一种合成手性苯酞的方法
本发明涉及有机合成技术领域,具体涉及一种合成手性苯酞的方法。
背景技术:
苯酞类化合物存在于自然界,其中丁苯酞(化学名为3-丁基-1(-3h)-异苯并呋喃酮,式ii,r1=n-c4h9)存在于芹菜、当归等植物中,具有抗惊厥、抗气喘、增加血流量等药理活性,临床上用于治疗缺血性脑卒中等疾病。临床结果表明,手性l-丁苯酞比外消旋丁苯酞有更好的疗效[1]。
目前,l-丁苯酞的合成方法多为由外消旋丁苯酞通过拆分得到。这样,会产生大量的无效体(d-丁苯酞),降低了产物的利用效率。carpentier、林国强等通过[rucl2(p-cymene)]2-手性二胺配合物分别催化异丙醇、甲酸钠对邻戊酰基苯甲酸酯的还原反应,对映体选择性地合成了手性丁苯酞[2-4],但反应时间过长,催化剂也较为昂贵。周其林等采用[ir(cod)cl]2-手性螺环氨基膦催化h2对邻戊酰基苯甲酸酯的还原反应,高选择性地生成l-丁苯酞[5,6]。但是,反应需采用到贵金属催化剂,在10atmh2下进行。
上述制备手性苯酞的方法仍存在着缺陷,或需要贵金属,或需要高压,或反应时间长,不利于大规模、低成本工业化生产。铜作为一种较为廉价的金属,利用铜盐-手性膦配合物在常压下催化合成手性苯酞对大规模合成苯酞有重要价值。
技术实现要素:
为弥补现有技术的不足,本发明提供了一种合成手性苯酞的方法,以有效解决背景技术中所提及的技术问题,通过以下技术方案实现:
本发明在铜盐-手性膦配体形成的催化剂作用下,使用硅烷作为还原剂,对邻酰基苯甲酸酯进行不对称还原/内酯化反应。该方法使用催化量的cu-手性膦催化剂,在反应体系中依次进行对羰基的不对称还原和内酯化反应,不需分离还原反应形成的中间体,并且反应条件温和,具有对映体选择性高、产率高等优点。
一种合成手性苯酞的方法,合成路线是:
合成方法具体为:在休良克瓶中分别加入铜化合物、手性膦配体,抽真空,氮气置换;保持氮气氛围加入反应溶剂搅拌,再加入硅烷搅拌,将式ⅰ化合物加入休良克瓶中,即可合成式ⅱ化合物手性苯酞。
所述式ⅰ化合物与式ⅱ化合物中的r1为c1-c10的烷基、c1-c10的环烷基、c1-c10的带取代基的环烷基中的一种;r2为c1-c10的直链烷基、c1-c10的直链环烷基、c1-c10的支链烷基、c1-c10的支链环烷基中的一种。
进一步的,所述的铜化合物为cuf(pph3)3·2meoh、cu(r3co2)2、cu(r3co2)2·mh2o、cuor3中的一种,或cux、cux2、cui中的一种与mor3的混合物,其中x为f、cl、br中的一种;m=1或2或3;r3为c1-c12的烷基或c1-c12的取代烷基;m为na或k。
进一步的,所述的手性膦配体为有如下结构的旋光纯的膦化合物l1-l5的一种,或l1-l5的对映体的一种;
l1-l3中,r4为ph、2-mec6h4、4-mec6h4、t-bu、c-c6h11、3,5-me2-c6h3、3,5-me2-4-meo-c6h2、3,5-(t-bu)2-c6h3、4-meo-3,5-(t-bu)2-c6h2、3,4,5-(meo)3-c6h2中的一种;l4-l5中r5为甲基、乙基、异丙基、苯基、取代苯基的一种。
进一步的,所述的硅烷包括phsih3、ph2sih2、ph3sih、ph2mesih、(sihme2)2o、et3sih、(meo)3sih、(sihme2)2nh、r6(osihme)nor6中的一种,n=1,2,3,…,100的整数;r6为h或si(ch3)3或si(ch3)2but。
进一步的,所述的反应溶剂为醚类或芳烃类或卤代烷或酯类,包括但并不限于乙醚、丁醚、四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、苯、甲苯、乙苯、二甲苯、二氯甲烷、三氯甲烷、1,2-二氯乙烷、乙酸乙酯、乙酸丙酯、乙酸丁酯中的一种以上的混合物。
进一步的,反应溶剂的体积用量:式i化合物的质量=(1-1000)ml:1g。
进一步的,所述的铜化合物、手性膦配体、硅烷、式i化合物的摩尔比为(0.02~0.08):(0.02~0.08):(2~5):1。
进一步的,在休良克瓶中分别加入铜化合物、手性膦配体时,休良克瓶内的温度为-20-0℃。
进一步的,所述的合成方法具体为:在休良克瓶中分别加入铜化合物、手性膦配体,抽真空,氮气置换3次;保持氮气氛围加入反应溶剂搅拌10-20min,再加入硅烷搅拌20min,将式ⅰ化合物加入休良克瓶中,即可合成式ⅱ化合物手性苯酞。
本发明与现有技术相比的有益效果是:
本发明可以对映体选择性地合成手性苯酞,其中l-构型为优势异构体,产物的ee值高达93%,通过后处理可得到单一构型的化合物,提供了一种对映选择性的方法合成具有更高活性的l-苯酞。通过调整配体的构型也可得到d-构型为优势异构体的苯酞。
本发明可以较高产率制得手性苯酞,所用原料皆为市售原料,原料易得且价格低廉,该方法只使用了催化量的cu-手性膦催化剂,不存在外消旋体拆分的问题、不需要贵金属催化剂、对环境友好、成本低。
附图说明
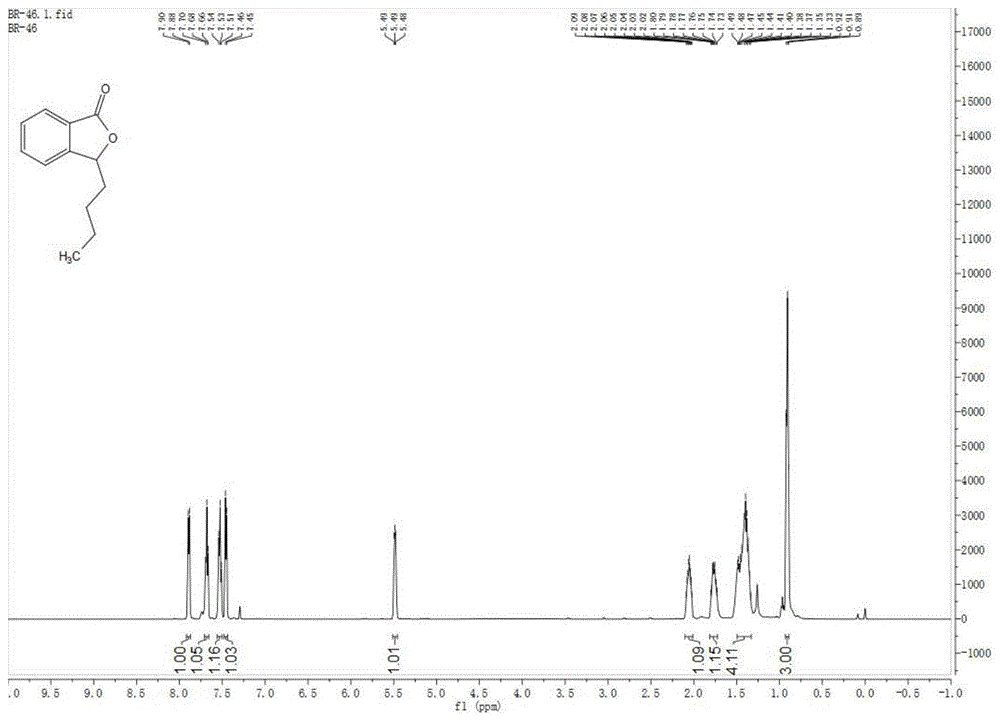
图1为实施例1的1hnmr图谱;
图2为实施例1的13cnmr图谱;
图3为实施例2的1hnmr图谱;
图4为实施例2的13cnmr图谱;
图5为实施例3的1hnmr图谱;
图6为实施例3的13cnmr图谱;
图7为实施例1的hplc色谱图。
具体实施方式
下面通过具体实施例详述本发明,但不限制本发明的保护范围。如无特殊说明,本发明所采用的实验方法均为常规方法,所用实验器材、材料、试剂等均可从商业途径获得。
实施例1
在干燥的休良克瓶中依次加入0.0098gcu(pph3)3f·2meoh和0.0057gl4(r5=ph),抽真空并氮气置换3次,加入1.5ml甲苯,搅拌10min,加入3equiv的pmhs,搅拌20min,溶液呈橙色,降温至0℃;加入0.0450g邻戊酰基苯甲酸甲酯/0.5ml甲苯溶液。反应3h后加入3ml饱和氯化铵水溶液、搅拌1h,分液,水相用3ml乙酸乙酯萃取2次。合并的有机相经2ml饱和氯化钠溶液洗涤2次,无水硫酸钠干燥、旋蒸、柱层析纯化,得到产物手性l-丁苯酞,呈黄色油状液体,0.0310g,产率78%,ee93%。其中rf=0.55,ea:pe=1:5;1h-nmr(500mhz,cdcl3):δ7.86(d,j=7.6hz,1h),7.65(dd,j=7.6hz,1h),7.49(dd,j=7.6hz,1h),7.42(d,j=7.6hz,1h),5.49–5.42(m,1h),2.02(ddt,j=14.5,9.7,4.7hz,1h),1.80–1.67(m,1h),1.49–1.31(m,4h),0.87(t,j=6.1hz,3h);13c-nmr(126mhz,cdcl3)δ170.56,150.01,133.85,128.90,126.01,125.51,121.65,81.33,34.32,26.77,22.32,13.76;hplc分析(ad-h柱,2%ipainhexanes,210nm,0.5ml/min),tr(major)=24.76min,tr(minor)=28.92min;
实施例2
在干燥的休良克瓶中依次加入0.0120gcu(pph3)3f·2meoh和0.0074gl4(r5=ph),抽真空并氮气置换3次,加入1ml甲苯,搅拌10min,加入2equiv的pmhs,搅拌20min,溶液呈橙色,降温至0℃;加入0.0699g邻丁酰基苯甲酸辛酯/1ml甲苯溶液。反应4h后经与实施例1相同的后处理,得到产物手性l-丙苯酞,呈无色液体,0.0330g,产率82%,ee77%。rf=0.46,ea:pe=1:5;1h-nmr(500mhz,cdcl3)δ7.90(d,j=7.7hz,1h),7.67(dd,j=7.6hz,1h),7.53(dd,j=9.9,5.4hz,1h),7.44(d,j=7.7hz,1h),5.49(dd,j=8.0,3.9hz,1h),2.02(dddd,j=14.2,10.3,6.3,3.8hz,1h),1.76(qd,j=9.6,8.9,4.6hz,1h),1.52(t,j=8.2hz,2h),0.99(t,j=7.5hz,3h);13c-nmr(126mhz,cdcl3)δ170.65,150.07,133.88,128.96,126.04,125.62,121.67,81.21,36.76,18.20,13.77;hplc分析(ad-h柱,2%ipainhexanes,210nm,0.5ml/min),tr(major)=33.08min,tr(minor)=42.16min;
实施例3
在干燥的休良克瓶中依次加入0.0114gcu(pph3)3f·2meoh和0.0063gl4(r5=ph),抽真空并氮气置换3次,加入1ml无水四氢呋喃,搅拌20min,加入3equiv的pmhs,搅拌20min,溶液呈橙色,降温至0℃;加入0.0497g邻异戊酰基苯甲酸异丙酯/0.5ml四氢呋喃溶液。反应5h后经与实施例1相同的后处理,得到产物手性l-异丁基苯酞,呈无色液体,0.0260g,产率68%,ee90%。其中rf=0.52,ea:pe=1:5;1h-nmr(500mhz,cdcl3)δ7.32(d,j=7.9hz,2h),7.13(d,j=7.9hz,2h),6.31(s,1h),5.87(s,1h),3.81(s,3h),2.48(d,j=7.1hz,2h),1.87(dt,j=13.5,6.7hz,1h),0.91(d,j=6.6hz,6h);13c-nmr(126mhz,cdcl3)δ167.44,141.82,141.07,133.92,128.83,127.92,126.07,79.99,52.11,45.08,30.12,22.35;hplc分析(ad-h柱,2%ipainhexanes,210nm,0.5ml/min),tr(major)=27.46min,tr(minor)=36.42min;
实施例4
在干燥的休良克瓶中依次加入0.0151gcu(pph3)3f·2meoh和0.0063gl1(r4=4-mec6h4),抽真空并氮气置换3次,加入1ml无水四氢呋喃,搅拌20min,加入3equiv的(me2sih)2o,搅拌20min,溶液呈红色,降温至0℃;加入0.0551g邻戊酰基苯甲酸丁酯/0.5ml四氢呋喃溶液。反应5h后经与实施例1相同的后处理,得到产物手性l-丁苯酞,呈黄色油状液体,0.0310g,产率78%,ee69%。所得表征数据与实施例1相同。
实施例5
在干燥的休良克瓶中依次加入0.0022gcu(oac)2和0.0077gl3(r2=ph),抽真空并氮气置换3次,加入1ml无水甲基叔丁基醚,搅拌20min,加入5equiv的pmhs,搅拌20min,溶液呈黄色,降温至-20℃;加入0.0662g邻戊酰基苯甲酸戊酯/0.5ml甲基叔丁基醚溶液。反应4.5h后经与实施例1相同的后处理,得到产物手性l-丁苯酞,呈黄色油状液体,0.0210g,产率46%,ee69%)。所得表征数据与实施例1相同。
实施例6
在干燥的休良克瓶中依次加入0.0110gcu(pph3)3f·2meoh和0.0063gl4(r5=ph),抽真空并氮气置换3次,加入1ml乙醚,搅拌10min,加入3equiv的ph2sih2,搅拌20min,溶液呈橙色,降温至-20℃;最后加入0.0603g邻戊酰基苯甲酸环戊酯/0.5ml乙醚溶液。反应6h后经与实施例1相同的后处理,得到产物l-丁苯酞,呈黄色油状液体,0.0260g,产率62%,ee86%。所得表征数据与实施例1相同。
实施例7
在干燥的休良克瓶中依次加入0.0015gcubr、0.0057gl4(r5=ph)和0.0470gkobut,抽真空并氮气置换3次,加入1ml甲苯,搅拌10min,加入3equiv的pmhs,搅拌20min,溶液呈橙色,降温至-20℃;加入0.047g邻戊酰基苯甲酸甲酯/0.5ml甲苯溶液。反应6h后经与实施例1相同的后处理,得到产物l-丁苯酞,呈黄色油状液体,0.0250g,产率61%,ee74%。所得表征数据与实施例1相同。
以上所述,仅为本发明创造较佳的具体实施方式,但本发明创造的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明创造披露的技术范围内,根据本发明创造的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明创造的保护范围之内。
参考文献:
[1]刘振涛;牛占旗;申东民;王荣端;冯小龙,左旋丁苯酞在制备预防和治疗脑缺血疾病药物中的应用.cn200410001748.x.
[2]everaere,k.;scheffler,j.l.;mortreux,a.;carpentier,j.f.tetrahedronlett.2001,42,1899.
[3]zhang,b.;xu,m.h.;lin,g.q.orglett.2009,11,4712.
[4]林国强;徐明华;张波,一种制备高光学纯度3-取代手性苯酞类化合物的方法.cn200810041638.4.
[5]xie,j.h.;liu,x.y.;xie,j.b.;wang,l.x.;zhou,q.l.angew.chem.,int.ed.2011,50,7329.
[6]周其林;谢建华;刘晓艳;谢剑波;王立新,手性螺环吡啶胺基膦配体化合物与合成方法及其应用.cn201010550836.0.
- 还没有人留言评论。精彩留言会获得点赞!