一种科里内酯衍生物的制备方法
1.本发明属于有机合成技术领域,具体涉及一种科里内酯衍生物的制备方法。
背景技术:
2.前列腺素(缩写为pg)是一类生物活性物质,对于生殖、心血管、泌尿及神经系统均有生理作用,其具有天然来源少、提取困难的不足之处,因此难以广泛应用于临床治疗中。多年来,科研人员已合成了一系列前列腺素物质或类似物,但仍不可避免地存在合成路线长、合并收率低的问题。科里内酯及其衍生物是制备前列腺素类化合物的重要中间体,由于前列腺素类化合物多具有特定的构型,因此如何将科里内酯转化为具有特定结构的衍生物,对于后续能否高效地制备前列腺素类化合物具有至关重要的意义,但有关的合成文献报道很少。
3.yankee等人报道了苯甲酸酯保护的烯酮与格氏试剂的反应,但制备得到的产物其差向异构体比例为1:1,再经过还原、水解、色谱分离等步骤制备得到卡前列素甲酯(“total synthesis of 15-methylprostaglandins”,journal of american chemical society,96(18),5865-5876)。
4.申请号为cn201710476945.4的专利申请公开了用三乙基氯硅烷保护(-)-科里内酯的伯醇、再与苯甲酸进行酯化反应后脱除伯醇保护基团从而得到(-)-苯甲酰科里内酯的路线,但其收率和纯度仍有较大的提升空间,并且仅适用于制备(-)-苯甲酰科里内酯、而不适用于制备(-)-联苯甲酰科里内酯,仍会产生相当量的二取代杂质。
5.因此,迫切希望有一种新的科里内酯衍生物的制备方法,能解决当前收率低、杂质含量高的问题。
技术实现要素:
6.本发明针对现有的制备科里内酯衍生物的方法中收率低的问题,以及杂质含量高的问题,提出了新的制备方法,通过使科里内酯的伯醇被保护后再与苯甲酸或联苯甲酸进行酯化反应,得到伯醇被保护的苯甲酰科里内酯或联苯甲酰科里内酯,脱除伯醇保护基团后得到科里内酯衍生物的方法,实现了高收率、高纯度地制备得到科里内酯衍生物,可用于后续制备前列腺素类药物的过程中。
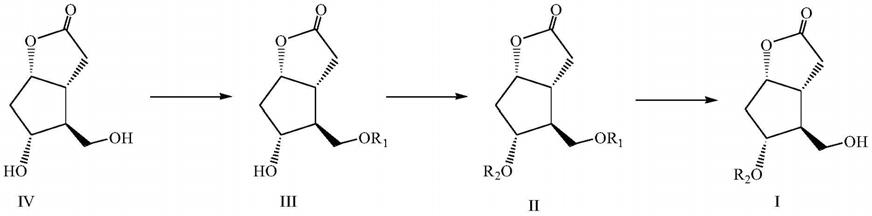
7.本发明为了实现上述技术目的,提供如下技术方案:一种科里内酯衍生物的制备方法,具体步骤如下:
8.9.使式iv化合物与r
1-cl化合物反应得到式iii化合物,使式iii化合物与r
2-oh反应得到式ii化合物,式ii化合物水解后经后处理得到式i化合物;
10.式i、ii、iii及iv中,r1为c
3-6
的杂环基取代的c
1-6
烷基,所述c
3-6
的杂环基中杂原子数目至多为2个;r2为取代或未取代的芳基甲酰基。
11.进一步地,所述为c
3-6
的杂环基中杂原子分别独立地选自o、s或n。
12.进一步地,r1为
13.再进一步地,iv化合物与r
1-cl化合物的摩尔比为1:1-1.5;优选的,为1:1.05。
14.进一步地,r2为苯甲酰基或联苯基甲酰基。
15.再进一步地,式iii化合物与r
2-oh的摩尔比为1:1-1.5;优选的,为1:1-1.05。
16.进一步地,由式iv制备式iii的步骤中存在或不存在碱性试剂。
17.再进一步地,由式iv制备式iii的步骤中不存在碱性试剂。
18.再进一步地,所述碱性试剂为三乙胺或吡啶。
19.进一步地,所述水解为在酸性或碱性条件下进行。
20.进一步地,所述后处理包括干燥有机相、过滤、结晶。
21.由于采用了以上技术,本发明与现有技术相比,其显著优点为:
22.1)操作简便,具有极佳的选择性,便于大规模生产;
23.2)实现了高收率、高纯度制备得到科里内酯衍生物,可用于后续前列腺素类化合物如卡前列素等的生产中;
24.3)在伯醇保护过程中无需使用碱性试剂、可有效降低减少酸性试剂使用量,减少含盐废水的产生量,更加环保,能实现试剂的再利用。
具体实施方式
25.为了使本发明的技术方案及优点更加清楚明白,以下结合具体的实施例对本发明进行进一步详细说明,但并不因此而限定本发明的范围。
26.实施例1
27.1)科里内酯的伯醇保护反应:
[0028][0029]
室温下在四口瓶中加入100ml二氯甲烷,加入如式iv所示的科里内酯17.2g,再加入17.2g n-(3-氯丙基)-吗啉,搅拌混合,室温条件下反应2小时,将所得反应液倾入冰水20ml中,分去水层,有机相加入na2so4干燥后过滤,减压脱溶,浓缩得无定型物状态的式iii化合物29.9g,高效液相色谱(hplc)检测纯度99.3%。1h nmr(500mhz,chloroform-d)δ4.32-4.29(m,1h),3.70-3.66(m,4h),3.36(t,2h),3.34(s,2h),3.22(t,1h),2.39-2.36(m,
6h),2.27-2.26(d,2h),2.07-2.05(d,1h),2.02(s,1h),1.93-1.90(m,3h),1.57-1.55(m,2h).
[0030]
2)伯醇被保护的科里内酯的仲醇酯化反应:
[0031][0032]
向四口瓶内加入上步得到的无定形物状态的式iii化合物和50ml二氯甲烷,再加入14.6g苯甲酸和0.7gdmap,边搅拌边升温至40℃,保温1小时后降至室温,在室温条件下反应10小时,减压浓缩得淡黄色无定型物状态的式ii化合物40.1g,hplc检测纯度98.7%。1h nmr(500mhz,chloroform-d)δ8.03-8.00(d,2h),7.47-7.44(t,1h),7.38-7.36(m,2h),4.32-4.30(m,1h),3.70-3.67(m,4h),3.37(t,2h),3.34(s,2h),3.22(t,1h),2.39-2.36(m,6h),2.27-2.25(d,2h),2.07-2.05(d,1h),1.93-1.91(m,3h),1.58-1.55(m,2h).
[0033]
3)水解制备科里内酯衍生物:
[0034][0035]
向四口瓶内加入上步得到的无定形物状态的式ii化合物和50ml二氯甲烷,盐酸调ph至2.5-3,搅拌反应5小时,分去水层,有机相加入na2so4干燥后过滤,减压脱溶,加入石油醚40ml,析出如式i化合物所示的晶体27.2g,收率为95.81%(基于式iv所示的科里内酯),hplc检测纯度99.2%,ee值:99.96%。1h nmr(500mhz,chloroform-d)δ8.04-8.01(d,2h),7.48-7.45(t,1h),7.38-7.36(m,2h),4.32-4.29(m,1h),3.98(t,1h),3.52-3.50(d,2h),2.27-2.25(d,2h),2.18-2.15(m,3h),2.07-2.05(d,1h),2.02(s,1h).
[0036]
实施例2
[0037]
1)科里内酯的伯醇保护反应:
[0038][0039]
室温下在四口瓶中加入100ml二氯甲烷,加入如式iv所示的科里内酯17.2g和
15.5g三乙胺,搅拌均匀,再加入17.2g n-(3-氯丙基)-吗啉,搅拌混合,室温条件下反应1小时,将所得反应液倾入冰水40ml中,分去水层,有机相加入na2so4干燥后过滤,减压脱溶,浓缩得无定型物状态的式iii化合物29.9g,高效液相色谱(hplc)检测纯度99.5%。1h nmr同实施例1。
[0040]
2)伯醇被保护的科里内酯的仲醇酯化反应:
[0041][0042]
向四口瓶内加入上步得到的无定形物状态的式iii化合物和50ml二氯甲烷,再加入14.6g苯甲酸和0.7gdmap,边搅拌边升温至40℃,保温2小时后降至室温,在室温条件下反应10小时,减压浓缩得淡黄色无定型物状态的式ii化合物40.2g,hplc检测纯度98.5%。1h nmr同实施例1。
[0043]
3)水解制备科里内酯衍生物:
[0044][0045]
向四口瓶内加入上步得到的无定形物状态的式ii化合物和50ml二氯甲烷,盐酸调ph至2.5-3,搅拌反应5小时,分去水层,有机相加入na2so4干燥后过滤,减压脱溶,加入石油醚40ml,析出如式i化合物所示的晶体27.3g,收率为96.27%(基于式iv所示的科里内酯),hplc检测纯度99.3%,ee值:99.97%。1h nmr同实施例1。
[0046]
实施例3
[0047]
1)科里内酯的伯醇保护反应:
[0048][0049]
室温下在四口瓶中加入100ml二氯甲烷,加入如式iv所示的科里内酯17.2g,再加入15.7g n-(2-氯乙基)-吗啉,搅拌混合,室温条件下反应2小时,将所得反应液倾入冰水
20ml中,分去水层,有机相加入na2so4干燥后过滤,减压脱溶,浓缩得无定型物状态的式iii化合物28.3g,高效液相色谱(hplc)检测纯度98.4%。1h nmr(500mhz,chloroform-d)δ4.32-4.29(m,1h),3.70-3.66(m,4h),3.36(t,2h),3.34(s,2h),3.22(t,1h),2.39-2.36(m,6h),2.27-2.26(d,2h),2.07-2.05(d,1h),2.02(s,1h),1.93-1.90(m,3h).
[0050]
2)伯醇被保护的科里内酯的仲醇酯化反应:
[0051][0052]
向四口瓶内加入上步得到的无定形物状态的式iii化合物和50ml二氯甲烷,再加入25.8g对苯基苯甲酸和0.7g dmap,边搅拌边升温至50℃,保温1小时后降至室温,在室温条件下反应10小时,减压浓缩得类白色无定型物状态的式ii化合物43.8g,hplc检测纯度98.2%。1h nmr(500mhz,chloroform-d)δ8.07-8.05(d,2h),7.62-7.60(d,2h),7.48-7.45(d,2h),7.35-7.33(m,2h),7.23-7.20(m,1h),4.32-4.30(m,1h),3.96(t,1h),3.70-3.67(m,4h),3.46(t,2h),3.34(d,2h),2.55-2.53(t,2h),2.44-2.42(t,1h),2.39-2.36(m,4h),2.27-2.25(d,2h),2.17-2.15(t,2h),2.07-2.05(m,1h).
[0053]
3)水解制备科里内酯衍生物:
[0054][0055]
向四口瓶内加入上步得到的无定形物状态的式ii化合物和50ml二氯甲烷,盐酸调ph至2.5-3,搅拌反应5小时,分去水层,有机相加入na2so4干燥后过滤,减压脱溶,加入石油醚40ml,析出如式i化合物所示的晶体32.4g,收率为87.1%(基于式iv所示的科里内酯),hplc检测纯度97.9%,ee值:99.86%。1h nmr(500mhz,chloroform-d)δ8.08-8.05(d,2h),7.63-7.61(d,2h),7.48-7.45(d,2h),7.36-7.33(m,2h),7.23-7.21(m,1h),4.32-4.29(m,1h),3.98(t,1h),3.52-3.50(d,2h),2.27-2.25(d,2h),2.18-2.16(m,3h),2.08-2.05(d,1h),2.03(s,1h).
[0056]
实施例4
[0057]
1)科里内酯的伯醇保护反应:
[0058][0059]
室温下在四口瓶中加入100ml二氯甲烷,加入如式iv所示的科里内酯17.2g和17.2g三乙胺,搅拌均匀,,再加入15.7g n-(2-氯乙基)-吗啉,搅拌混合,室温条件下反应1小时,将所得反应液倾入冰水40ml中,分去水层,有机相加入na2so4干燥后过滤,减压脱溶,浓缩得无定型物状态的式iii化合物28.4g,高效液相色谱(hplc)检测纯度98.9%。1h nmr同实施例3。
[0060]
2)伯醇被保护的科里内酯的仲醇酯化反应:
[0061][0062]
向四口瓶内加入上步得到的无定形物状态的式iii化合物和50ml二氯甲烷,再加入25.8g对苯基苯甲酸和0.7g dmap,边搅拌边升温至50℃,保温1小时后降至室温,在室温条件下反应10小时,减压浓缩得类白色无定型物状态的式ii化合物44.3g,hplc检测纯度98.5%。1h nmr同实施例3。
[0063]
3)水解制备科里内酯衍生物:
[0064][0065]
向四口瓶内加入上步得到的无定形物状态的式ii化合物和50ml二氯甲烷,盐酸调ph至2.5-3,搅拌反应5小时,分去水层,有机相加入na2so4干燥后过滤,减压脱溶,加入石油醚40ml,析出如式i化合物所示的晶体32.5g,收率为89.6%(基于式iv所示的科里内酯),hplc检测纯度98.2%,ee值:99.91%。1h nmr同实施例3。
[0066]
上述实施例仅为本发明较佳的具体实施方案,不应视为对于本发明的限制,本发
明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等同替换方案为保护范围,任何熟悉本技术领域的技术人员在本发明披露的技术范围之内,根据本发明的技术方案及其发明构思进行等同替换或改进,都涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1