一种具有抗癌活性的嘧啶并哌啶衍生物的新合成方法
1.本发明属于化学、医学等领域,尤其涉及小分子抗癌药物的合成方法。
背景技术:
2.癌症日益严重的影响人们的健康,日益升高的癌症发病率已成为人类寿命延长的重要隐患。在抗癌药物的研发中心,小分子抗癌药物一直在传统抗癌药物中占据着稳定领先的地位,虽然不同种类的抗癌药物和手段在不断发展,但小分子药物仍然在靶点、制剂、成本等多个方向上保持着自己的优势。
3.ras为一组紧密相关的单体球状蛋白质(21kda分子量),其具有188-189个氨基酸且与鸟昔二磷酸gdp或鸟昔三磷酸gtp结合。ras亚家族成员包括hras、kras和nras。ras起分子开关作用,当ras含有所结合的gdp时,其处于休眠或关闭位置且“无活性”。当细胞暴露于某些促生长性刺激物时,ras经诱导而使其所结合的gdp转化为gtp,当与gtp结合时,ras“接通”且能够与其他下游标靶蛋白质相互作用并活化这些蛋白质。ras蛋白自身使gtp水解而恢复为gdp(从而使其自身转换为关闭状态)的固有能力极低。需要外源性蛋白gtp酶活化蛋白(gap)将其恢复为关闭状态,gap与ras相互作用极大地加速了gtp转化为gdp。
4.ras中的任何突变将影响ras与gap的相互作用,以及gtp转化为gdp的能力,这种突变将导致蛋白质活化时间的延长,从而延长细胞信号传导,继而导致细胞继续生长和分裂。由于这种信号传导引起细胞生长和分裂,因此过度活化的ras信号传导最终可导致癌症。
5.在肺癌中,约32%的肺癌中确认有ras基因的突变,ras(hras、nras或kras)基因的三种主要亚型中的任意一个突变可导致人肿瘤的发生。有报道指出,在ras基因中突变频率最高的为kras基因,在25-30%肿瘤中检测到kras突变。与之相比较,nras及hras家族成员中发生致癌性突变的比率低得多(分别为8%及3%)。最常见的kras突变发现于p环中的残基g12及g13上以及残基q61上。g12d突变为kras基因的频繁突变(甘氨酸-12突变为半胱氨酸)。在约13%的癌症,约43%的肺癌及几乎100%的myh相关息肉病(家族性结肠癌症候群)中已发现此突变。
6.因此开发选择性抑制kras突变的抑制剂是一个较好的方向,为了提高对kras突变抑制活性的同时降低对野生型kras的抑制活性,开发活性更高,选择性更好,毒性更低的新型ras突变体选择性抑制剂具有重要的意义。
7.本发明的有益效果
8.本发明的整个过程工艺流程简单、反应收率较高、生产成本较低、环境友好。
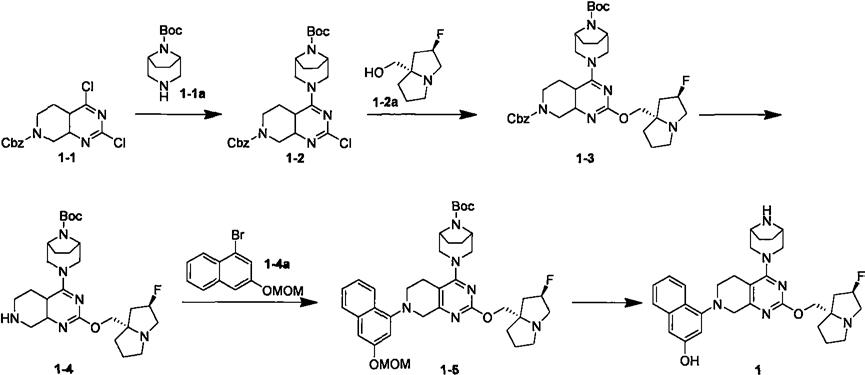
9.具体实施实例
10.将化合物1-1(7.5g,22.12mmol)溶于80ml乙腈中,分别加入n,n-二异丙基乙胺(5.72g,44.25mmol)和1-1a(5.6g,26.57mmol),回流17小时。反应液减压浓缩,残留物中加入30ml饱和碳酸氢钠溶液,乙酸乙酯萃取(50ml
×
3),合并有机相,无水硫酸钠干燥,减压浓缩,用硅胶柱层析色谱法以洗脱剂体系b纯化得到1-2(10.3g)。ms m/z(esi):515.2[m+1]
[0011]
化合物1-3的合成:
[0012]
将化合物1-2a(4.4g,27.62mmol)溶于100ml四氢呋喃中,冷却到0℃,滴加入双(三甲基硅基)氨基钠(2m,14ml),搅拌15分钟,再加入化合物1-2(9g,17.52mmol),室温反应1小时。加入20ml饱和氯化钠溶液,分液,水相用乙酸乙酯萃取(100ml
×
3),合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱层析色谱法以洗脱剂体系a纯化得到1-3(3.2g)。ms m/z(esi):638.4[m+1]。
[0013]
化合物1-4的合成:
[0014]
在氢气氛下,将化合物1-3(110mg,172.98umol)加入10ml四氢呋喃和甲醇的混合溶剂中(v/v=1∶1),加入钯碳(37mg,17.38umol,5%纯度),搅拌2小时。过滤,滤液减压浓缩,得到1-4(96mg)。ms m/z(esi):504.3[m+1]。
[0015]
化合物1-5的合成:
[0016]
在氩气氛下,将化合物1-4(142mg,282.14umol)、1-4a(150mg,563.79umol)、甲磺酸(2-二环己基膦基-2
′
,6
′‑
二异丙氧基-1,1
′‑
联苯基)(2-氨基-1,1
′‑
联苯-2-基)钯(ii)(71mg,84.79umol)、碳酸铯(276mg,847.1umol)加入到20ml甲苯中,在95℃搅拌17小时。冷却到室温,反应液减压浓缩,用薄层色谱法以展开剂体系dcm∶meoh=20∶1纯化得到1-5(120mg)。ms m/z(esi):688.4[m+1]。
[0017]
化合物1的合成:
[0018]
将化合物1-5(320mg,506umol)溶于2ml二氯甲烷中,加入0.5ml三氟乙酸,室温搅拌2小时。反应液减压浓缩,加饱和碳酸氢钠溶液调ph大于8,二氯甲烷萃取(20ml
×
3),合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩,用薄层色谱法以展开剂体系dcm∶meoh=20∶1纯化得到化合物1(120mg)。
[0019]
ms m/z(esi):544.3[m+1]。1h nmr(500mhz,chloroform-d)δ8.11(dt,j=8.0,0.9hz,1h),7.92(s,1h),7.71(dt,j=7.5,1.7hz,1h),7.57-7.46(m,2h),6.99(t,j=2.1hz,1h),6.57(d,j=2.2hz,1h),4.81(dtt,j=46.3,4.9,3.1hz,1h),4.66(d,j=11.9hz,2h),4.51-4.27(m,2h),3.80(ddd,j=7.1,4.7,1.2hz,2h),3.77-3.60(m,4h),3.30-3.09(m,6h),2.94-2.76(m,2h),2.57(t,j=5.7hz,1h),2.17-2.04(m,1h),2.02-1.64(m,9h).
技术特征:
1.小分子嘧啶并哌啶衍生物的合成路线化合物1-2的合成:将化合物1-1(7.5g,22.12mmol)溶于80ml乙腈中,分别加入n,n-二异丙基乙胺(5.72g,44.25mmol)和1-1a(5.6g,26.57mmol),回流17小时。反应液减压浓缩,残留物中加入30ml饱和碳酸氢钠溶液,乙酸乙酯萃取(50ml
×
3),合并有机相,无水硫酸钠干燥,减压浓缩,用硅胶柱层析色谱法以洗脱剂体系b纯化得到1-2(10.3g)。ms m/z(esi):515.2[m+1]化合物1-3的合成:将化合物1-2a(4.4g,27.62mmol)溶于100ml四氢呋喃中,冷却到0℃,滴加入双(三甲基硅基)氨基钠(2m,14ml),搅拌15分钟,再加入化合物1-2(9g,17.52mmol),室温反应1小时。加入20ml饱和氯化钠溶液,分液,水相用乙酸乙酯萃取(100ml
×
3),合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱层析色谱法以洗脱剂体系a纯化得到1-3(3.2g)。ms m/z(esi):638.4[m+1]。化合物1-4的合成:在氢气氛下,将化合物1-3(110mg,172.98umol)加入10ml四氢呋喃和甲醇的混合溶剂中(v/v=1∶1),加入钯碳(37mg,17.38umol,5%纯度),搅拌2小时。过滤,滤液减压浓缩,得到1-4(96mg)。ms m/z(esi):504.3[m+1]。化合物1-5的合成:在氩气氛下,将化合物1-4(142mg,282.14umol)、1-4a(150mg,563.79umol)、甲磺酸(2-二环己基膦基-2
′
,6
′‑
二异丙氧基-1,1
′‑
联苯基)(2-氨基-1,1
′‑
联苯-2-基)钯(ii)(71mg,84.79umol)、碳酸铯(276mg,847.1umol)加入到20ml甲苯中,在95℃搅拌17小时。冷却到室温,反应液减压浓缩,用薄层色谱法以展开剂体系dcm∶meoh=20∶1纯化得到1-5(120mg)。ms m/z(esi):688.4[m+1]。化合物1的合成:将化合物1-5(320mg,506umol)溶于2ml二氯甲烷中,加入0.sml三氟乙酸,室温搅拌2小时。反应液减压浓缩,加饱和碳酸氢钠溶液调ph大于8,二氯甲烷萃取(20ml
×
3),合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩,用薄层色谱法以展开剂体系dcm∶meoh=20∶1纯化得到化合物1(120mg)。ms m/z(esi)∶544.3[m+1]。1h nmr(500mhz,chloroform-d)δ8.11(dt,j=8.0,0.9hz,
1h),7.92(s,1h),7.71(dt,j=7.5,1.7hz,1h),7.57-7.46(m,2h),6.99(t,j=2.1hz,1h),6.57(d,j=2.2hz,1h),4.81(dtt,j=46.3,4.9,3.1hz,1h),4.66(d,j=11.9hz,2h),4.51-4.27(m,2h),3.80(ddd,j=7.1,4.7,1.2hz,2h),3.77-3.60(m,4h),3.30-3.09(m,6h),2.94-2.76(m,2h),2.57(t,j=5.7hz,1h),2.17-2.04(m,1h),2.02-1.64(m,9h).
技术总结
发明公开了一种嘧啶并哌啶衍生物合成的新工艺,其主要是以嘧啶并哌啶为母核在催化剂的一系列催化作用下得到一种具有抗癌活性的小分子嘧啶并哌啶衍生物。本发明方法简单,不仅大大降低了生产成本、提高了反应收率,同时也优化了工艺流程。也优化了工艺流程。
技术研发人员:王忠长 武松宇 张卿 雷德维
受保护的技术使用者:南京大学人工智能生物医药技术研究院
技术研发日:2021.10.31
技术公布日:2022/2/18
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1