一种烷氧基取代的环对苯撑类化合物的制备方法
本发明涉及一种烷氧基取代的环对苯撑类化合物的制备方法,具体涉及用于制备烷氧基取代的环对苯撑类化合物的弯曲合成子及其制备方法,以及用于制备上述弯曲合成子的前驱体及其制备方法。
背景技术:
碳存在多种同素异形体,这些同素异形体表现出明显的结构和性质差异,从而对材料性质具有极大影响。纳米碳环材料例如共轭发光的环对苯撑类化合物在材料科学领域具有极大的潜在应用价值,尤其是在有机和生物电子学(如人造皮肤和神经)、合成生物学(例如人造细胞和病毒)、生物成像、光电功能材料以及多孔材料等领域具有突出的应用潜力。环对苯撑类化合物中引入功能化基团可以得到具有不同性质和功能的结构。而且,建立能够在环对苯撑类化合物结构上修饰多个功能基团的方法,最终可以获得性能更广泛的环形共轭结构。
现有技术cn108546229a公开了一种环对苯撑类化合物的制备方法,但是该制备方案制备的环对苯撑类化合物的环上没有功能基团,进而限制了其进一步的应用。kenichiroitami在论文“η6-cycloparaphenylenetransitionmetalcomplexes:synthesis,structure,photophysicalproperties,andapplicationtotheselectivemonofunctionalizationofcycloparaphenylenes”(j.am.chem.soc.,2015,137,1356-1361)中利用六羰基铬(cr(co)6)与环对苯撑形的配位反应形成过渡金属配合物,接着进行去质子化、亲核取代以及解配位反应,实现在环对苯撑结构上选择性的引入单取代基。但是,由于形成环对苯撑的过渡金属配合物较为困难,而且该配合物对光极为敏感,该方法对反应条件要求比较苛刻,从而限制了该方法的广泛使用。
因此,对于建立能够简单、快速制备带有功能化基团的环对苯撑类化合物的新方法是该研究领域的重大挑战。
技术实现要素:
本发明的目的在于提供一种烷氧基取代的环对苯撑类化合物的制备方法,从而实现烷氧基取代环对苯撑类化合物的简单、快速合成,从而为合成带有功能化基团的环对苯撑类化合物提供简便的新方法。
为此,一方面,本发明提供了一种用于制备弯曲合成子的前驱体,其结构如式(i)所示:
其中,式(i)中r1、r2、r3、r4各自独立地为氢、c1-20烷基或c1-20烷氧基;r5、r6、r7各自独立地为c1-20烷基;x为氯原子且y为溴原子或碘原子,或x为溴原子且y为碘原子。
优选地,式(i)中r1、r2、r3、r4各自独立地为氢,r5、r6、r7各自独立地为c1烷基;x为氯原子,y为碘原子。
此外,本发明还公开了一种上述前驱体的制备方法,包括以下步骤:
(1)在-78℃~-10℃的温度下,在有机溶剂中,将式(iv)的化合物用烷基锂化合物处理以得到相应的芳基锂化合物式(v)。
(2)使用所述芳基锂化合物式(v)与式(vi)的化合物发生缩合反应,得到式(vii)的化合物。
(3)在-15℃~25℃的温度下,在碱催化剂条件下,在有机溶剂中,使所述式(vii)的化合物与烷基卤化物r7n反应,从而得到所述式(i)的化合物,其中,n为溴原子或碘原子。
优选地,在所述式(iv)的化合物用烷基锂化合物处理之后,加入所述式(vi)的化合物以发生缩合反应,其中在步骤(2)中得到的所述式(vii)的化合物未经分离而直接用于步骤(3)的反应;
此外,本发明公开了一种采用所述的前驱体制备的弯曲合成子,其结构如式(ii)或(iii)所示:
其中,所述式(ii)中的x为氯原子或溴原子,所述式(ii)中r1、r2、r3、r4、r8、r9、r12、r13各自独立地为氢、c1-20烷基或c1-20烷氧基;r5、r6、r7、r10、r11各自独立地为c1-20烷基。
其中,所述式(iii)中r1、r2、r3、r4、r8、r9、r12、r13各自独立地为氢、c1-20烷基或c1-20烷氧基;r5、r6、r7、r10、r11各自独立地为c1-20烷基。
优选地,式(ii)中r1、r2、r3、r4、r8、r9、r12、r13各自独立地为氢,r5、r6、r7、r10、r11各自独立地为c1烷基,x为氯原子;式(iii)中r1、r2、r3、r4、r8、r9、r12、r13各自独立地为氢,r5、r6、r7、r10、r11各自独立地为c1烷基
此外,本发明还公开了一种上述弯曲合成子的制备方法,当所述弯曲合成子为式(ii)时,包括以下步骤:
(4),在-78℃~-10℃的温度下,在有机溶剂中,将式(i)的化合物用烷基锂化合物处理以得到相应的芳基锂化合物式(viii)。
(5),使用所述芳基锂化合物式(viii)与式(ix)的化合物发生缩合反应,得到式(x)的化合物。
(6),在-15℃~25℃的温度下,在碱催化剂条件下,在有机溶剂中,使所述式(x)的化合物与烷基卤化物r10m反应,从而得到所述式(ii)的化合物,其中,m为溴原子或碘原子。
当所述弯曲合成子为式(iii)时,还包括步骤(7),在80℃~100℃的温度下,在含钯催化剂和碱催化剂条件下,在有机溶剂中,使所述式(ii)的化合物与联硼酸频那醇酯进行miyaura硼酸酯化反应,从而得到所述式(iii)的化合物。
优选地,在所述式(i)的化合物用烷基锂化合物处理之后,加入所述式(ix)的化合物以发生缩合反应,其中在步骤(5)中得到的所述式(x)的化合物未经分离而直接用于步骤(6)的反应。
优选地,步骤(3)和步骤(6)所述有机溶剂是四氢呋喃或乙醚;所述碱催化剂是氢化钠或氢化钾;所述烷基卤化物是烷基溴代物或者烷基碘代物,例如碘甲烷或溴甲烷。
优选地,步骤(7)所述含钯催化剂是乙酸钯(pd(oac)2)或1,1'-双二苯基膦二茂铁二氯化钯(pdcl2(dppf));所述催化剂配体是有机膦配体,例如2-双环己基膦-2',6'-二甲氧基联苯(sphos);所述无机碱是乙酸钾或磷酸钾;所述有机溶剂是1,4-二氧六环或甲苯。
最后,本发明提供一种利用上述弯曲合成子制备含烷氧基取代环对苯撑类化合物的方法,所述方法包括:
(1),在含钯催化剂条件下,使用通式(iii)的化合物与式(xi)或(xii)所示的化合物进行suzuki偶联反应,得到相应的环状化合物;
(2),在还原剂条件下,使所得到的环状化合物中的环己二烯结构单元进行还原芳构化反应,从而得到相应的环对苯撑类化合物。
所述式(xi)或(xii)如下所示:
本发明具有但不限于以下优势:
1、相较于现有的方法,本发明所述的弯曲合成子合成步骤少,操作简单,环境污染小,易于快速、大量制备。
2、所述弯曲合成子可与不同长度或不同结构的匹配单元发生suzuki偶联反应,可用于制备不同结构类型的含烷氧基取代的环对苯撑类化合物。
3、所述前驱体和由其制备的弯曲合成子的烷氧基中的r易于变换,使所得环苯撑化合物的性能易于调节,例如不同碳链长度的r基团可以显著影响环苯撑化合物的凝固点,溶解度等;另一方面,烷氧基取代可以改变环苯撑类化合物的光物理性质。
4、本发明建立了制备含烷氧基取代的环对苯撑类化合物的新方法,操作简单,收率高。
附图说明
图1是根据本发明一个实施例合成的弯曲合成子(i)在氘代氯仿(cdcl3)中的核磁共振氢谱(1h-nmr);
图2是根据本发明一个实施例合成的弯曲合成子(i)在氘代氯仿(cdcl3)中的核磁共振碳谱(13c-nmr);
图3是根据本发明一个实施例合成的弯曲合成子(ii)在氘代氯仿(cdcl3)中的核磁共振氢谱(1h-nmr);
图4是根据本发明一个实施例合成的弯曲合成子(ii)在氘代氯仿(cdcl3)中的核磁共振碳谱(13c-nmr);
图5是根据本发明一个实施例合成的弯曲合成子(iii)在氘代氯仿(cdcl3)中的核磁共振氢谱(1h-nmr);
图6是根据本发明一个实施例合成的弯曲合成子(iii)在氘代氯仿(cdcl3)中的核磁共振碳谱(13c-nmr);
图7是根据本发明一个实施例合成的环对苯撑类化合物[10]cpp-2ome在氘代氯仿(cdcl3)中的核磁共振氢谱(1h-nmr);
图8是根据本发明一个实施例合成的环对苯撑类化合物[10]cpp-2ome在氘代氯仿(cdcl3)中的核磁共振碳谱(13c-nmr);
图9是根据本发明一个实施例合成的环对苯撑类化合物[10]cpp-2ome的基质辅助激光解析串联飞行时间质谱(maldi-tof-ms)谱图(实线)和模拟谱图(虚线);
图10是根据本发明一个实施例合成的环对苯撑类化合物[12]cpp-2ome在氘代氯仿(cdcl3)中的核磁共振氢谱(1h-nmr);
图11是根据本发明一个实施例合成的环对苯撑类化合物[12]cpp-2ome在氘代氯仿(cdcl3)中的核磁共振碳谱(13c-nmr);
图12是根据本发明一个实施例合成的环对苯撑类化合物[12]cpp-2ome的基质辅助激光解析串联飞行时间质谱(maldi-tof-ms)谱图(实线)和模拟谱图(虚线)。
具体实施方式
本发明提供了一种烷氧基取代的环对苯撑类化合物的制备方法,具体涉及用于制备烷氧基取代的环对苯撑类化合物的弯曲合成子及其制备方法,以及用于制备上述弯曲合成子的前驱体及其制备方法,其中前驱体式(i),弯曲合成子(ii)、(iii)的结构如下所示:
其中,式(i)中r1、r2、r3、r4各自独立地为氢、c1-20烷基或c1-20烷氧基,优选为氢;r5、r6、r7各自独立地为c1-20烷基,优选为c1烷基;x为氯原子且y为溴或碘原子,或x为溴原子且y为碘原子,优选的,x为氯原子,y为碘原子;
其中,式(ii)中r1、r2、r3、r4、r8、r9、r12、r13各自独立地为氢、c1-20烷基或c1-20烷氧基,优选为氢;r5、r6、r7、r10、r11各自独立地为c1-20烷基,优选为c1烷基;x为氯或溴原子。
其中,式(iii)中r1、r2、r3、r4、r8、r9、r12、r13各自独立地为氢、c1-20烷基或c1-20烷氧基,优选为氢;r5、r6、r7、r10、r11各自独立地为c1-20烷基,优选为c1烷基。
式(i)、(ii)、(iii)的化合物中,烷氧基团(or7、or10、or11)连接于环己二烯单元上。当该环己二烯单元在还原剂条件下进行还原芳构化转变为苯基的反应过程中,该烷氧基团可以被消除。
在本发明中,c1-20烷基的实例可以是甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、正己基、异己基等。
在本发明中,c1-20烷氧基的实例可以是甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、异戊氧基、新戊氧基、正己氧基、异己氧基等。
本发明提供的弯曲合成子的前驱体以及弯曲合成子的制备方法,包括以下步骤:
(1)在-78℃~-10℃(例如-78℃)的温度下,在无水有机溶剂(例如无水四氢呋喃)中,将式(iv)的化合物用烷基锂化合物(例如正丁基锂的己烷溶液)处理以得到相应的芳基锂化合物式(v);
(2)使用所述芳基锂化合物式(v)与式(vi)的化合物发生缩合反应,得到式(vii)的化合物;
(3)在-15℃~25℃的温度下(例如在冰水浴条件下),在碱催化剂(例如氢化钠)条件下,在无水有机溶剂(例如无水四氢呋喃)中,使所述式(vii)的化合物与烷基卤化物r7n(例如碘甲烷或溴甲烷)反应,从而得到所述式(i)的化合物;
(4)在-78℃~-10℃(例如-78℃)的温度下,在无水有机溶剂(例如无水四氢呋喃)中,将式(i)的化合物用烷基锂化合物(例如正丁基锂的己烷溶液)处理以得到相应的芳基锂化合物式(viii);
(5)使用所述芳基锂化合物式(viii)与式(ix)的化合物发生缩合反应,得到式(x)的化合物;
(6)在-15℃~25℃的温度下(例如在冰水浴条件下),在碱催化剂(例如氢化钠)条件下,在无水有机溶剂(例如无水四氢呋喃)中,使所述式(x)的化合物与烷基卤化物r10m(例如碘甲烷或溴甲烷)反应,从而得到所述式(ii)的化合物;
(7)在80℃~100℃(例如90℃)的温度下,在含钯催化剂(例如乙酸钯(pd(oac)2))、催化剂配体(例如2-双环己基膦-2',6'-二甲氧基联苯(sphos))和无机碱(例如磷酸钾)条件下,在有机溶剂(例如无水1,4-二氧六环)中,使所述式(ii)的化合物与联硼酸频那醇酯进行miyaura硼酸酯化反应,从而得到所述式(iii)的化合物。
其中,r1、r2、r3、r4、r5、r6、r7、r8、r9、r10、r11、r12、r13、x和y如上述所定义。
优选地,在所述式(iv)的化合物用烷基锂化合物处理之后,加入所述式(vi)的化合物以发生缩合反应,其中在步骤(2)中得到的所述式(vii)的化合物未经分离而直接用于步骤(3)的反应;在所述式(i)的化合物用烷基锂化合物处理之后,加入所述式(ix)的化合物以发生缩合反应,其中在步骤(5)中得到的所述式(x)的化合物未经分离而直接用于步骤(6)的反应。
优选地,步骤(3)和步骤(6)所述有机溶剂是四氢呋喃或乙醚;所述碱催化剂是氢化钠或氢化钾;所述烷基卤化物是烷基溴代物或者烷基碘代物,例如碘甲烷或溴甲烷。
优选地,步骤(7)所述含钯催化剂是乙酸钯(pd(oac)2);所述催化剂配体是2-双环己基膦-2',6'-二甲氧基联苯(sphos);所述无机碱是磷酸钾;所述有机溶剂是1,4-二氧六环。
本发明还提供了利用上述弯曲合成子来制备烷氧基取代环对苯撑类化合物的方法,所述方法包括:
(1)在含钯催化剂条件下,使用通式(iii)的化合物与以下式(xi)、(xii)所示的化合物进行suzuki偶联反应,得到相应的环状化合物;
(2)在还原剂条件下,使所得到的环状化合物中的环己二烯结构单元进行还原芳构化反应,从而得到相应的环对苯撑类化合物。
在一个具体实施方案中,本发明的弯曲合成子前驱体(i)和弯曲合成子(ii)和弯曲合成子(iii)可以按如下步骤合成:
在-78℃条件下,将式(iv)的化合物用等当量的正丁基锂处理,以生成相应的芳基锂化合物(v);在-78℃条件下,将式(vi)的化合物用过量的氢化钠处理,以生成相应的钠盐;然后将上述两者混合发生缩合反应,以生成式(vii)的化合物。对所得到的式(vii)的化合物经简单分离后,置于诸如烧瓶的反应器中,在冰水浴温度下,先与过量的氢化钠反应,接着与碘甲烷反应,对式(vii)中的羟基进行甲基化保护,从而得到弯曲合成子(i),总体收率为30%(三步反应的总收率)。
在-78℃条件下,将式(i)的化合物用等当量的正丁基锂处理,以生成相应的芳基锂化合物(viii);然后加入式(ix)的化合物发生缩合反应,以生成式(x)的化合物。对所得到的式(x)的化合物经简单分离后,置于诸如烧瓶的反应器中,在冰水浴温度下,先与过量的氢化钠反应,接着与碘甲烷反应,对式(x)中的羟基进行甲基化保护,从而得到弯曲合成子(ii),总体收率为48%(三步反应的总收率)。
在无水1,4-二氧六环溶剂中,在乙酸钯(pd(oac)2)、2-双环己基膦-2',6'-二甲氧基联苯(sphos)和磷酸钾条件下,将式(ii)的化合物与过量的联硼酸频那醇酯发生miyaura硼酸酯化反应,经分离纯化后得到弯曲合成子(iii),该步收率为81%。
下面通过结合具体实施例和附图对本发明做进一步说明,但本发明不限于这些实施例。
实施例1:弯曲合成子的前驱体(i)的合成(其中,r1、r2、r3、r4均为氢,r5、r6、r7均为甲基,x为氯原子,y为碘原子):
在200ml干燥圆底烧瓶中,将式(iv)的化合物(5.1g,13.08mmol)(其中,r5、r6均为甲基,y为碘原子,即1,4-二碘-2,5-二甲氧基苯,购买于北京伊诺凯科技有限公司)溶于65ml无水四氢呋喃(thf)(北京伊诺凯科技有限公司)中,将反应瓶置于-78℃的低温恒温搅拌反应浴(型号:dhjf-8005,厂商:郑州长城科工贸有限公司)中,向其中缓慢滴加7.7ml浓度为1.6m的正丁基锂溶液(北京伊诺凯科技有限公司)(12.32mmol),保持在-78℃的温度下继续反应1小时以生成芳基锂化合物(a瓶);同时,在另一个200ml干燥圆底烧瓶中,依次加入氢化钠(含量60%,混合于矿物油中)(北京伊诺凯科技有限公司)609mg(15.23mmol)和无水四氢呋喃7.5ml,将反应瓶置于-78℃的低温恒温搅拌反应浴中,然后将溶解在30ml无水四氢呋喃中的的式(vi)的化合物2.24g(10.15mmol)缓慢滴加到该反应容器中,保持在-78℃的温度下继续反应1小时(b瓶);使用u型钢针将上述a瓶中的芳基锂化合物在氩气气压下转移至b瓶,并保持在-78℃的温度下继续反应3小时。反应结束后,加入蒸馏水淬灭反应。使用旋转蒸发仪(型号r-1001vn,厂商:郑州长城科工贸有限公司)除去四氢呋喃,用二氯甲烷(江苏强盛功能化学股份有限公司)萃取(3×50ml),将有机相合并,用无水硫酸钠(江苏强盛功能化学股份有限公司)干燥,旋转蒸发除去溶剂并真空干燥,得到相应的式(vii)的化合物(其中,r1、r2、r3、r4、r7均为氢,r5、r6均为甲基,x为氯原子,y为碘原子)。在未对该式(vii)的化合物作进一步分离和纯化的情况下,将其直接用于下一步反应。
在冰水浴控温下,将上述干燥后的式(vii)的化合物溶解在30ml无水四氢呋喃中,缓慢滴加到含有570mg氢化钠(含量60%,混合于矿物油中)(14.25mmol)和10ml无水四氢呋喃的100ml干燥圆底烧瓶中,搅拌反应1小时。然后,将无水n,n-二甲基甲酰胺(北京伊诺凯科技有限公司)(4ml)与碘甲烷(北京伊诺凯科技有限公司)(1.16ml,18.54mmol)的混合溶液缓慢滴加到反应体系中,移除冰水浴,使反应缓慢升至室温,并保持在室温条件下反应24小时。反应结束后,加入蒸馏水淬灭反应。使用旋转蒸发仪除去溶剂,用二氯甲烷萃取(3×50ml),将有机相合并,用无水硫酸钠干燥,旋转蒸发除去溶剂,剩余物经硅胶柱层析分离纯化(石油醚:乙酸乙酯10:1),得到式(i)为制备弯曲合成子的前驱体1.56g(白色固体粉末,产率30%)。
这里使用的式(vi)的化合物(其中,r1、r2、r3、r4、r7均为氢,x为氯原子)制备方法如下:在500ml干燥圆底烧瓶中,将4-溴氯苯(北京伊诺凯科技有限公司)(15.45g,80.73mmol)溶于200ml无水四氢呋喃中并置于-78℃的低温恒温搅拌反应浴中,然后缓慢向其中滴加27.3ml浓度为2.5m的正丁基锂溶液(68.32mmol)(北京伊诺凯科技有限公司),保持在-78℃的温度下继续反应0.5小时,再缓慢加入4,4-二甲氧基-2,5-环己二烯-1-酮(该化合物可以参考tetrahedron2010,66(31),5852-5862合成得到)(9.58g,62.1mmol),保持在-78℃的温度下继续反应1小时。反应结束后,加入蒸馏水淬灭反应。旋转蒸发除去四氢呋喃,用二氯甲烷萃取(3×50ml),将有机相合并,用无水硫酸钠干燥,旋转蒸发除去溶剂得到棕色油状物,将其溶解在23ml丙酮(国药试剂)中,然后加入88ml的10%乙酸(国药试剂)水溶液,室温条件下搅拌16小时,得到的沉淀固体通过过滤收集,并用少量二氯甲烷洗涤沉淀,真空干燥得10.9g式(vi)的化合物(白色粉末状固体,产率80%)。
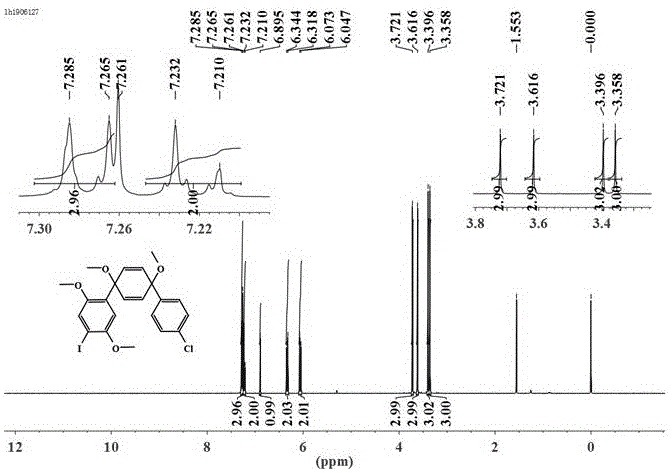
所得式(i)的弯曲合成子的的前驱体通过基质辅助激光解吸飞行时间质谱仪(maldi-tof-ms)(型号:atouflexspeed,岛津集团英国kratos分析仪器公司)进行表征(m/z):理论值:c22h22clio4[m]+:512.0251,实验值:511.9689;通过核磁共振氢谱(型号:avanceaviii400,厂商:瑞士布鲁克公司)进行表征:1hnmr(400mhz,cdcl3)δ7.20-7.27(m,3h),7.22(d,2h),6.89(s,1h),6.33(d,2h),6.06(d,2h),3.72(s,3h),3.62(s,3h),3.40(s,3h),3.36(s,3h)(图1);通过核磁共振碳谱(型号:avanceaviii400,厂商:瑞士布鲁克公司)进行表征:13cnmr(101mhz,cdcl3)δ152.66,152.52,141.83,133.21,131.81,131.75,128.25,127.62,124.04,110.96,85.34,74.36,73.91,57.09,56.62,51.99,51.57(图2)。
实施例2:弯曲合成子(ii)的合成(其中,r1、r2、r3、r4、r8、r9、r12、r13均为氢,r5、r6、r7、r10、r11均为甲基,x为氯原子):
在200ml干燥圆底烧瓶中,将式(i)的化合物(1.88g,3.67mmol)溶于110ml无水四氢呋喃中并置于-78℃的低温恒温搅拌反应浴中,然后缓慢向其中滴加2.5ml浓度为1.6m的正丁基锂溶液(4.0mmol),保持在-78℃的温度下继续反应40分钟,然后加入式(ix)的化合物(947mg,4.0mmol)(其中,r8、r9、r12、r13均为氢,r11为甲基,x为氯原子)使其与上述所得的芳基锂化合物发生缩合反应,保持在-78℃的温度下继续反应3小时后通过加入蒸馏水淬灭反应。旋转蒸发除去四氢呋喃,用二氯甲烷萃取(3×50ml),将有机相合并,用无水硫酸钠干燥,旋转蒸发除去溶剂并真空干燥,得到相应的式(x)的化合物(其中,r1、r2、r3、r4、r8、r9、r12、r13均为氢,r5、r6、r7、r11均为甲基,x为氯原子)。在未对该式(x)的化合物作进一步分离和纯化的情况下,将其直接用于下一步反应。
在冰水浴控温下,将上述干燥后的式(x)的化合物溶解在30ml无水四氢呋喃中,缓慢滴加到含有225mg氢化钠(含量60%,混合于矿物油中)(5.6mmol)和10ml无水四氢呋喃的干燥圆底烧瓶中,搅拌反应1小时。然后,将无水n,n-二甲基甲酰胺(1.7ml)与碘甲烷(0.45ml,7.3mmol)的混合溶液缓慢滴加到反应体系中,移除冰水浴,使反应缓慢升至室温,并保持在室温条件下反应24小时。反应结束后,加入蒸馏水淬灭反应。使用旋转蒸发仪除去溶剂,用二氯甲烷萃取(3×50ml),将有机相合并,用无水硫酸钠干燥,旋转蒸发除去溶剂,剩余物经硅胶柱层析分离纯化(石油醚:乙酸乙酯5:1),得到式(ii)的弯曲合成子1.43g(白色固体粉末,产率48%)。
这里使用的式(ix)的化合物(其中,r8、r9、r12、r13均为氢,r11为甲基,x为氯原子)制备方法如下:在200ml干燥圆底烧瓶中,将4-溴氯苯(6.13g,32mmol)溶于80ml无水四氢呋喃中并置于-78℃的低温恒温搅拌反应浴中,然后缓慢向其中滴加17.1ml浓度为1.6m的正丁基锂溶液(27.4mmol),保持在-78℃的温度下继续反应0.5小时,再缓慢加入4,4-二甲氧基-2,5-环己二烯-1-酮(3.83g,25mmol),保持在-78℃的温度下继续反应1小时。然后,将无水n,n-二甲基甲酰胺(8.0ml)与碘甲烷(3.4ml,55mmol)的混合溶液缓慢滴加到反应体系中,移除冰水浴,使反应缓慢升至室温,并保持在室温条件下反应24小时。反应结束后,加入蒸馏水淬灭反应。旋转蒸发除去四氢呋喃,用二氯甲烷萃取(3×50ml),将有机相合并,用无水硫酸钠干燥,旋转蒸发除去溶剂得到棕色油状物,将其溶解在9ml丙酮中,然后加入35ml的10%乙酸水溶液,室温条件下搅拌16小时,得到的沉淀固体通过过滤收集,并用少量二氯甲烷洗涤沉淀,真空干燥得3.5g式(ix)的化合物(白色粉末状固体,产率60%)
所得式(ii)的弯曲合成子通过基质辅助激光解吸飞行时间质谱仪(maldi-tof-ms)(型号:atouflexspeed,岛津集团英国kratos分析仪器公司)进行表征(m/z):理论值:c36h36cl2o6[m]+:634.1889,实验值:634.1113;通过核磁共振氢谱(型号:avanceaviii400,厂商:瑞士布鲁克公司)进行表征:1hnmr(400mhz,cdcl3)δ7.29(dt,4h),7.20(dt,4h),6.96(s,2h),6.38(d,4h),6.03(d,4h),3.55(s,6h),3.40(s,6h),3.36(s,6h)(图3);通过核磁共振碳谱(型号:avanceaviii400,厂商:瑞士布鲁克公司)进行表征:13cnmr(101mhz,cdcl3)δ151.86,142.01,133.09,132.89,132.19,131.04,128.19,127.62,113.49,74.43,73.85,56.60,51.98,51.58(图4)。
实施例3:弯曲合成子(iii)的合成(其中,r1、r2、r3、r4、r8、r9、r12、r13均为氢,r5、r6、r7、r10、r11均为甲基):
将式(ii)的化合物(850mg,1.34mmol)、磷酸钾(1.8g,8.48mmol)(北京伊诺凯科技有限公司)和联硼酸频那醇酯(2.1g,8.8mmol)(北京伊诺凯科技有限公司)加入到100ml圆底烧瓶中,接着加入无水1,4-二氧六环(50ml)(国药试剂),然后向溶液中通入氮气脱气50分钟,之后加入乙酸钯(58mg,0.26mmol)(北京伊诺凯科技有限公司)和2-双环己基膦-2',6'-二甲氧基联苯(sphos)(210mg,0.51mmol)(北京伊诺凯科技有限公司),继续向溶液中通入氮气10分钟。反应在氮气保护下于90oc反应24小时。旋转蒸发去除有机溶剂,剩余物经硅胶柱层析分离纯化(石油醚:乙酸乙酯3:1),得到式(iii)的化合物890mg(白色粉末状固体,产率81%)。
所得式(iii)的弯曲合成子通过基质辅助激光解吸飞行时间质谱仪(maldi-tof-ms)(型号:atouflexspeed,岛津集团英国kratos分析仪器公司)进行表征(m/z):理论值:c48h60b2o10[m]+:818.4373,实验值:818.4104;通过核磁共振氢谱(型号:avanceaviii400,厂商:瑞士布鲁克公司)进行表征:1hnmr(400mhz,cdcl3)δ7.69(d,4h),7.37(d,4h),6.97(s,2h),6.38(d,4h),6.06(d,4h),3.57(s,6h),3.41(s,6h),3.36(s,6h),1.31(s,24h)(图5);通过核磁共振碳谱(型号:avanceaviii400,厂商:瑞士布鲁克公司)进行表征:13cnmr(101mhz,cdcl3)δ151.80,146.49,134.69,132.95,131.86,130.88,125.45,113.28,83.73,74.84,73.92,56.56,51.88,51.54,24.85.(图6)。
实施例4:合成具有以下结构的环对苯撑化合物[10]cpp-2ome(二甲氧基十苯基环对苯撑)
将式(iii)的化合物(133mg,0.165mmol)、氢氧化钾(118mg,2.1mmol)(国药试剂)和式(xi)的化合物(参考chem.sci.2012,3(10),3018-3021.合成)(120mg,0.18mmol)加入到500ml圆底烧瓶中,接着加入四氢呋喃(200ml)和水(20ml),然后向溶液中通入氮气脱气50分钟,之后加入四(三苯基膦)钯(pd[p(c6h5)3]4)(27mg,0.023mmol)(北京伊诺凯科技有限公司),继续向溶液中通入氮气10分钟。反应在氮气保护下于75oc反应48小时。旋转蒸发去除有机溶剂,二氯甲烷萃取(3×100ml),无水硫酸钠干燥,旋转蒸发除去溶剂并干燥得粗产物。
在50ml圆底烧瓶中加入氯化亚锡二水合物(sncl2·2h2o)(357mg,1.58mmol)(北京伊诺凯科技有限公司)和四氢呋喃(30ml),向溶液中通入氮气脱气30分钟,之后加入264μl盐酸(浓度为36.0~38.0%)(国药试剂),室温条件下继续搅拌30分钟,向其中加入上步粗产物,室温条件下继续搅拌2小时。以饱和碳酸氢钠水溶液淬灭反应,二氯甲烷萃取(3×60ml),无水硫酸钠干燥,旋转蒸发除去溶剂,剩余物经硅胶柱层析分离纯化(石油醚:二氯甲烷2:1),得28mg[10]cpp-2ome(浅黄色粉末状固体,产率10%)。
通过基质辅助激光解吸飞行时间质谱仪(maldi-tof-ms)(型号:atouflexspeed,岛津集团英国kratos分析仪器公司)进行表征(m/z):理论值:c62h44o2[m]+:820.3358,实验值:820.3177(图7);通过核磁共振氢谱(型号:avanceaviii400,厂商:瑞士布鲁克公司)进行表征:1hnmr(400mhz,cdcl3)δ7.45-7.67(m,36h),6.86(s,2h),3.73(s,6h).(图8);通过核磁共振碳谱(型号:avanceaviii400,厂商:瑞士布鲁克公司)进行表征:13cnmr(101mhz,cdcl3)δ151.80,138.57,138.40,138.28,138.21,138.11,138.09,138.02,137.92,136.51,129.47,129.00,127.45,127.42,127.41,127.34,127.31,127.22,127.19,126.63,115.11,56.63.(图9)。
实施例5:合成具有以下结构的环对苯撑化合物[12]cpp-2ome(二甲氧基十二苯基环对苯撑)
将式(iii)的化合物(133mg,0.165mmol)、氢氧化钾(118mg,2.1mmol)和式(xii)的化合物(参考angew.chem.int.ed.2019,58(19),6244-6249.合成)(153mg,0.17mmol)加入到500ml圆底烧瓶中,接着加入四氢呋喃(200ml)和水(20ml),然后向溶液中通入氮气脱气50分钟,之后加入四(三苯基膦)钯(pd[p(c6h5)3]4)(27mg,0.023mmol),继续向溶液中通入氮气10分钟。反应在氮气保护下于75oc反应48小时。旋转蒸发去除有机溶剂,二氯甲烷萃取(3×100ml),无水硫酸钠干燥,旋转蒸发除去溶剂并干燥得粗产物。
在50ml圆底烧瓶中加入氯化亚锡二水合物(sncl2·2h2o)(471mg,2.1mmol)和四氢呋喃(30ml),向溶液中通入氮气脱气30分钟,之后加入348μl盐酸(浓度为36.0~38.0%),室温条件下继续搅拌30分钟,向其中加入上步粗产物,室温条件下继续搅拌2小时。以饱和碳酸氢钠水溶液淬灭反应,二氯甲烷萃取(3×60ml),无水硫酸钠干燥,旋转蒸发除去溶剂,剩余物经硅胶柱层析分离纯化(石油醚:二氯甲烷3:2),得32mg[12]cpp-2ome(浅黄色粉末状固体,产率10%)
通过基质辅助激光解吸飞行时间质谱仪(maldi-tof-ms)(型号:atouflexspeed,岛津集团英国kratos分析仪器公司)进行表征(m/z):理论值:c74h52o2[m]+:972.3980,实验值:972.4055(图10);通过核磁共振氢谱(型号:avanceaviii400,厂商:瑞士布鲁克公司)进行表征:1hnmr(400mhz,cdcl3)δ7.54-7.66(m,44h),6.90(s,2h),3.75(s,6h).(图11);通过核磁共振碳谱(型号:avanceaviii400,厂商:瑞士布鲁克公司)进行表征:13cnmr(101mhz,cdcl3)δ151.59,138.91,138.65,138.63,138.58,138.52,138.51,138.38,136.80,129.70,129.35,127.46,127.43,127.39,127.36,127.31,127.27,126.64,115.21,56.67.(图12)。
本发明制备的弯曲合成子易大规模制备,合成步骤少,操作方便,环境污染小。利用本发明的弯曲合成子制备的含烷氧基取代环对苯撑类化合物具有良好的化学稳定性,荧光光谱范围广,发光强,在材料科学领域具有潜在的应用价值,尤其是在有机和生物电子学、合成生物学及生物成像等领域具有突出的应用前景。
以上所述仅是本发明的优选实施方式,应当指出,在本技术领域,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!