一种构建高检测性能捕获文库的方法和试剂盒与流程
1.本发明属于分子生物学领域,具体涉及构建杂交捕获文库的方法和试剂盒。
背景技术:
2.外显子捕获是利用探针捕获并富集外显子区域的dna序列的技术,在科研和临床检测领域被广泛应用。与全基因组测序相比,其费用更低、周期更短、覆盖度更好、更加经济、高效。传统的外显子捕获文库的构建一般包括以下步骤:将基因组dna片段化,进行末端修复和末端加a,然后连接接头和标签序列,并通过第一轮pcr扩增获得预文库;将预文库与杂交探针进行杂交,纯化后通过第二轮pcr扩增以获得最终的捕获文库(参见图1:传统外显子捕获流程示意图)。
3.通过pcr反应实现dna富集是二代测序技术(next
‑
generation sequencing,ngs)领域的常用技术。pcr应用于外显子捕获,在实现了捕获产物扩增,得到上机所需的文库量的同时,也带来了扩增错误与偏好性,不能对原始基因组序列进行完美呈现。例如对某些特点的dna优势扩增,造成dna无法实现均衡扩增,进而造成捕获数据对目标区域的覆盖不均一,最终造成检测错误或漏检。pcr
‑
free技术的应用可以完美地规避以上不足,并且省去扩增等步骤,精简了实验流程且降低建库成本。传统的pcr
‑
free技术更多的被应用于片段化的dna与接头连接后直接上机测序。由于pcr
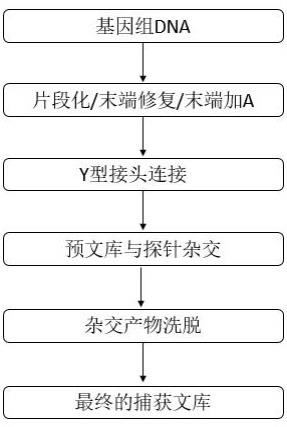
‑
free技术对dna模板投入要求较高,且探针捕获流程要考虑到捕获效率问题,故会造成dna模板的大量浪费。目前普遍涵盖探针捕获实验方法的流程中,起始的dna投入量需高达500ng
‑
3μg之大,且在探针捕获步骤前后都要涉及pcr扩增。在本发明前,发明人通过进行技术优化,实现了低dna起使量条件下,捕获前无pcr反应(参见图2:本实验室优化后外显子捕获流程示意图),但依然尚未解决杂交捕获后仍需pcr扩增的问题。
4.因此,仍需要建立一种简单经济、可以规避扩增错误与偏好性、实现检测性能的提升与有效的数据利用率,且能实现微量dna投入的pcr
‑
free杂交捕获流程。
技术实现要素:
5.鉴于对捕获数据性能检测的更高追求,对进一步节约成本和简化文库构建流程的要求,发明人在在先发明(专利申请公布文本cn110409001a中已描述,在此通过全文引用并入本文),即捕获前无需pcr的基础上,提出了一种捕获前后均无需pcr扩增的捕获文库构建方法(参见图3:本发明全建库流程pcr
‑
free文库构建方法流程示意图)。
6.本发明是基于发明人发现的以下事实:(1)使用无捕获前pcr的原发明实验流程,将捕获后pcr循环数减少,indel的检测性能显著提高。进而想到若能完全去掉pcr,能否达到最优的检测性能。
7.(2)理论计算的50
‑
100ng dna起使,且捕获前后无pcr时,杂交捕获的文库总量够上机。
8.(3)连接长y型接头的预文库,杂交捕获得到的单链dna已具备上机所需全部序列。
9.(4)低浓度的双链dna文库在碱变性并进行中和反应后,可以在
‑
20℃稳定保存十天以上,单链文库稳定性可满足上机要求。
10.(5)碱变性可以打开探针与杂交产物的dna双链、cot
‑
1与杂交产物的dna双链,也可以打开链霉亲和素与生物素的连接。碱变性处理后,大量带有生物素标记的探针、cot
‑
1和未成功连接的接头序列都会残留到文库中,在后续上机过程中会带来哪些影响是不确定的。发明人通过实验发现,这些残留探针并不会影响测序数据质量。
11.因此,在第一个方面,本发明提供一种构建适用于二代测序平台的捕获文库的方法,包括以下步骤:(1)获得片段化的dna;(2)将片段化的dna与y型接头连接,获得预文库;(3)将预文库与杂交探针进行杂交,获得杂交产物;(4)杂交产物洗脱,获得捕获文库;其中所述步骤(4)中,包括步骤(4a)对杂交产物进行碱变性。
12.在一些实施方案中,所述方法用于构建二代测序捕获文库。
13.在一些实施方案中,其中所述片段化的dna选自天然存在的短片段dna或通过人工打断基因组dna获得的短片段dna。
14.在另一些实施方案中,其中所述天然存在的短片段dna选自外周血游离dna、肿瘤游离dna或自然降解的基因组dna;所述人工打断基因组dna通过超声处理、机械打断或通过酶切实现。
15.在一些实施方案中,其中所述片段化的dna源自下组的样本:血液、血清、血浆、关节液、精液、尿液、汗液、唾液、粪便、脑脊液、腹水、胸水、胆汁或胰腺液。
16.优选地,其中所述片段化的dna长度为150
‑
400bp,更优选为180
‑
230bp。
17.在一些实施方案中,其中所述方法还包括在步骤(1)后,将获得的片段化的dna进行末端修复和/或末端加a的步骤。优选地,所述末端修复和末端加a在一个反应体系中进行。更优选地,dna片段化、末端修复和末端加a在一个反应体系中进行。
18.在一些实施方案中,其中步骤(2)所述y型接头是长y型接头,包括扩增引物、index标签序列、read 1/read 2测序引物和index read 测序引物。
19.在一些实施方案中,所述步骤(3)在液相系统中进行。
20.在一些实施方案中,其中步骤(4)中,在步骤(4a)后还包括:步骤(4b):孵育;步骤(4c):去除磁珠;和步骤(4d):中和。
21.在优选的实施方案中,其中步骤(4a)中碱变性所用的碱试剂选自下组的一种或多种:naoh、koh、氢氧化锂、氢氧化钙、氢氧化镁、氨水、碳酸锂、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、碳酸氢铵、甲醇钠、甲醇钾、乙醇钠、乙醇钾、叔丁醇钠或叔丁醇钾。进一步优选地,所述碱试剂为naoh或koh,优选其水溶液。
22.在优选的实施方案中,所述碱试剂浓度为0.05m
‑
1m,优选0.1
‑
0.5m,更优选0.2m。
23.优选地,其中步骤(4a)中,在杂交产物中加入碱试剂,进行碱变性,其中,加入碱试剂后的体系ph值为11
‑
14,进一步优选为12
‑
14,最优选为12
‑
13。
24.进一步优选地,所述中和步骤采用中和剂中和;优选地,所述中和剂选自tris
‑
hcl、乙酸、柠檬酸盐缓冲液、磷酸盐缓冲液或乙酸盐缓冲液中的一种或多种。优选地,所述中和剂选自tris
‑
hcl。
25.在另一方面,本发明还涉及一种用于构建捕获文库的试剂盒,其包括:(1)用于连接接头的试剂,包括y型接头;(2)用于杂交的试剂;和(3)用于杂交产物洗脱的试剂。
26.在一些实施方案中,其中所述y型接头是长y型接头;长y型接头包括扩增引物序列、index标签序列、read 1/read 2测序引物序列和index read测序引物序列。
27.在一些实施方案中,还包括用于进行末端修复和/或末端加a的试剂。
28.在一些实施方案中,其中所述用于杂交的试剂包括杂交缓冲液、cot
‑
1 dna和杂交探针;优选地,所述用于杂交的试剂不包括封闭序列。
29.在一些实施方案中,其中所述封闭序列包括设计为与接头和/或标签序列反向互补的序列。
30.在一些实施方案中,其中所述用于杂交产物洗脱的试剂,包括变性剂和中和剂。优选地,所述变性剂为碱变性试剂。
31.在优选的实施方案中,所述碱变性试剂选自下组的一种或多种:naoh、koh、氢氧化锂、氢氧化钙、氢氧化镁、氨水、碳酸锂、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、碳酸氢铵、甲醇钠、甲醇钾、乙醇钠、乙醇钾、叔丁醇钠或叔丁醇钾;优选naoh或koh,更优选其水溶液。所述中和剂选自下组的一种或多种:tris
‑
hcl、乙酸、柠檬酸盐缓冲液、磷酸盐缓冲液或乙酸盐缓冲液;优选tris
‑
hcl。
32.在另一方面,本发明还包括根据上文所述的方法构建的捕获文库,或根据上文所述的试剂盒构建的捕获文库。在优选的实施方案中,所述捕获文库用于二代测序平台。
33.本发明的优异技术效果在于:(1)起始dna的含量要求较低,甚至可以低至25ng,这大大提高了罕见样本的利用率,扩展了本发明的应用范围,例如本发明的方法和试剂盒可以应用于干血斑、口腔拭子等因dna提取量少而无法满足普通外显子捕获流程的样本类型。
34.(2)建库流程简单,本发明的方法彻底实现了捕获前后全流程pcr
‑
free,因此简化并缩短了实验流程。
35.(3)由于本发明不涉及pcr扩增因此节约了与扩增相关的建库成本, pcr
‑
free文库自身的低冗余优势增加了有效数据利用率进而避免了数据量的浪费,实现测序成本的降低。
36.(4)pcr
‑
free文库可以完美地规避扩增错误与偏好性,展现dna序列真实面貌,使得变异检测性能得到有效提高。
37.下面将参考附图并结合实例来详细说明本发明。需要说明的是,本领域的技术人员应该理解本发明的附图及其实施例仅仅是为了例举的目的,并不能对本发明构成任何限制。在不矛盾的情况下,本技术中的实施例及实施例中的特征可以相互组合。
附图说明
38.图1:传统外显子捕获流程示意图。
39.图2:经发明人优化后外显子捕获流程示意图(专利申请cn110409001a中已描述)。
40.图3:本发明全建库流程pcr
‑
free文库构建方法流程示意图。
41.图4:y型接头结构示意图。
42.图5:杂交捕获体系微观示意图。
43.图6:文库浓度测量结果。
具体实施方式
44.构建捕获文库的方法在第一个方面,本发明提供一种构建捕获文库的方法。
45.在传统的捕获文库构建的最后,往往会对杂交产物进行pcr扩增,一方面是为了通过扩增的方式把探针捕获回来的片段复制到液相体系中(链酶亲和素磁珠上的亲和素与探针上的生物素共价结合较为强力,同时探针与目的片段也通过碱基互补配对形成双链结构,参见图5:杂交捕获体系微观示意图),另一方面是为了提高终文库产量,进而满足上机需求。杂交后pcr的引入不仅增加了实验成本与操作复杂程度,而且会带来pcr扩增的种种弊端(扩增错误与偏好性,不利于变异检测,冗余较高增加测序成本)。
46.为克服以上弊端,实现真正的全建库流程pcr
‑
free,本发明人尝试研发一种可以直接将目的片段(探针捕获的文库)从链酶亲和素磁珠上脱离至液相体系中的方法(杂交反应后)。
47.首先考虑到的是通过使用游离生物素的方法与探针上的生物素进行竞争进而得到一部分脱落的探针与目的片段形成的双链,但经过测试发现,亲和素与探针上生物素的结合过于紧密,实验所得文库浓度极小。
48.其次采用了具有链置换活性的deep vent dna聚合酶,试图将目的片段从探针上置换下来,并且可以得到稳定性更高的双链文库,通过测试发现,由于在链置换步骤需添加酶与单端引物,故需在置换反应后引入xp磁珠纯化步骤,使得终文库的总量被浪费,所得到的文库无法满足多次上机需求,并且在总体的实验流程上并不能带来简化以及成本的节约。
49.最后考虑通过变性的方法(双链基对的氢键断裂)解开探针与目的片段形成的双链。目前,常见的变性方法有高温与极端的ph。经梯度测试,使用高温手段使双链变性的流程较难操作且不好控制,实验所获得文库的浓度与稳定性不佳。
50.排除综上实验条件,本发明最终选用了碱变性的实验流程,通过加入碱试剂,为双链解开创造了强碱性条件,通过孵育及涡旋,促进双链解开,在实验流程的最后,中和以降低体系的碱性,并且利于单链文库的保存。最终所得文库满足多次上机与长期保存需求。
51.由此,本发明提供一种构建高检测性能捕获文库的方法,包括以下步骤:(1)获得片段化的dna;(2)将片段化的dna与y型接头连接,获得预文库;(3)预文库与探针进行杂交,获得杂交产物;(4)杂交产物洗脱,获得捕获文库;
其中步骤(4)中,包括步骤(4a):对杂交产物进行碱变性。
52.在一些实施方案中,本发明的方法可用于构建二代测序捕获文库。
53.在一些实施方案中,所述片段化的dna是指天然存在的短片段dna或通过人工打断基因组dna获得的短片段dna。
54.在一些实施方案中,片段化的dna可以源自血液、血清、血浆、关节液、精液、尿液、汗液、唾液、粪便、脑脊液、腹水、胸水、胆汁或胰腺液等样本。
55.在优选的实施方案中,天然短片段dna是外周血游离dna、肿瘤游离dna或自然降解的基因组dna。
56.在另一些实施方案中,基因组dna可以有各种来源,例如来自口腔拭子、羊水、干血片、组织、外周血等。本领域技术人员知晓打断基因组dna的方法,例如通过超声处理、机械打断或通过酶切等。由于超声处理和机械打断相对而言会损失较多的dna,因此在起始dna含量较少(例如,低至50ng)的情况下,优选用酶切的方法使dna片段化。
57.在一些实施方案中,所述片段化的dna长度为150
‑
400bp,优选180
‑
230bp。
58.在一些实施方案中,本发明的方法在与y型接头连接(即,步骤(2))之前,还包括步骤(1’):将片段化的dna进行末端修复和/或末端加a。在该实施方案中,可以用本领域技术人员已知的任何适用于末端修复的酶对dna进行末端修复,例如t4 dna聚合酶、klenow酶或其混合物。在该实施方案中,可以用本领域技术人员已知的任何适用于末端加a的酶对dna进行末端加a。这种酶的实例包括但不限于taq酶、klenow ex
‑
酶或其混合物。在该实施方案中,末端修复和末端加a可以在两个反应体系中进行,即,在末端修复后,经过纯化再进行末端加a。可替换地且为优选地,末端修复和末端加a在一个反应体系中进行,即,末端修复和末端加a同时完成,之后再对核酸进行纯化。或者,更加优选地,将dna片段化、末端修复和末端加a三者在一个反应体系中进行,之后再连接接头。上述方法不仅简化了操作步骤、节约成本,同时也降低了样本间的污染。
59.在一些实施方案中,末端补平和末端加a所用的温育时间和温度可以根据具体需要由本领域技术人员根据常规技术确定。
60.在一些实施方案中,步骤(2)可以用本领域技术人员已知的任何适用于连接接头的酶进行。这种酶的实例包括但不限于:t4 dna 连接酶、t7 dna连接酶或其混合物。进行连接反应的条件是本领域技术人员熟知的。
61.在本发明的上下文中,“y型接头”是指不完全互补的两条链形成的接头,所述接头的一端由于两条链的碱基互补形成双链,而另一端由于两条链的碱基之间不完全互补,没有形成双链。本发明适用于普通y型接头(true seq接头),如图4:y型接头结构示意图。
62.普通y型接头主要包括扩增引物序列(p5/p7)、index标签序列、read 1/read 2测序引物序列和index read 测序引物序列,其中read 1/read 2测序引物序列和index read 测序引物的序列不完全互补形成一部分双链。
63.举例而言,可用于本发明的y型接头包含的两条链的序列如下:
其中,下划线示出了两条链中碱基互补的部分。
64.对寡核苷酸进行磷酸化修饰的方法是本领域技术人员熟知的。例如,可以通过多核苷酸激酶使寡核苷酸5’端磷酸化,或在合成引物时直接在5’端加上磷酸基团。
65.在一些实施方案中,本发明方法的步骤(3)在液相杂交系统中进行。
66.通过本实验室以前的技术优化,在无需pcr预扩增制备预文库并且采用y型接头的情况下,杂交体系中不需要加入任何封闭序列也能实现较好的捕获效率,从而实现了省略杂交前预文库扩增建库流程,优化了分析性能且减小了实验成本。
67.因此,在一些实施方案中,用于杂交的体系包括杂交缓冲液、cot
‑
1 dna和杂交探针,但不包括封闭序列。本领域技术人员根据实际需要可以调节杂交的条件,例如杂交温度、杂交时间等。设计和制备杂交探针的一般原理也是本领域技术人员熟知的。
68.在一些实施方案中,本发明方法的步骤(4)还包括杂交产物进行洗脱进而获得终文库的步骤,具体地,所述步骤(4)包括:步骤(4a):对杂交产物进行碱变性;步骤(4b):孵育;步骤(4c):去除磁珠;和步骤(4d):中和。
69.在优选的实施方案中,所述碱变性的过程中,能够使用的碱变性试剂及其浓度是本领域技术人员熟知并选择的。优选地,例如,所述碱变性使用的试剂为无机碱或有机碱。进一步地,所述无机碱选自naoh、koh、氢氧化锂、氢氧化钙、氢氧化镁、氨水、碳酸锂、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、碳酸氢铵中的一种或多种;所述有机碱选自甲醇钠、甲醇钾、乙醇钠、乙醇钾、叔丁醇钠或叔丁醇钾中的一种或多种。
70.在进一步优选的实施方案中,所述碱变性试剂选自无机碱,优选选自naoh或koh;更优选地,为上述试剂的水溶液。进一步地,所述碱变性试剂浓度范围为0.05m
‑
1m,优选0.1
‑
0.5m,更优选0.2m。
71.在优选的实施方案中,所述碱变性步骤中,加入碱试剂调整体系的ph值,以使得杂交产物变性。所述体系ph值调整范围是本领域技术人员熟知的。例如,优选地,可调整体系ph值为11
‑
14,更优选12
‑
14,最优选12
‑
13。
72.在本发明的上下文中,术语“ph值”包括该术语的常规用法,如氢离子浓度倒数的对数。在本文中,“ph值”还包括观察ph值,即通过使介质与已知类型的,用已知方法适当校准的常规酸碱计接触而测量的ph值。通常,本发明测量方法中使用的介质会含一些水。如本发明所用,所述溶液的“ph值”通常是指,例如,室温下(25℃)下的ph值。
73.在一些实施方案中,加入碱变性试剂后还包括以下过程:将反应体系孵育;孵育结束后,进行吸附以去除磁珠,最终加入中和剂以降低体系碱性。优选地,例如,所述孵育在室温下进行。本领域技术人员能够选择任何适合使用的、以中和上述碱变性试剂的试剂,并确
定其浓度范围,例如tris
‑
hcl、乙酸、柠檬酸盐缓冲液、磷酸盐缓冲液或乙酸盐缓冲液等。
74.在优选的实施方案中,所述中和剂选自tris
‑
hcl;所述tris
‑
hcl浓度范围优选为100nm
‑
10mm,更优选100
‑
500nm,最优选400nm。
75.试剂盒在第二个方面,本发明还提供一种用于构建捕获文库的试剂盒,其包括:(1)用于连接接头的试剂,包括y型接头;(2)用于杂交的试剂;和(3)用于杂交产物洗脱的试剂。
76.在一些实施方案中,其中所述y型接头是长y型接头;长y型接头包括扩增引物序列、index标签序列、read 1/read 2测序引物序列和index read 测序引物序列。
77.在一些实施方案中,还包括用于进行末端修复和/或末端加a的试剂。
78.在一些实施方案中,其中所述用于杂交的试剂包括杂交缓冲液、cot
‑
1 dna和杂交探针;优选地,所述用于杂交的试剂不包括封闭序列。其中,上述封闭序列包括设计为与接头和/或标签序列反向互补的序列。
79.在一些实施方案中,其中所述用于杂交产物洗脱的试剂,包括变性剂和中和剂。优选地,所述变性剂为碱变性试剂。
80.在优选的实施方案中,所述碱变性试剂选自下组的一种或多种:naoh、koh、氢氧化锂、氢氧化钙、氢氧化镁、氨水、碳酸锂、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、碳酸氢铵、甲醇钠、甲醇钾、乙醇钠、乙醇钾、叔丁醇钠或叔丁醇钾。所述中和剂选自下组的一种或多种:tris
‑
hcl、乙酸、柠檬酸盐缓冲液、磷酸盐缓冲液或乙酸盐缓冲液。
81.在进一步优选的实施方案中,所述碱变性试剂选自naoh或koh,更优选其水溶液,所述中和剂选自tris
‑
hcl。具体地,所述naoh或koh水溶液浓度为0.1
‑
0.5m,更优选0.2m;所述tris
‑
hcl浓度优选100nm
‑
500nm,更优选400nm。
82.在一些实施方案中,根据本发明方法制备的捕获文库,或本发明试剂盒制备的捕获文库适用于多种二代测序平台,包括但不限于,例如roche/454 flx、illumina/hiseq、miseq、nextseq和life technologies/solid system、pgm、proton等。
实施例
83.实施例1:使用不同的模板投入量构建文库,上机测序,数据分析,比较不同投入量的测序数据质控以及性能分析结果。
84.该实施例以标准细胞系gm24385基因组dna为模板,根据本发明的方法构建起始dna投入量分别为25、50、75、100、150、200ng的捕获文库。将文库在 nextseq cn500测序平台进行测序(150bp双端测序)。利用生物信息学对测序结果进行分析,分析不同模板投入量的文库质量。
85.步骤1:获得片段化的dna、末端修复和末端加a根据制造商的说明,用5
×
wgs fragmentation mix试剂盒(enzymatics,货号y9410l)和配套的10
×
fragmentation buffer试剂盒(enzymatics,b0330)制备如表1所示的反应体系以一步完成片段化、末端修复和末端加a。
86.将该反应体系按下表2程序进行反应。
87.步骤2:连接接头本发明所构建的文库接头为普通y型接头(true seq接头),使用wgs ligase试剂盒(enzymatics,货号l6030
‑
600000),用步骤1的反应产物制备如表3所示的连接体系,并将该连接体系在20℃温育15分钟,然后保持在4℃。
88.连接反应结束后,用beckman agencourt ampure xp试剂盒(beckman,货号a63882)对连接产物进行纯化。
89.步骤3:捕获杂交使用xgen lockdown reagents试剂盒(idt,货号1072281),和streptavidin dynbeads m270磁珠(thermo fisher scientific,货号35302),在步骤2的纯化产物中加入14.5μl杂交试剂(9.5μl xgen 2
×
杂交缓冲液、3μl xgen杂交缓冲液增强剂和2μl的cot
‑
1dna),充分混匀后于室温孵育10分钟。孵育结束后,取12.75μl上清液到新的低吸附的 0.2ml离心管中,然后加入4.25μl杂交探针。孵育结束后,充分混匀后瞬离,并按照如下表4所述程序进行捕获杂交。
90.杂交结束后,根据制造商的说明,用xgen lockdown reagents试剂盒(idt,货号 1072281)清洗并纯化杂交产物(即,与目标序列结合的磁珠)。
91.步骤4:目的片段洗脱在步骤3的纯化杂交产物(带beads)中加入3.5μl naoh溶液(0.2m)和15μl无酶水,充分混匀后于室温孵育10分钟,期间交替涡旋混匀。孵育结束后,置于磁力架上吸附5分钟后取18μl上清液到已加入4
µ
l tris
‑
hcl(400nm)的低吸附的 0.2ml离心管中,涡旋混匀,即得到最终的捕获文库。
92.将捕获文库进行qpcr定量,然后按照测序仪标准操作流程,用 nextseq cn500测序平台对文库进行测序(150bp双端测序),每个样本测得2.5g数据。测序结果包括基本质控与标准细胞系snv&indel性能分析如表5所示。
93.由上表可见,相同数据量下,在25ng
‑
200ng的dna模板投入范围内,根据本发明方法构建的捕获文库除qpcr浓度以外,在基本质控与性能分析没有显著差异。这表明,根据本发明的方法可以使用起始dna含量低至25ng的样本,所制备的捕获文库完全满足上机测序和后续数据分析的要求。表中的“均匀性”即iqr(四分位距,interquartile range,又称四分差),在本文中是指,密度深度分布25%
‑
75%之间的序列覆盖率差异。该值是结果变异性的量度,反映了整个数据集覆盖范围的不均匀性。高iqr表示基因组覆盖率的高度变化性,而低iqr则反映了更均匀的序列覆盖率。
94.对照实施例1:在实施例1流程中的步骤3结束后使用pcr扩增步骤(12个循环,具体
见表7)代替实施例1中的步骤(4),并使用不同的模板投入量构建文库,上机测序,数据分析,比较不同投入量(分别为50ng、75ng和100ng)的测序数据质量以及性能分析结果并用以上结果与实施例1中对应相同模板投入量的分析结果进行对比。
95.该对照实施例以标准细胞系gm24385基因组dna为模板,在实施例1流程中的步骤3结束后,对纯化杂交产物(带beads)进行pcr扩增制备终文库,并构建起始dna投入量分别为50ng、75ng、100ng的捕获文库。将文库在 nextseq cn500测序平台进行测序(150bp双端测序)。利用生物信息学对测序结果进行分析,分析不同模板投入量的文库质量。
96.具体地,根据制造商的说明,用2
×
kapa hifi hot start ready mix试剂盒(kapa,货号kk2602),制备如表6所示的扩增体系,预扩增引物的序列如下:k2602),制备如表6所示的扩增体系,预扩增引物的序列如下:并按照以下表7的程序进行pcr。
97.pcr程序完成之后,用beckman agencourt ampure xp试剂盒(beckman,货号a63882)对进行纯化,即获得最终的捕获文库。
98.将对照实施例1制备的捕获文库进行qpcr定量,然后按照测序仪标准操作流程,用nextseq cn500测序平台对文库进行测序(150bp双端测序),每个样本测得2.5g数据。测序结果如表8所示。
99.由上表可见,相同数据量下,本发明所构建的文库(表8中pcr
‑
free)与引入pcr扩增文库在不同模板量投入下的数据波动并不明显。pcr
‑
free文库在重复率上明显低于引入pcr的流程(在模板量为50/75/100ng下的重复率均值为1.73%,引入pcr的重复率均值为4.35%),致使上机数据可以得到有效利用。且本发明所构建的文库在变异检测性能方面有提升,尤其在对于indel的检出性能上有明显改善(在模板量为50、75、100ng下的灵敏度与精确度均值分别为93.37%与97.91%,而引入pcr流程所建文库在模板量为50、75、100ng下的灵敏度与精确度均值分别为81.57%与90.63%)。
100.对照实施例2:在实施例1流程中的步骤3结束后使用游离生物素(又名d
‑
生物素)饱和溶液进行目的片段洗脱,代替实施例1中的步骤(4),使用100ng的模板投入量构建文库后进行qpcr浓度定量,并用以上结果与实施例1中对应相同模板投入量的文库浓度结果进行对比。
101.该对照实施例以标准细胞系gm24385基因组dna为模板,在实施例1流程中的步骤3结束后,向纯化杂交产物(带beads)中加入游离亲和素饱和溶液并加热孵育,最终达到终文库洗脱的目的。
102.具体地将游离生物素(thermofisher,货号b1595)溶于无酶水制成饱和溶液(约为0.2mg/ml),在步骤3的纯化杂交产物(带beads)中加入22.5μl游离生物素饱和溶液,充分混匀后于37℃孵育30分钟,孵育结束后,置于磁力架上吸附5分钟后取22μl上清液到0.2ml低吸附离心管中,涡旋混匀,即得到最终的捕获文库。
103.将对照实施例2制备的捕获文库进行qpcr定量,文库浓度如表9所示。
104.由上表可见,在相同模板dna投入量时,使用游离生物素置换法所得文库浓度过低且不满足上机需求。
105.对照实施例3:在实施例1流程中的步骤3结束后使用deep vent dna聚合酶进行目的片段的链置换反应,代替实施例1中的步骤(4),使用100ng的模板投入量构建文库后进行
qpcr浓度定量,并用以上结果与实施例1中对应相同模板投入量的文库浓度结果进行对比。
106.该对照实施例以标准细胞系gm24385基因组dna为模板,在实施例1流程中的步骤3结束后,对纯化杂交产物(带beads)进行链置换反应制备终文库。
107.具体地,根据制造商的说明,用deep vent
®ꢀ
dna polymerase(neb,货号m0258s),制备如表10所示的链置换体系,链置换引物的序列如下:制备如表10所示的链置换体系,链置换引物的序列如下:并按照以下表11的程序进行反应。
108.pcr程序完成之后,用beckman agencourt ampure xp试剂盒(beckman,货号a63882)对进行纯化,即获得最终的捕获文库。
109.将对照实施例3制备的捕获文库进行qpcr定量,文库浓度如表12所示。
110.由上表可见,在相同模板dna投入量时,使用链置换法所得文库浓度约为碱变性法文库浓度的31.66%,文库浓度相差较大,且不满足多次上机需求。
111.对照实施例4:在实施例1流程中的步骤3结束后使用高温热变性手法,在不同孵育时间内(2分钟、5分钟)将目的片段洗脱,代替实施例1中的步骤(4),使用100ng的模板投入量构建文库后进行qpcr浓度定量,并用以上结果与实施例1中对应相同模板投入量的文库浓度结果进行对比。
112.该对照实施例以标准细胞系gm24385基因组dna为模板,在实施例1流程中的步骤3结束后,向纯化杂交产物(带beads)中加入22.5μl 无酶水,在高温条件下进行dna双链解旋变性后快速去除磁珠,最终达到终文库洗脱的目的。
113.具体地,在步骤3的纯化杂交产物(带beads)中加入22.5μl 无酶水,充分混匀后于98℃分别孵育2分钟与5分钟,孵育结束后,置于磁力架上吸附1分钟后取22μl上清液到0.2ml低吸附离心管中,涡旋混匀,即得到最终的捕获文库。
114.将对照实施例4制备的捕获文库进行qpcr定量,文库浓度如表13所示。
115.由上表可见,在相同模板dna投入量时,使用热变性法所得文库浓度过低且不满足上机需求。
116.实施例2:采用实施例1中100ng起始dna投入量所构建的文库与对照实施例1中引入pcr扩增的相同模板dna投入量所构建的文库上机测序,分别截取2、2.5、3、3.5、4、4.5、5g的数据量,进行数据分析,比较两种不同建库流程在不同测序深度下的测序数据质控及性能分析结果。分析结果分别见表14(a)、14(b)。
117.由以上两表可知,随着测序数据量的增加,样本的测序深度会随之产生较大幅度改变,测序深度的改变可以在一定程度上优化snp以及indel的检出性能。对比以上两表数据可以进一步看出本发明对于indel的检出性能占据较大优势,且本发明流程在2.5g数据量下即可得到较为稳定的检出性能。
118.实施例3:该实施例以标准细胞系gm24385基因组dna为模板,构建起始dna投入量为100ng的捕获文库。本实施例的文库构建方法与实施例1相同,区别仅在于步骤4目的片段洗脱时室温孵育时间分别为5分钟、10分钟与15分钟。将文库在 nextseq cn500测序平台进行测序(150bp双端测序)。利用生物信息学对测序结果进行分析,分析不同孵育时间的文库质量。测序结果如表15所示。
119.由上表可见,文库浓度随孵育时间的延长而增加,3种孵育条件所得文库均满足3次上机需求。在相同数据量下,3种孵育条件所得文库的基本质控以及对于snp/indel的检出性能相差不大,孵育时间条件为10min的文库比对率及关键基因20
×
覆盖度较高,且针对于实验操作时间角度上更为合理,故本发明推荐采用室温孵育时间为10分钟的流程。
120.实施例4:本实施例的文库构建方法与实施例1相同(构建起始dna投入量为100ng的捕获文库),区别在于将dna模板由原来的标准细胞系改为了临床阳性样本。分别采用口腔拭子dna、羊水dna、干血片dna、组织dna、外周血dna制备捕获文库。将捕获文库进行qpcr定量,然后按照测序仪标准操作流程,用 nextseq cn500测序平台进行测序(150bp双端测序),每个样本测得2.5g数据。测序结果如表16所示。
121.上表所采用的5例临床阳性样本,文库基本质控均达标,且正确检出。由此可见,本发明的测序文库的构建方法可适用于多种样本类型,尤其是dna含量较少的样本。
122.实施例5:文库qpcr浓度稳定性探究根据实施例1所述的方法(构建起始dna投入量为100ng的捕获文库),以六例标准细胞系基因组dna为模板(gm24385、 gm24694、gm12878、gm24631、gm24143和gm24695),构建文库,并相隔0天、20天、40天进行文库qpcr浓度测定(文库置于
‑
20℃保存)。探究存放时间对文库qpcr浓度的影响。浓度测量结果如表17、图6所示。
123.由表17、图6可见,使用本发明流程所构建的文库,在
‑
20℃保存40天内,文库浓度无明显变化,稳定性较好。
124.实施例6:文库重上机检出稳定性探究使用实施例5所构建的六例标准细胞系基因组dna文库,按照测序仪标准操作流程,用nextseq cn500测序平台对文库进行测序(150bp双端测序),每个样本测得2.5g数据。并在文库
‑
20℃存放25天后重新上机(相同数据量),以探究存放时间对文库重上机检出稳定性的影响。测序结果如表18所示。
125.由上表可见,使用本发明流程所构建的文库,在
‑
20℃保存25天内,上机测序质量无明显变化。结合实施例5可知,由本发明流程构建的文库较为稳定,可存放较长时间。
126.需要说明的是,以上仅为本发明的优选实施例,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。本领域技术人员理解的是,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1