别以水、25%、50%、75%、95%乙醇进行梯度洗脱。在本发明应用较佳的实施方式中,上述收集50%乙醇溶液洗脱部位经过旋转蒸发法获得粗提物。在本发明应用较佳的实施方式中,上述粗提取物经硅胶柱色谱体积比为200-1∶1的氯仿:甲醇梯度洗脱,合并并收集得到fr.1~fr.6共6个组份;fr.3组份经硅胶柱色谱的洗脱是依次以体积比为50-1:1的氯仿:甲醇梯度洗脱,合并收集获得fr.3-1~fr.3-8共8个组份,将其中的fr.3-2组份经硅胶柱,依次以体积比为200-8:1的氯仿:甲醇梯度洗脱,合并收集获得fr.3-2-1~fr.3-2-7共7个组份,将fr.3-2-4组份经ods柱色谱,依次用20%、40%、60%乙腈水溶液梯度洗脱。在本发明应用较佳的实施方式中,上述fr.3-2-4组份经ods柱色谱并以乙腈水溶液梯度洗脱后,旋蒸法去除乙腈和水,然后将去除乙腈和水的产物经过液相柱色谱分离,以体积比为20-40:1的乙腈和水为流动相进行梯度洗脱,检测波长为265nm和/或300nm,流速为8-12ml/min,保留时间为65.72±10min处制备得到半日花烷型二萜类化合物。在本发明应用较佳的实施方式中,在乙醇回流提取步骤中,以60-70%的乙醇回流提取至少2次,每次1.5-4h,合并提取液,浓缩制得总提取物;回流提取为连续回流提取法。本发明还提供了一种半日花烷型二萜类化合物或由上述的制备方法制备的半日花烷型二萜类化合物在制备神经保护组合物中的应用。本发明还提供了一种神经保护组合物,其包括半日花烷型二萜类化合物或由上述的制备方法制备的半日花烷型二萜类化合物。在本发明应用较佳的实施方式中,上述神经保护组合物还包括药学上可接受的添加剂或辅料;优选地,神经保护组合物的剂型选自片剂、丸剂、粉剂、混悬剂、凝胶、乳液、乳膏、颗粒剂、纳米颗粒、胶囊、栓剂、注射剂、喷雾或针剂。在本发明应用较佳的实施方式中,上述神经保护组合物用于治疗或预防神经退行性疾病,优选地,神经退行性疾病包括如下疾病中的至少一种:阿尔茨海默病、轻度认知障碍、亨廷顿症、朊病毒引起的疾病、额颞痴呆、路易体痴呆、血管性痴呆、肌萎缩性侧索硬化症、慢性损伤性脑病、进行性核上性麻痹、多系统萎缩症、皮质基底节变性、皮克氏病和橄榄体脑桥小脑萎缩。本发明具有以下有益效果:本发明从裸花紫珠叶中分离获得一种半日花烷型二萜类化合物,该化合物具有神经保护的功效,可以用于制备治疗或预防神经退行性疾病的神经保护组合物。该制备方法简单易行,具有良好的经济效益和应用前景。附图说明为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。图1为实施例1分离获得的半日花烷型二萜类化合物的结构以及关键的hmbc、1h-1hcosy信号;图2为分离获得半日花烷型二萜类化合物的液相色谱图;图3为化合物1的1h-nmr图谱(氘代甲醇);图4为化合物1的13c-nmr图谱(氘代甲醇);图5为化合物1的hmqc图谱(氘代甲醇);图6为化合物1的hmbc图谱(氘代甲醇);图7为化合物1的1h-1hcosy图谱(氘代甲醇);图8为化合物1的noesy图谱(氘代甲醇);图9为化合物1的hr-esi-ms图谱(氘代甲醇)。具体实施方式现将详细地提供本发明实施方式的参考,其一个或多个实例描述于下文。提供每一实例作为解释而非限制本发明。实际上,对本领域技术人员而言,显而易见的是,可以对本发明进行多种修改和变化而不背离本发明的范围或精神。例如,作为一个实施方式的部分而说明或描述的特征可以用于另一实施方式中,来产生更进一步的实施方式。现有研究表明裸花紫珠中的化学成分以黄酮类、苯乙醇苷类、二萜类、三萜类、环烯醚萜及其苷类等为主,发明人在前人研究基础上,继续对其进行化学成分研究,发现并分离获得一种新的化合物—半日花烷型二萜类化合物。经验证表明该化合物具有神经保护作用,启示该化合物可以用于新的制药用途。具体地,本发明提供了一种半日花烷型二萜类化合物。半日花烷型二萜类化合物的名称为:(5e)-4-methyl-5-[((1r,2s,3s,4r,4as,8as)-decahydro-2,3,4-trihydroxy-2,5,5,8a-tetramethyl-1-naphthalenyl)methylene]-2(5h)-furanone,(5e)-4-甲基-5-[((1r,2s,3s,4r,4as,8as)-十氢-2,3,4–三羟基-2,5,5,8a-四甲基-1-萘基)亚甲基]-2(5h)-呋喃酮,半日花烷型二萜类化合物具有如下的结构式:发明人将上述的半日花烷型二萜类化合物命名为callicapenem6。hr-esi-ms显示有m/z:373.1981[m+na]+(计算值373.1985),723.4072[2m+na]+(计算值723.4078),再结合该化合物的1h-nmr与13c-nmr谱数据,该化合物的分子式为c20h30o5,不饱和度为6。本发明还提供半日花烷型二萜类化合物的制备方法,其包括如下步骤:以裸花紫珠叶为原料,用乙醇回流提取,浓缩制得总提取物;将获得的总提取物经吸附树脂,分别以水、25%-100%的乙醇进行梯度洗脱,收集乙醇洗脱液,经过除醇除水处理获得粗提取物;将粗提取物经硅胶柱色谱分离,以洗脱液梯度洗脱得到fr.1~fr.6共6个组份,将fr.3组份经硅胶柱色谱,经梯度洗脱得到fr.3-1~fr.3-8共8个组份,将其中的fr.3-2组份经硅胶柱,梯度洗脱后得到fr.3-2-1~fr.3-2-7共7个组份,将fr.3-2-4组份经ods柱色谱,依次用20%、40%、60%乙腈水溶液梯度洗脱,并将乙腈水溶液梯度洗脱后的洗脱液经液相柱色谱分离得到半日花烷型二萜类化合物。在其他实施方式中,也可根据需要选择其他的柱子对粗提取物进行分离。需要说明的是,将洗脱液经液相柱色谱分离后可同时得到半日花烷型二萜类化合物和sterebina(甜叶菊素a)。根据液相柱色谱洗脱出的产物顺序即可实现二者的分离。乙腈水溶液洗脱液经过液相柱色谱分离,以体积比为40:1的乙腈和水为流动相,检测波长为265nm和/或300nm,流速为10ml/min,保留时间为51.56min处制备得到sterebina。需要说明的是,可以在265nm或300nm的检测波长处单独收集sterebina,也可以同时在265nm和300nm的检测波长处收集sterebina。在本发明应用较佳的实施方式中,上述吸附树脂为da-201大孔吸附树脂,分别以水、25%、50%、75%、95%乙醇进行梯度洗脱。通过da-201大孔吸附树脂能够吸附难溶于水,而又高度溶解于乙醇有机溶剂中各类带极性的有机化合物,可以达到富集苯乙醇苷类化合物的目的。在其他实施方式中,也可选自水、25%、50%、60%、70%、80%、95%的乙醇进行梯度洗脱。需要说明的是,上述指出的以水、25%-100%的乙醇进行梯度洗脱并不限于本发明列举的几种梯度洗脱的实施方式。在本发明应用较佳的实施方式中,上述收集50%乙醇溶液洗脱部位经过旋转蒸发法获得粗提物。通过旋蒸仪实现50%乙醇溶液洗脱部位脱醇脱水。在其他实施方式中,也可选择收集25%和50%的乙醇溶液洗脱部位进行脱醇脱水处理,并将脱醇脱水处理后的产物进行混合得到相应的粗提物。上述收集50%乙醇溶液洗脱部位进行脱醇脱水处理仅为最优的一种实施方式。在另一种实施方式中,也可选择收集25%、50%和75%的乙醇溶液洗脱部位进行脱醇脱水处理,并将脱醇脱水处理后的产物进行混合得到相应的粗提物。粗提取物经硅胶柱色谱体积比为200-1∶1的氯仿:甲醇梯度洗脱,合并并收集得到fr.1~fr.6共6个组份。例如可以设置200:1、180:1、150:1、120:1、100:1、70:1、40:1、10:1、1:1的氯仿:甲醇梯度洗脱。fr.3组份经硅胶柱色谱的洗脱是依次以体积比为50-1:1的氯仿:甲醇梯度洗脱,合并收集获得fr.3-1~fr.3-8共8个组份。例如可以设置50:1、40:1、30:1、20:1、10:1、1:1的氯仿:甲醇梯度洗脱。将其中的fr.3-2组份经硅胶柱,依次以体积比为200-8:1的氯仿:甲醇梯度洗脱,合并收集获得fr.3-2-1~fr.3-2-7共7个组份,将fr.3-2-4组份经ods柱色谱,依次用20%、40%、60%乙腈水溶液梯度洗脱。粗提取物进行硅胶柱色谱目的在于对粗提取物进行初步的分离处理,以去除部分的杂质。硅胶柱色谱所采用的洗脱液选择氯仿和甲醇的混合液是由于该混合液的极性与目的产物的极性比较相符,有利于斑点以较好程度的分开,发明人发现若采用乙酸乙酯与甲醇的混合洗脱液则无法实现各点的分离,分离效果较差。在本发明应用较佳的实施方式中,上述fr.3-2-4组份经ods柱色谱并以乙腈水溶液梯度洗脱后,旋蒸法去除乙腈和水,然后将去除乙腈和水的产物经过液相柱色谱分离,以体积比为20-40:1的乙腈和水为流动相进行梯度洗脱,检测波长为265nm和/或300nm,流速为8-12ml/min,保留时间为65.72±10min处制备得到半日花烷型二萜类化合物。需要说明的是,旋蒸去除乙腈和水后,需要加入少量的溶剂进行溶解,以便于后续上色谱柱进行分离。发明人发现,可以选择在265nm波长处检测收集,也可选择在300nm波长处检测收集,也可以同时选择265nm和300nm波长处检测收集洗脱液。在一种实施方式中,只要出峰即可选择收集,保留时间与色谱分离的色谱柱类型以及色谱条件相关。在一种实施方式中,流速可设置为10ml/min。采用乙腈水溶液作为ods柱色谱的洗脱液可以更好的实现目的产物的分离。在本发明应用较佳的实施方式中,上述乙醇回流提取步骤中,以60-70%的乙醇回流提取至少2次,每次1.5-4h,合并提取液,浓缩制得总提取物;回流提取为连续回流提取法。回流提取为连续回流提取法。在其他实施方式中,上述针对原料的提取步骤,也可以根据实际需要选择超声提取法等其他方法。回流提取的次数可以是2次、3次、4次或者多次。本发明还提供了一种半日花烷型二萜类化合物或由上述的制备方法制备的半日花烷型二萜类化合物在制备神经保护组合物中的应用。需要说明的是,上述的神经保护组合物可以药物,制剂或冻干粉。可以是作为脑部神经、视神经等神经保护的药物的制备中的应用。本发明还提供了一种神经保护组合物,其包括半日花烷型二萜类化合物或由上述的制备方法制备的半日花烷型二萜类化合物。在本发明应用较佳的实施方式中,上述神经保护组合物还包括药学上可接受的添加剂或辅料;优选地,药物的剂型选自片剂、丸剂、粉剂、混悬剂、凝胶、乳液、乳膏、颗粒剂、纳米颗粒、胶囊、栓剂、注射剂、喷雾或针剂。在本发明应用较佳的实施方式中,上述神经保护组合物用于治疗或预防神经退行性疾病,优选地,神经退行性疾病包括如下疾病中的至少一种:阿尔茨海默病(alzheimer’sdisease,ad)、帕金森症(pakinson’disease,pd)、轻度认知障碍(mci)、亨廷顿症(hd)、朊病毒引起的疾病、额颞痴呆(ftd)、路易体痴呆、血管性痴呆、肌萎缩性侧索硬化症(amyotuophiclateralsclerosis,als)、慢性损伤性脑病(cte)、进行性核上性麻痹(psp)、多系统萎缩症(msa)、皮质基底节变性(cbgd)、皮克氏病和橄榄体脑桥小脑萎缩(opca)。在其他实施方式中,上述的神经退行性疾病(neurodegenerativediseases,ndd)临床表现为患者常有记忆力下降、认知困难、痴呆、动作平衡失调、运动能力丧失等症状。需要说明的是,上述列举的仅为发明人列举的几种可行的疾病,在其他实施方式中,只要能实现神经退行性病症的缓解、治愈等效果均在本发明的保护范围内,而并不限于上述列举的几种疾病的类型。为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。以下结合实施例对本发明的特征和性能作进一步的详细描述。实施例所采用的设备:brukeravanceiiihd600mhz型核磁共振波谱仪(瑞士bruker公司),absciextripleesi5600+型高分辨飞行时间质谱联用仪(美国absciex公司),agilent1260-ii型高效液相(美国安捷伦公司),依利特p3500半制备液相色谱仪(大连依利特分析仪器有限公司),ymc-triartc18半制备色谱柱(50mm×250mm,10μm,日本ymc公司),da-201大孔吸附树脂(郑州和成新材料科技有限公司),100~200目、200~300目柱色谱硅胶(青岛海洋化工厂),薄层硅胶板gf254(青岛海洋化工厂),ms204s/01型分析天平(瑞士mettlertoledo公司),milliq超纯水机(millipore公司),reprostar3型camag薄层色谱扫描影像系统(瑞士camag公司),tlcplateheater(瑞士camag公司),所用试剂均为色谱纯或分析纯。实施例1本实施例提供了半日花烷型二萜类化合物的制备方法,本实施例中的裸花紫珠药材购自海南白沙恒生源种植专业合作社,经江西中医药大学范崔生教授鉴定为裸花紫珠(callicarpanudiflorahook.etarm),凭证标本(no.l201905z)存放于江中药谷科研中心。该制备方法包括如下步骤:取上述的裸花紫珠干燥叶5.0kg,用15倍量65%乙醇加热回流提取2次,每次2h,趁热过滤合并提取液,减压浓缩至无醇味(即制得总提取物)。离心后取上清液经da-201大孔吸附树脂柱色谱进行分离,依次用水、25%、50%、75%、95%的乙醇进行梯度洗脱,减压回收溶剂分别得到25%乙醇、50%乙醇(250.93g)、75%乙醇、95%乙醇共四部分。将50%乙醇部位采用旋转蒸发仪去除醇与水制得50%乙醇浸膏(250.93g粗提物)。将上述浸膏(250.93g)经硅胶柱色谱(8.0cm×100.0cm)分离,依次以体积比为200:1~1:1的氯仿∶甲醇进行梯度洗脱(本实施例中的梯度洗脱,依次以200:1、50:1、10:1、5:1、1:1洗脱。为达到粗分目的,使得洗脱液极性跨度大),点板合并其中较为一致的主斑点,得到fr.1~fr.6共6个组份。将其中的fr.3组份(本实施例中的fr.3组份由上述10:1洗脱液收集得到,21.52g)过硅胶柱,依次以体积比为50∶1~1∶1的氯仿:甲醇进行梯度洗脱(本实施例中的梯度洗脱体积比为50:1,25:1,15:1,10:1,8:1,4:1,1:1),点板合并其量多且一致的主斑点部位,得到fr.3-1~fr.3-8共8个组份。将其中收集获得的6.12g的fr.3-2(本实施例中的fr.3-2组份由前述对fr.3组份的50:1洗脱液收集得到)过硅胶柱,以氯仿∶甲醇(分别以体积比为200:1,100:1,50:1,20:1,10:1,8:1梯度洗脱)梯度洗脱得到fr.3-2-1~fr.3-2-7共7个组份。取其中的fr.3-2-4(fr.3-2-4组份由前述对fr.3-2组份的50:1洗脱液收集得到,0.82g)过ods柱色谱,依次用20%、40%、60%乙腈水溶液梯度洗脱,点板合并干净且一致的主斑点并经半制备液相柱色谱进行分离得到半日花烷型二萜类化合物(2.8mg)、甜叶菊素a(即sterebina,18.2mg)。本实施例中,fr.3-2-4组份经ods柱色谱依次用体积百分比为20:80、40:60、40:60、50:50的乙腈水溶液进行梯度洗脱(洗脱调节如下表所示),点板合并干净且一致的主斑点,将乙腈水溶液洗脱液经旋转蒸发仪蒸发去除乙腈和水,旋蒸产物中加入能使旋蒸产物部分溶解的溶剂(溶剂为甲醇),经过液相柱色谱分离,以体积比为20:1、30:1、40:1的乙腈和水为流动相,检测波长为265nm和300nm,流速为10ml/min,保留时间为65.72min处制备得到半日花烷型二萜类化合物,在保留时间为51.56min处制备得到sterebina,液相色谱图参照图2所示。时间(min)流速(ml/min)乙腈(%)水(%)0102080601040607010406085105050结构鉴定:主要利用核磁共振(1h-nmr与13c-nmr谱数据)鉴定其结构,进一步分析该化合物的hmbc图谱数据同时结合hmqc图谱数据,从而构建了该化合物的平面结构(参照图1所示),半日花烷型二萜类化合物呈淡黄色固体,分子式为c20h30o5,不饱和度为6。hr-esi-ms显示有m/z:373.1981[m+na]+(计算值373.1985),723.4072[2m+na]+(计算值723.4078)。其波谱数据见表1。表1化合物1的1h和13c-nmr数据(600/125mhz,cd3od)a表示重叠的氢。图3为化合物1的1h-nmr图谱(氘代甲醇);图4为化合物1的13c-nmr图谱(氘代甲醇);图5为化合物1的hmqc图谱(氘代甲醇);图6为化合物1的hmbc图谱(氘代甲醇);图7为化合物1的1h-1hcosy图谱(氘代甲醇);图8为化合物1的noesy图谱(氘代甲醇);图9为化合物1的hr-esi-ms图谱(氘代甲醇)。表1数据显示有5个单峰甲基信号[δh:2.20(3h,s,h-16)、1.23(3h,s,h-17)、1.16(3h,s,h-18)、1.01(3h,s,h-19)、1.02(s,3h,h-20)],两个烯氢质子信号[δh:5.55(1h,d,j=11.2hz,h-11)、5.99(1h,s,h-14)],2个连氧次甲基质子信号[δh:3.61(1h,dd,j=8.6,2.4hz,h-6)、3.35(1h,d,j=9.5hz,h-7)]。13c-nmr谱(表1)中显有20个碳原子的位移信号,其中有5个甲基碳信号[δc:11.8(c-16)、19.1(c-17)、37.0(c-18)、22.5(c-19)、17.5(c-20)],3个连氧碳信号[δc:73.0(c-6)、85.5(c-7)、76.9(c-8)],4个烯碳信号[δc:111.1(c-11)、154.8(c-12)、157.1(c-13)、117.0(c-14)],1个酯羰基碳信号[δc:171.8(c-15)]。在hmbc图谱中显示单峰甲基δh:2.20(3h,s,h-16)与c-12/13/14/15远程相关,烯烃质子δh:5.55(1h,d,j=11.2hz,h-11)与c-8/10/12/13远程相关,烯烃质子δh:5.99(1h,s,h-14)与c-12/13/15远程相关,可知具有两个双键且共轭,c-16位的甲基连在c-13位上,结合1个酯羰基碳信号[δc:171.8(c-15)]可推测该结构中具有一个-4-甲基-5-亚甲基]-2(5h)-呋喃酮结构片段。在1h-1hcosy图谱中显示了h-9/h-11相关,可知c-11与c-9相连,同时还能观察到h-1/h2/h3相关,h-6与h-5/h-7相关。进一步分析该化合物的hmbc(1h的异核多碳相关谱)图谱数据同时结合hmqc图谱数据,从而构建了该化合物的平面结构(参照图1所示)。经sci-finder检索可知半日花烷型二萜类化合物为1个新化合物。在noesy谱图中可以看出h-6与h-9/11/16/17/18/20相互远程偶合。结合文献(oshimay,sterebinsa,b,candd,bisnorditerpenoidsofsteviarebaudianaleaves,tetrahedron,1986),可知该化合物与sterebina-d四个化合物旋光度均为正值,将c-6定位r构型,c-7定位为s构型,所以c-9为r构型,c-5/8/10为s构型,从而构建了该化合的相对构型。因此将该化合物命名为:(5e)-4-methyl-5-[((1r,2s,3s,4r,4as,8as)-decahydro-2,3,4-trihydroxy-2,5,5,8a-tetramethyl-1-naphthalenyl)methylene]-2(5h)-furanone,(5e)-4-甲基-5-[((1r,2s,3s,4r,4as,8as)-十氢-2,3,4–三羟基-2,5,5,8a-四甲基-1-萘基)亚甲基]-2(5h)-呋喃酮。以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1