一类含吩噻嗪的柱芳烃荧光吸附材料及其制备方法
本发明属于有机化学合成技术领域,具体涉及一类含吩噻嗪的柱芳烃荧光吸附材料及其制备方法。
背景技术:
柱芳烃及其衍生物是一类新型大环化合物,具有高度对称性和刚性,相比葫芦脲和环糊精等传统大环更容易被修饰,因其对卤代烃、烷烃、芳香烃等化合物具有良好的选择性吸附,被认为是非常有潜力的小分子吸附材料。但是柱芳烃本身没有荧光,在实际应用中难以通过简单的观察来判断吸附情况。通过烷基链连接荧光分子的方法,对于卤代烃、烷烃、芳香烃等简单小分子,无法判断是否有结合,也难以得到紧凑、刚性的晶体,限制了该类分子的进一步应用。而母体结构带有荧光单元的柱芳烃,可以通过简单直接的目视判断柱芳烃在吸附过程中空腔的变化,但是合成方法往往冗长繁琐,提纯分离困难,限制了对柱芳烃及其衍生物的研究与应用。通过巧妙的分子设计以及简单高效的合成方法,将荧光单元融合到柱芳烃分子中,具有重要的研究意义和实用价值。
技术实现要素:
本发明的第一个目的是提供一类含吩噻嗪的柱芳烃荧光吸附材料。
本发明的第二个目的是提供一种所述含吩噻嗪的柱芳烃荧光吸附材料的制备方法。
为了实现上述目的,本发明采用的技术方案如下:
本发明的第一方面提供了一类含吩噻嗪的柱芳烃荧光吸附材料,结构通式如下所示:
其中:r1为
r2为氢或c1~c3烷基。
较优选的,所述结构通式中,r2为氢、甲基、乙基。
最优选的,所述含吩噻嗪的柱芳烃荧光吸附材料的结构为以下结构中的一种:
本发明的第二方面提供了一种所述含吩噻嗪的柱芳烃荧光吸附材料的制备方法,包括以下步骤:
在手套箱中,将化合物6、碱、钯催化剂、配体、卤代芳烃、甲苯混合,温度为100~110℃,氩气氛围下,搅拌18~24h,得到化合物ii,即通式中r2=h的所述含吩噻嗪的柱芳烃荧光吸附材料;
所述碱为叔丁醇钾或叔丁醇钠;所述钯催化剂为醋酸钯或氯化钯;所述配体为三叔丁基膦或三叔丁基膦四氟硼酸盐;所述卤代芳烃为2-溴-9-芴酮、2-溴蒽、7-溴异喹啉;所述化合物6和碱的摩尔比为1:2至1:3;所述化合物6和钯催化剂的摩尔比为1:0.01至1:0.05;所述钯催化剂和配体的摩尔比为1:2;所述化合物6和卤代芳烃的摩尔比为1:1至1:3。
将化合物ii、碘化物或溴化物、碱和溶剂混合,温度为50~80℃的条件下,搅拌18~24h,得到式i所示化合物即所述含吩噻嗪的柱芳烃荧光吸附材料;
所述碱为碳酸钾或碳酸钠;
所述溶剂为丙酮、乙腈、二甲基甲酰胺中的一种;
所述化合物ii和碱的摩尔比为1:1至1:3;
所述化合物ii和碘化物或溴化物的摩尔比为1:5至1:15。
所述碘化物或溴化物为碘甲烷、碘乙烷或溴乙烷。
所述化合物6的制备方法包括以下步骤:
第一步,在室温下,将化合物1溶于二氯乙烷中,向该溶液中加入多聚甲醛和三氟乙酸,回流搅拌1~10h,得到化合物2;其中,二氯甲烷和三氟乙酸体积比为20:1,化合物1和多聚甲醛的摩尔比为1:2;
第二步,在室温下,将化合物2溶于二氯甲烷和四氢呋喃的混合液中,向该溶液中滴加硝酸铈铵的水溶液,滴毕,搅拌1~24h,得到化合物3;其中,二氯甲烷和四氢呋喃体积比1:1,化合物2和硝酸铈铵的摩尔比为1:2.2;
第三步,在室温下,将化合物3溶于二氯甲烷和甲醇的混合液中,向该溶液中加入巯基苯胺,搅拌1~24h,得到化合物4;其中,二氯甲烷和甲醇体积比1:1,化合物3和巯基苯胺的摩尔比为1:1.6;
第四步,在室温下,将化合物4溶于二氯甲烷和甲醇的混合液中,向该溶液中加入碳酸钾,搅拌1~24h,得到化合物5;其中,二氯甲烷和甲醇体积比1:2,化合物4和碳酸钾的摩尔比为1:0.3;
第五步,在室温下,将化合物5溶于二甲基甲酰胺和水中,加入连二亚硫酸钠,在70~90℃的条件下,搅拌1~12h,得到化合物6;二甲基甲酰胺与水体积比10:1,化合物5与连二亚硫酸钠摩尔比为1:10。
由于采用上述技术方案,本发明具有以下优点和有益效果:
为了使柱芳烃空腔变化能够以荧光发射变化的方式被观测到,本发明提供的含吩噻嗪的柱芳烃荧光吸附材料将吩噻嗪融合到柱芳烃的母体结构中,由于吩噻嗪单元的一部分构成柱芳烃本身,同时芳环取代基(r1)是否能自由运动、能运动的幅度以及荧光单元堆积方式的改变都会影响分子荧光发射,客体进入大环分子后,能使化合物产生荧光颜色的变化。实现了具有荧光响应的柱芳烃设计合成,通过更改取代基,可以实现不同结构的柱芳烃合成,也为设计具有特定吸附能力的柱芳烃分子铺平了道路。
如实施例4可见,化合物10的固体对于气体二甲苯具有吸附能力,吸附过后荧光发生改变,可被肉眼直接观察。如实施例5可见,化合物10的溶液对分子结构类似但尺寸不同的客体化合物产生差异性识别,可通过荧光光谱分辨。如实施例5和6可见,改变该化合物的取代基r1和r2,所得到的化合物10、11的荧光发射范围和客体化合物的响应能力会发生很大的改变。
本发明使用了后修饰的方法,使用易得的柱芳烃衍生物制备了含吩噻嗪的柱芳烃荧光吸附材料,合成路线较短,纯化容易,收率较高,方法具有普适性,易于大量制备。
本发明的含吩噻嗪的柱芳烃荧光吸附材料分子设计合理,合成目标物新颖,能够实现特异性检测、吸附并分离结构相似的有机物,这一过程可以通过材料荧光的变化进行监测。该类分子溶解性良好,合成、纯化简单,分子结构可变,合成方法具有普适性,便于根据需要设计结构、性能不同的功能化合物。
附图说明
图1为本发明实施例1制备的化合物7的1h-nmr图。
图2为本发明实施例1制备的化合物8的1h-nmr图。
图3为本发明实施例2制备的化合物9的1h-nmr图。
图4为本发明实施例2制备的化合物10的1h-nmr图。
图5为本发明实施例3制备的化合物11的1h-nmr图。
图6为本发明实施例4演示化合物10的效果图。
图7为本发明实施例5中在化合物10的二氯甲烷溶液中加入丁二腈后的荧光光谱示意图。
图8为本发明实施例5中在化合物10的二氯甲烷溶液中加入己二腈后的荧光光谱示意图。
图9为本发明实施例6中在化合物11的二氯甲烷溶液中加入丁二腈后的荧光光谱示意图。
图10是对比例1的化合物ix的晶体识别客体的前后荧光变化示意图。
具体实施方式
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
本发明实施例所需的化合物6的合成路线如下:
化合物6的制备方法包括以下步骤:
(1)在室温下,将化合物1(21.0g,0.15mol)(1,4-二甲氧基苯,购于adamas,规格500g,纯度99%)溶于二氯乙烷(600ml)中,向该溶液中加入多聚甲醛(9.0g,0.30mmol)和三氟乙酸(30ml),回流搅拌5h,得到化合物2;其中,二氯甲烷和三氟乙酸体积比20:1,化合物1和多聚甲醛的摩尔比为1:2。
(2)在室温下,将化合物2(7.000g,9.30mmol)溶于二氯甲烷(350ml)和四氢呋喃(350ml)的混合液中,向该溶液中滴加硝酸铈铵(11.235g,20.5mmol)水溶液(42ml),滴毕,搅拌24h,得到化合物3;其中,二氯甲烷和四氢呋喃体积比1:1,化合物2和硝酸铈铵的摩尔比为1:2.2。
(3)在室温下,将化合物3(0.900g,1.24mmol)溶于二氯甲烷(25ml)和甲醇(25ml)的混合液中,向该溶液中加入巯基苯胺(0.25g,1.98mmol),搅拌15h,得到化合物4;其中,二氯甲烷和甲醇体积比1:1,化合物3和巯基苯胺的摩尔比为1:1.6。
(4)在室温下,将化合物4(1.00g,1.18mmol)溶于二氯甲烷(10ml)和甲醇(20ml)的混合液中,向该溶液中加入碳酸钾(48.8mg,0.354mmol),搅拌18h,得到化合物5;其中,二氯甲烷和甲醇体积比1:2,化合物4和碳酸钾的摩尔比为1:0.3。
(5)在室温下,将化合物5(826mg,1.00mmol)溶于二甲基甲酰胺(5ml)和水(0.5ml)中,加入连二亚硫酸钠(1.74g,10.0mmol),在70~90℃的条件下,搅拌8h,得到化合物6;二甲基甲酰胺与水体积比10:1,化合物5与连二亚硫酸钠摩尔比为1:10。
实施例1
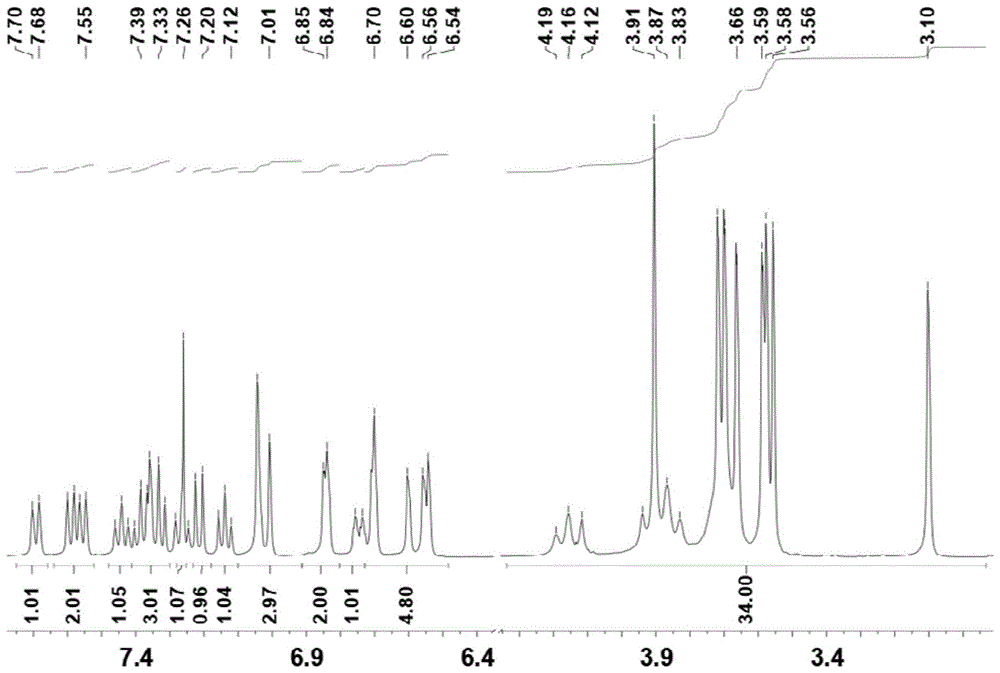
在手套箱中,氩气氛围下,将化合物6(414mg,0.5mmol)、叔丁醇钠(170mg,1.5mmol)、醋酸钯(5mg,0.01mmol)、三叔丁基膦四氟硼酸盐(12mg,0.02mmol)、2-溴-9-芴酮(259mg,1.0mmol)、甲苯(3ml)混合,温度为100~110℃,氩气氛围下,搅拌23h。将反应液冷却到室温,加入水50ml,二氯甲烷(50ml×3)萃取,合并有机相,硫酸钠干燥,柱色谱纯化得到327mg化合物7,产率为65.0%。
图1为本发明实施例1制备的化合物7的1h-nmr图,1hnmr(400mhz,cdcl3)δ=7.69(d,j=8.0hz,1h),7.59(d,j=8.0hz,1h),7.56(d,j=4.0hz,1h),7.44(t,j=8.0hz,1h),7.32-7.40(m,3h),7.27(t,j=6.0hz,1h),7.21(d,j=6.0hz,1h),7.14(t,j=8.0hz,1h),7.04(s,2h),7.01(s,1h),6.85(m,2h),6.75(m,1h),6.54-6.70(m,5h),3.10-4.19(m,34h)。
将化合物7(300mg,0.30mmol)、碳酸钾(62mg,0.45mmol)、碘甲烷(0.187ml,3mmol)、丙酮(50ml)混合,52℃搅拌24h。将反应液冷却到室温,减压蒸馏除去溶剂,重结晶得到298mg化合物8,产率为98%。
图2为本发明实施例1制备的化合物8的1h-nmr图,1hnmr(400mhz,cdcl3)δ=7.93(s,1h),7.66(d,j=12.0hz,1h),7.57(d,j=8.0hz,1h),7.53(t,j=8.0hz,1h),7.30-7.40(m,3h),7.25(t,j=8.0hz,1h),7.18(d,j=8.0hz,1h),7.13(t,j=8.0hz,1h),6.98(d,j=2.4hz,1h),6.80(s,1h),6.73(s,1h),6.72(s,1h),6.68(s,1h),6.61(s,1h),6.58(s,1h),6.53(s,1h),6.43(s,1h),6.42(s,1h),3.17-4.18(m,37h)。
实施例2
在手套箱中,氩气氛围下,将化合物6(455.4mg,0.55mmol)、叔丁醇钠(187mg,1.65mmol)、醋酸钯(5mg,0.01mmol)、三叔丁基膦四氟硼酸盐(12mg,0.02mmol)、2-溴蒽(257mg,1.0mmol)、甲苯(3.2ml)混合,温度为100~110℃,氩气氛围下,搅拌22h。将反应液冷却到室温,加入水50ml,二氯甲烷(50ml×3)萃取,合并有机相,硫酸钠干燥,柱色谱纯化得到338mg化合物9,产率为61.2%。
图3为本发明实施例2制备的化合物9的1h-nmr图,1hnmr(400mhz,cdcl3)δ=8.24(s,1h),8.06(s,1h),7.88(d,j=8.0hz,1h),7.82(d,j=8.0hz,1h),7.79(d,j=4.0hz,1h),7.77(d,j=4.0hz,1h),7.59(d,j=8.0hz,1h),7.45(t,j=8.0hz,1h),7.34-6.24(m,4h),7.10(d,j=4.0hz,j=8.0hz,1h),7.07(s,1h),7.00(d,j=2.4hz,1h),6.99(s,1h),6.82(s,1h),6.80(s,1h),6.73(s,1h),6.67(s,1h),6.57(s,1h),6.52(s,2h),3.10-4.19(m,34h)。
将化合物9(250mg,0.25mmol)、碳酸钾(41mg,0.3mmol)、碘甲烷(0.200ml,3.2mmol)、丙酮(45ml)混合,52℃搅拌24h。将反应液冷却到室温,减压蒸馏除去溶剂,重结晶得到240mg化合物10,产率为94.7%。
图4为本发明实施例2制备的化合物10的1h-nmr图,1hnmr(400mhz,cdcl3)δ=8.24(s,1h),8.02(s,1h),7.90(d,j=8.0hz,1h),7.81(d,j=8.0hz,1h),7.77(d,j=8.0hz,1h),7.68(d,j=12.0hz,1h),7.58(d,j=8.0hz,1h),7.44(t,j=8.0hz,1h),7.38-6.24(m,4h),7.01(d,j=4.0hz,j=8.0hz,1h),7.98(s,1h),6.80(s,1h),6.76(s,1h),6.71(s,1h),6.71(s,1h),6.66(s,1h),6.61(s,1h),6.59(s,1h),6.47(s,1h),6.40(s,1h),3.70-4.25(m,4h),3.67(s,3h),3.58(s,3h),3.53(s,3h),3.50(s,3h),3.49(s,2h),3.48(s,2h),3.47(s,2h),3.46(s,2h),3.38(s,3h),3.26(s,3h),3.18(s,3h)。
实施例3
在手套箱中,氩气氛围下,将化合物6(405mg,0.49mmol)、叔丁醇钠(141mg,1.47mmol)、醋酸钯(10mg,0.02mmol)、三叔丁基膦四氟硼酸盐(24mg,0.04mmol)、7-溴异喹啉(208mg,1.0mmol)、甲苯(3ml)混合,温度为100~110℃,氩气氛围下,搅拌22h。将反应液冷却到室温,加入水50ml,二氯甲烷(50ml×3)萃取,合并有机相,硫酸钠干燥,柱色谱纯化得到293mg化合物11,产率为62.6%。
图5为本发明实施例3制备的化合物11的1h-nmr图,1hnmr(400mhz,cdcl3)δ=8.97(s,1h),8.29(d,j=4.0hz,1h),7.74(d,j=8.0hz,1h),7.68(d,j=8.0hz,1h),7.62(d,j=8.0hz,1h),7.46(d,j=8.0hz,1h),7.39(s,1h),7.32-7.26(m,2h),7.09(d,j=4.0hz,j=8.0hz,1h),7.02(s,1h),7.00(s,1h),6.81-6.82(m,3h),6.71(s,1h),6.67(s,1h),6.56(s,1h),6.51(s,1h),6.50(s,1h),4.12-4.16(m,2h),3.79-3.95(m,7h),3.64-3.69(m,13h),3.54-3.56(m,9h),3.03(s,3h)。
实施例4
取1ml对二甲苯,置于一小玻璃瓶中。将5.2mg化合物10置于玻璃片上,然后将两者放置在一广口瓶中,盖上塞子。放置一段时间后,使用365nm的紫外光照射广口瓶中的化合物10,可观察到固体化合物10的荧光颜色由原先的绿色变为蓝绿色。如图6所示,图6为本发明实施例4演示化合物10的效果图,化合物10在吸附前后,使用365nm的紫外光照射下的荧光照片。从图中可以看出,化合物10吸附对二甲苯蒸汽实物与荧光图(左为吸附前,右为吸附后),吸附对二甲苯蒸汽后颜色轻微蓝移。
实施例5
配置化合物10的二氯甲烷溶液(10-5m),取2.5ml溶液,分别加入丁二腈或己二腈,每次加入量为10mg,测溶液的荧光光谱。图7为本发明实施例5中在化合物10的二氯甲烷溶液中加入丁二腈后的荧光光谱示意图。从图7可以看出,化合物10在470nm处有一明显的荧光发射峰。加入丁二腈后,470nm处荧光强度增加。加入己二腈后(见图8,图8为本发明实施例5中在化合物10的二氯甲烷溶液中加入己二腈后的荧光光谱示意图。),除了470nm处荧光强度增加外,400nm处产生一个新的荧光发射峰。这是由于两种客体化合物的分子长度不同,导致对其荧光发射产生的影响不同。这表明化合物10的溶液可以对分子尺寸不同、结构类似的客体化合物产生差异性识别,用于检测分子尺寸有差异的客体。
实施例6
配置化合物11的二氯甲烷溶液(10-5m),取2.5ml溶液,加入丁二腈,每次加入量为10mg,测溶液的荧光光谱。图9为本发明实施例6中在化合物11的二氯甲烷溶液中加入丁二腈后的荧光光谱示意图。从图9可以看出,化合物11在600nm和400nm处有两个明显的荧光发射峰。加入丁二腈后,600nm处荧光发射峰发生蓝移,荧光强度明显下降。同时,400nm处荧光发射峰蓝移至355nm,荧光强度明显提高。这表明化合物11的溶液可以对客体化合物产生识别。通过与图8对比可以看到,化合物11的荧光发射范围与响应能力与化合物10有显著差异。
对比例1
文献(chem.commun.,2018,54,837-840,doi:10.1039/c7cc08561c)报道了一种柱芳烃衍生的荧光分子,化学结构式如式ix所示:
如图10所示,图10是对比例1的化合物ix的晶体识别客体的前后荧光变化示意图。化合物ix的晶体具有荧光,当该化合物分子内部进入客体1,4-二氰基丁烷后,其分子构象发生变化,荧光发生淬灭。
本发明的实施例4、5所测试的化合物10,不仅在固态条件下对客体具有荧光响应,在溶液中还可以对不同尺寸的化合物产生不同的响应:荧光光谱具有显著差异。该化合物不仅具有识别客体的能力,还能根据客体的不同产生差异性识别。
此外,本发明提出了一种具有普适性的合成方法:通过化合物6可以经一到两步,以较高的收率得到一系列化合物。依据本发明,可以得到具有多色荧光发射的化合物,具有多样性。
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!