一种基于微通道反应技术快速制备环丙烷衍生物的方法与流程
本发明属于环丙烷类化合物的合成方法,具体的说是涉及一种基于微通道反应技术快速制备环丙烷衍生物的方法。
背景技术:
环丙烷类化合物作为一类重要的有机中间体,在医药分子、精细化学品、功能材料等领域中有着广泛的应用,具有极高的附加价值。传统环丙烷类化合物的制备一般以目标环丙烷对应的烯烃为原料,以重氮甲烷作为烷基化试剂,在釜式反应器中进行加成烷基化反应。但该方法因生成的重氮甲烷制备过程中性质非常活泼、易爆且剧毒而导致重氮甲烷的制备和转移在工业生产中非常困难,因此其使用成本较高,受到很大的限制。发明专利201910589163.0公开了一种环丙烷类化合物连续合成的方法及装置,但该发明的缺陷在于第一反应器中的反应产物还需进入分离器中进行分层,增加了其得到的重氮甲烷前体溢出的隐患。
技术实现要素:
本发明为了克服现有技术存在的不足,提供一种基于微通道反应技术快速制备环丙烷衍生物的方法,从而可以迅速、高效、安全地合成环丙烷类化合物。
本发明是通过以下技术方案实现的:本发明公开了一种基于微通道反应技术快速制备环丙烷衍生物的方法,该制备方法包括碱水解反应和亲电加成反应共两步反应,具体步骤如下:
(1)碱水解反应:
将n-甲基-n-亚硝基脲溶液和氢氧化钾溶液按一定当量比例泵入第一段微通道反应器中,在一定温度下进行碱水解反应,反应一定时间生成重氮甲烷;
(2)亲核加成反应:
将步骤(1)中得到的重氮甲烷通入第二段微通道反应器中,与一定当量比例的烯烃溶液混合,在一定温度下反应一定时间生成环丙烷类化合物产品;
反应路线如下:
将下式化合物用作步骤(1)中的重氮甲烷:
其中
r1=硼酸酯;
r2=取代/未取代的芳基或杂芳基,其中取代基选自烷基、烷氧基、卤素、氰基或酯基中的至少一种;
r3=氢、c1-6烷基、酯基;
r4=氢或氘。
步骤(1)所述n-甲基-n-亚硝基脲溶液中所用溶剂为二乙二醇单乙醚、甲基叔丁基醚、甲醇、乙醇,或上述溶剂的任意组合;所述氢氧化钾溶液所用溶剂为乙醇、异丙醇、水,或上述溶剂的任意组合;所述烯烃化合物溶液所用溶剂为四氢呋喃、二乙二醇单乙醚、异丙醇、甲醇、乙醇、乙酸乙酯,或上述溶剂的任意组合。
步骤(1)中所述的一种基于微通道反应技术快速制备环丙烷衍生物的方法,其特征在于:步骤(1)中所述n-甲基-n-亚硝基脲与所述氢氧化钾的当量比为1~4:2;步骤(1)或(2)中第一段微通道反应器中和第二段微通道反应器中的内径为1~5mm。
作为优选,步骤(1)中所述n-甲基-n-亚硝基脲与氢氧化钾的当量比例为1.5:2;步骤(1)或(2)中第一段微通道反应器中和第二段微通道反应器中的内径1~3mm。
步骤(1)中碱水解的反应温度为15℃~35℃,反应停留时间为1~3min。
作为优选,步骤(1)中碱水解交换的反应温度为20℃~25℃,反应停留时间为1.5~2.5min。
步骤(2)中的亲电加成反应,所述重氮甲烷与烯烃化合物的当量比为1~10:1;步骤(2)的反应温度为15℃~35℃,反应停留时间为1~6min。
作为优选,步骤(2)中的亲核加成反应,所述重氮甲烷与烯烃化合物的当量比例为2:1;步骤(2)的反应温度为20℃~25℃,反应停留时间为2~5min。
本发明的有益效果是:本发明的创新点在于通过耦合微通道反应技术,为重氮甲烷及环丙衍生物的合成过程提供了一种绿色、高效、低成本廉、条件温和且底物普适性强的合成方法。本发明所述的微通道反应器的微尺度特征使得反应器具有本质安全性,大大降低了制备重氮甲烷的危险系数,且使得反应器的比表面积和质热传递系数远高于传统反应器,使反应在可控的连续化条件下可获得较高的环丙烷衍生物产物纯度,使得本发明工艺区别于现有的环丙烷衍生物制备工艺。
附图说明
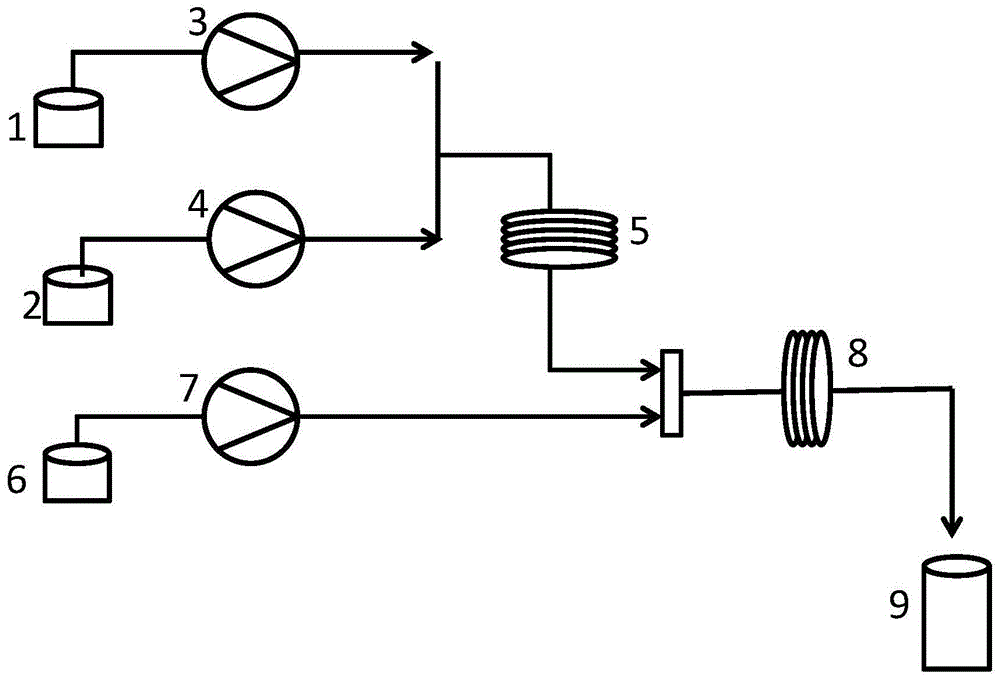
图1是本发明的合成工艺流程图:
图中:1-n-甲基-n-亚硝基脲储罐;2-氢氧化钾储罐;3-第一计量泵;4-第二计量泵;5-第一段微通道反应器;6-烯烃化合物储罐;7-第三计量泵;8-第二段微通道反应器;9-产品接收罐。
具体实施方式
以下结合附图和具体实施方式对本发明作详细描述。
本发明公开了一种基于微通道反应技术快速制备环丙烷衍生物的方法,该制备方法包括碱水解反应和亲电加成反应共两步反应,具体步骤如下:
(1)碱水解交换反应:
将n-甲基-n-亚硝基脲溶液和氢氧化钾溶液按一定当量比例泵入第一段微通道反应器中,在一定温度下进行碱水解反应,反应一定时间生成重氮甲烷;
(2)亲电加成反应:
将步骤(1)中得到的重氮甲烷通入第二段微通道反应器中,与一定当量比例的烯烃溶液混合,在一定温度下反应一定时间生成环丙烷类化合物,产物经柱层析后得到产品环丙烷类产品;
反应路线如下:
将下式化合物用作步骤(1)中的重氮甲烷:
其中
r1=硼酸酯;
r2=取代/未取代的芳基或杂芳基,其中取代基选自烷基、烷氧基、卤素、氰基或酯基中的至少一种;
r3=氢、c1-6烷基、酯基;
r4=氢或氘。
步骤(1)所述n-甲基-n-亚硝基脲溶液中所用溶剂为二乙二醇单乙醚、甲基叔丁基醚、甲醇、乙醇,或上述溶剂的任意组合;所述氢氧化钾溶液所用溶剂为乙醇、异丙醇、水,或上述溶剂的任意组合;所述烯烃化合物溶液所用溶剂为四氢呋喃、二乙二醇单乙醚、异丙醇、甲醇、乙醇、乙酸乙酯,或上述溶剂的任意组合。
步骤(1)中所述的一种基于微通道反应技术快速制备环丙烷衍生物的方法,其特征在于:步骤(1)中所述n-甲基-n-亚硝基脲与所述氢氧化钾的当量比为1~4:2;步骤(1)或(2)中第一段微通道反应器中和第二段微通道反应器中的内径为1~5mm。
作为优选,步骤(1)中所述n-甲基-n-亚硝基脲与氢氧化钾的当量比例为1.5:2;步骤(1)或(2)中第一段微通道反应器中和第二段微通道反应器中的内径1~3mm。
步骤(1)中碱水解的反应温度为15℃~35℃,反应停留时间为1~3min。
作为优选,步骤(1)中碱水解交换的反应温度为20℃~25℃,反应停留时间为1.5~2.5min。
步骤(2)中的亲电加成反应,所述重氮甲烷与烯烃化合物的当量比为1~10:1;步骤(2)的反应温度为15℃~35℃,反应停留时间为1~6min。
作为优选,步骤(2)中的亲核加成反应,所述重氮甲烷与烯烃化合物的当量比例为2:1;步骤(2)的反应温度为20℃~25℃,反应停留时间为2~5min。
实施例1:
反应式:
如图1所示,将1.0毫摩尔当量的n-甲基-n-亚硝基脲的甲基叔丁基醚溶液装入n-甲基-n-亚硝基脲储罐1中,1.5毫摩尔当量氢氧化钾的乙醇溶液装入氢氧化钾储罐2中,n-甲基-n-亚硝基脲的甲基叔丁基醚溶液和氢氧化钾的乙醇溶液分别由第一计量泵3和第二计量泵4打入第一段微通道反应器5中进行反应,控制第一段微通道反应器5在15℃~25℃下进行反应,反应停留时间约2min,反应生成的重氮甲烷通入第二段微通道反应器8中;同时将a1的四氢呋喃溶液装入烯烃化合物储罐6中,储罐6中的反式-2-苯乙烯硼酸频哪酯的四氢呋喃溶液(0.5毫摩尔当量烯烃化合物)由第三计量泵7打入第二段微通道反应器8中与重氮甲烷与混合进行亲电加成反应,第二段微通道反应器8在15℃~25℃中反应,停留时间为3min,将烯烃原位转化,在反应末端的产品接收罐9内收集样品,快速柱层析(石油醚/乙酸乙酯洗脱体系)后浓缩得到产品a2,纯度96.94%。
实施例2:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为b1,获得产品b2,纯度98.56%。
实施例3:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为c1,获得产品c2,纯度88.56%。
实施例4:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为d1,获得产品d2,纯度86.32%。
实施例5:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为e1,获得产品e2,纯度86.25%。
实施例6:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为f1,获得产品f2,纯度97.15%。
实施例7:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为g1,获得产品g2,纯度92.65%。
实施例8:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为g1,获得产品g2,纯度91.25%。
实施例9:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为h1,获得产品h2,纯度89.56%。
实施例10:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为i1,获得产品i2,纯度92.66%。
实施例11:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为j1,获得产品j2,纯度91.39%。
实施例12:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为k1,获得产品k2,纯度96.12%。
实施例13:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为l1,获得产品l2,纯度94.19%。
实施例14:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为m1,获得产品m2,纯度95.19%。
实施例15:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为n1,获得产品n2,纯度93.19%。
实施例16:
反应式:
具体制备过程同实施例1,将反应物烯烃化合物a1改为b1,第一段微通道反应器中,碱水解反应温度为20℃~35℃,停留时间2min;第二段微通道反应器中进行亲电加成反应,反应温度为20℃~35℃,停留时间为2min,获得b2产品的纯度为92.48%。
最后应当说明的是,以上内容仅用以说明本发明的技术方案,而非对本发明保护范围的限制,本领域的普通技术人员对本发明的技术方案进行的简单修改或者等同替换,均不脱离本发明技术方案的实质和范围。
- 还没有人留言评论。精彩留言会获得点赞!