一种β-丙氨酸生产菌、构建方法及应用
一种
β-丙氨酸生产菌、构建方法及应用
技术领域
1.本发明涉及一种β-丙氨酸生产菌、构建方法及应用,属于代谢工程领域。
背景技术:
2.β-丙氨酸(3-氨基丙酸)是一种非蛋白质氨基酸,存在于所有生物中。β-丙氨酸作为泛酸的重要组成部分,是辅酶a和酰基载体蛋白合成的前体。以往的研究表明,在微生物体内,缺乏β-丙氨酸合成途径是致命的,因为不能合成辅酶a。另外,β-丙氨酸也可作为补品,被体内吸收后,可与组氨酸偶联形成二肽,在人体肌肉和脑组织中起重要作用。因此,β-丙氨酸广泛应用于药物(如帕米膦酸和胍丙酸和巴尔)、饲料和食品添加剂等领域。
3.目前β-丙氨酸的合成方法主要有化学催化法、酶法和微生物发酵法,其中化学催化法是工业生产β-丙氨酸的主要方法。然而,以丙烯酰胺、丁二酰亚胺或β-氨基丙腈为原料的化学催化法,由于环境和社会的压力,是不可持续的。因此,利用酶法和微生物发酵法生产β-丙氨酸,引起了学者的兴趣。目前,许多生物的adc被表征,然后被用于酶法生产β-丙氨酸。虽然酶法合成β-丙氨酸取得了很大进展,并初步满足了工业应用的需要,但微生物发酵法仍是一种很有前途的工业生产方法。β-丙氨酸是以草酰乙酸和富马酸为前体合成的天门冬氨酸衍生物。在大肠杆菌中,草酰乙酸和富马酸可以通过三羧酸循环的还原/氧化分支来合成。因此通过tca循环的还原途径合成β-丙氨酸在理论产量和能量消耗方面更有优势。
技术实现要素:
4.本发明的目的提供一种β-丙氨酸生产菌、构建方法及应用,满足β-丙氨酸工业应用的需要。
5.为实现本发明的上述目的,本发明采用如下技术方案:一种β-丙氨酸生产菌,由如下方法构建获得:(1)以e. coli w3110为出发菌,将其基因组中pand基因的启动子替换为trc启动子,得到重组菌株e. coli w3110/trc-pand,记为ala1;(2)将源自bacillus subtilis的外源基因pand整合入质粒ptrc99a中,得到质粒ptrc99a-pand;将质粒ptrc99a-pand导入ala1中,增强pand基因的表达,得到重组菌株ala1/ptrc99a-pand,记为ala2;(3)将ala2基因组中的ppc基因的启动子替换为trc启动子,得到重组菌株ala2/trc-ppc,记为ala3;(4)将ala3基因组中的pyka基因敲除,得到重组菌株ala3/
△
pyka,记为ala4;(5)将ala4基因组中的aspa基因敲除,得到重组菌株ala4/
△
aspa,记为ala5,即为所述β-丙氨酸生产菌。
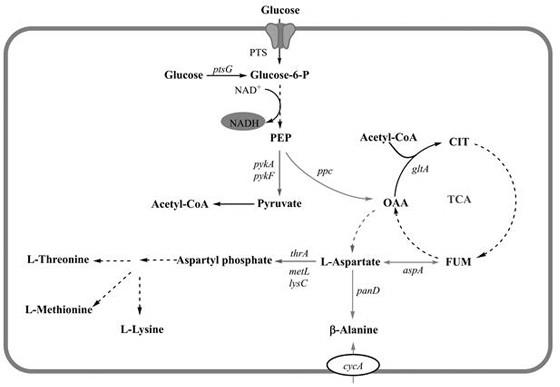
6.大肠杆菌中β-丙氨酸生物合成途径及本发明涉及的基因工程改造示意图参见图1。首先,在出发菌e. coli w3110的基础上,用trc启动子替换基因pand的原始启动子,促进天冬氨酸转化为β-丙氨酸;再将bacillus subtilis来源的pand整合到质粒ptrc99a质粒
中,并导入工程菌中,进一步促进天冬氨酸转化为β-丙氨酸;然后,通过启动子替换,强化基因ppc的表达,增强天冬氨酸合成的前体草酰乙酸的供应;再通过敲除基因pyka,降低碳通量流入tca循环;最后,通过敲除基因aspa阻断天冬氨酸降解为富马酸;最终得到β-丙氨酸生产菌株。
7.本发明还涉及构建所述β-丙氨酸生产菌的方法,所述方法如下:(1)运用crispr-cas9基因编辑技术,将出发菌e. coli w3110基因组中的pand基因的启动子替换成trc启动子,得到重组菌株w3110/trc-pand,记为ala1;(2)将源自bacillus subtilis的外源基因pand整合入质粒ptrc99a中,得到质粒ptrc99a-pand;将质粒ptrc99a-pand导入步骤ala1中,增强pand基因的表达,得到重组菌株ala1/ptrc99a-pand,记为ala2;(3)运用crispr-cas9基因编辑技术,将ala2基因组中的ppc基因的启动子替换为trc启动子,得到重组菌株ala2/trc-ppc,记为ala3;(4)运用crispr-cas9基因编辑技术,将ala3基因组中的pyka基因敲除,得到重组菌株ala3/
△
pyka,记为ala4;(5)运用crispr-cas9基因编辑技术,将ala4基因组中的aspa基因敲除,得到重组菌株ala4/
△
aspa,记为ala5,即为所述β-丙氨酸生产。。
8.具体的,步骤(1)trc启动子核苷酸序列如seq id no.1所示。
9.seq id no.1序列如下:ttgacaattaatcatccggctcgtataatgtgtggaattgtgagcggataacaatttcacacaggaaacagacc具体的,步骤(2)外源基因pand核苷酸序列如seq id no.2所示(编码氨基酸序列如seq id no.3所示)。
10.seq id no.2序列如下:atgtatcgaacaatgatgagcggcaagcttcacagggcaactgttacggaagcaaatctgaattatgtgggaagcattacaattgatgaagatctcattgatgcggtgggaatgcttcctaatgaaaaagtgcaaattgtgaataataataatggagcacgtctggaaacgtatattattcctggtaaacgcggaagcggcgtcatctgcttaaacggtgcagccgcacgccttgtacaggaaggagataaggtcattattatttcctacaaaatgatgtctgatcaagaagcggcaagccacgagccgaaagtggctgttttgaatgatcaaaacaaaattgaacaaatgctggggaacgaaccagcccgtacaattttgtagseq id no.3序列如下:myrtmmsgklhratvteanlnyvgsitidedlidavgmlpnekvqivnnnngarletyiipgkrgsgviclngaaarlvqegdkviiisykmmsdqeaashepkvavlndqnkieqmlgnepartil本发明还涉及所述β-丙氨酸生产菌在微生物发酵制备β-丙氨酸中的应用。
11.具体的,所述应用为:将所述β-丙氨酸生产菌接种至发酵培养基中,于28~32 ℃、400~800 rpm条件下进行发酵培养48~100h,发酵结束后取发酵液上清分离纯化得到β-丙氨酸;所述发酵培养基组成如下:葡萄糖10~30g/l、硫酸铵10~20g/l、酵母浸粉1~5g/l、kh2po
4 1~5g/l、mgso
4 0.1~2.0g/l、微量金属盐溶液0.5~5ml/l,ph 6.5~7.0,溶剂为去离子水;微量金属盐溶液组成为:10 g/l cucl2、10 g/l feso4·
7h2o、1 g/l znso4·
7h2o、0.20 g/l cuso4、0.02 g/l nicl2·
7h2o,溶剂为去离子水。。
12.优选的,所述发酵培养基组成如下:葡萄糖20 g/l,硫酸铵16 g/l,酵母粉2 g/l,
kh2po
4 2 g/l,mgso
4 0.5 g/l,盐溶液 2 ml/l,溶剂为去离子水。
13.通常,所述基因工程菌株发酵前,先接种至lb培养基中,于温度37℃、转速200 rpm的摇床上过夜培养,然后以体积浓度5~15%接种量接种到发酵培养基中培养。
14.本发明有益效果主要体现在:本发明通过启动子替换过表达基因pand,并引入b. subtilis来源的pand导入到工程菌中,强化天冬氨酸转化为β-丙氨酸。同时通过启动子替换,强化基因ppc的表达,增强天冬氨酸合成的前体草酰乙酸的供应。再通过敲除基因pyka,降低碳通量流入tca循环;最后,通过敲除基因aspa阻断天冬氨酸降解为富马酸;最终得到β-丙氨酸生产菌株,其在发酵过程中,β-丙氨酸产量可达29.35g/l,为后续高产β-丙氨酸工程菌的构建奠定了基础。
附图说明
15.图1为大肠杆菌中β-丙氨酸生物合成途径及本发明涉及的基因工程改造示意图;图2为质粒ptrc99a-pand图谱。
16.(五)具体实施方式下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:以下实施例中,所述卡那霉素(kana)的使用终浓度为50ng/μl。所述盐酸壮观霉素(sd)的使用终浓度为50ng/μl。实施例中所用大肠杆菌感受态细胞为商用购买的大肠杆菌dh5α,重组反应所用的一步法定向克隆无缝克隆试剂盒购于诺唯赞公司,但不局限于此公司。
17.实施例1:ala1菌株的构建构建β-丙氨酸生产菌株采用的基因编辑方法参照文献(zhang, b., et al., metabolic engineering of escherichia coli for d-pantothenic acid production. food chem, 2019. 294: p. 267-275.),该编辑方法使用的工具质粒为ptarget和pcas,具体进行方法如下:ala1菌株的构建是以e. coli w3110为出发菌株(1)ptarget-grna质粒突变设计定点突变引物,将质粒ptarget上20bp同源序列突变成靶基因pand启动子中pam位点前的20bp序列。以ptarget质粒为模板,pt-tpand-f和pt-tpandr为引物,pcr扩增ptarget突变质粒,扩增体系如下:
pcr扩增结束后,利用琼脂糖凝胶电泳检测pcr产物,然后向pcr产物中加入0.5μl的dpni,37℃消化1h,取消化产物10μl加入到100μl的dh5α感受态细胞中进行转化实验,然后将培养液涂布于lb固体培养基(sd抗性),37℃培养过夜。挑取单菌落接种于10ml的lb液体培养基(sd抗性)中,培养18h以上,取菌液送测序并提取突变质粒ptarget-pand-g备用。
18.(2)pcr扩增donor dna和线性化ptarget-pand-g质粒片段以大肠杆菌基因组为模板,分别以tpand-p1、tpand-p2和tpand-p3、tpand-p4为引物pcr扩增出靶基因上下游各约500bp序列作为同源臂(donor dna)。以pt+d cf com、pt+d cr com为引物,ptarget-pand-g为模板,pcr扩增ptarget-pand-g线性化质粒片段。扩增体系如下:pcr扩增结束后,利用琼脂糖凝胶电泳检测pcr产物,然后向线性化ptarget-pand-g质粒片段中加入1μl的dpni,37℃消化1h。然后使用dna清洁试剂盒对dpni消化产物和pcr扩增产物donor dna进行clean up,并检测清洁后产物的dna浓度,-20℃保存备用。
19.(3)一步克隆连接线性化ptarget-pand-g质粒片段和donor dna
取(2)中清洁后的产物,根据线性化ptarget-pand-g质粒片段和donor dna的浓度,利用一步法定向克隆无缝克隆试剂盒进行重组反应。取一步克隆产物10μl加入到100μl的dh5α感受态细胞中进行转化实验,然后将培养液涂布于lb固体培养基(sd抗性)上,37℃培养过夜。待长出单菌落,挑取单菌落接种到10ml的lb液体培养基(sd抗性)中,送测序并提取质粒ptarget-pand-pdg备用。
20.(4)ptarget-pand-pdg的电击转化制备含pcas质粒的w3110菌株的电转感受态。取一只电转感受态,冰浴放置,在超净台中加入2μl ptarget-pand-pdg,用移液器轻柔混匀,冰浴1min;用移液器将混匀液转移至预冷的2mm电击杯中(电击杯需提前在超净台风干),冰浴45s。用纸巾擦干电击杯外侧水雾,置于电转仪中,使用eco 2档电击。在超净台中向电击杯凹槽加入1ml预冷lb培养基,倾斜电击杯,从电击杯口吸取全部菌液,转移至2ml无菌ep管中。30℃,180rpm复苏2.5h以上;取200μl涂布于lb固体培养基(sd+kana抗性)上,30℃培养过夜。
21.(5)阳性克隆筛选和验证在各靶标基因上游同源臂外侧100bp处设计正向验证引物tpand-vf,替换的trc启动子上设计反向验证引物trc-vr,并以此为正反引物,挑取克隆子作为模板,进行菌落pcr,同时以原基因组为模板作阴性对照。菌落pcr有条带的视为阳性。
22.(6)ptarget-pdg与pcas消除与测序验证将阳性克隆接种于10 ml lb试管(kana抗性),并加入10μl iptg母液,30℃,180 rpm培养12h。取过夜培养菌液划线于lb固体培养基(kana抗性)上,30℃培养过夜。取过夜划线平板,将单菌落编号,并挑取编号的部分单菌落,划线于lb固体培养基(sd抗性)上所对应的区域,37℃培养过夜。在lb固体培养基(sd抗性)所对应的区域不能生长的单菌落为ptarget-pand-pdg成功消除的克隆。
23.将ptarget-pand-pdg成功消除的克隆接种于10 ml lb试管(无抗性),37℃,180rpm培养12h。取过夜培养菌液,划线于lb固体培养基(无抗性)上,37℃培养过夜。取过夜培养菌液,将单菌落编号,并挑取编号的部分单菌落,划线于lb固体培养基(kana抗性)上所对应的区域,30℃培养过夜。在lb固体培养基(kana抗性)所对应的区域不能生长的单菌落为pcas成功消除的克隆。
24.(7)阳性克隆测序验证挑取ptarget-pdg与pcas均成功消除的克隆作为菌落pcr模板,以验证引物进行菌落pcr,菌落pcr产物送测序,验证阳性克隆,得到ala1。
25.实施例2:ptrc99a-pand质粒的构建以ptrc99a质粒(实验室保藏)为模板,ptrc99a-f、ptrc99a-r为引物,pcr扩增线性化质粒片段,同时以pand-f、pand-r为引物,b. subtilis基因组为模板,扩增pand片段(seq id no.2,编码氨基酸序列如seq id no.3所示)。利用dpni消化扩增的线性化质粒片段,然后使用clean up试剂盒对消化后的线性化ptrc99a载体片段和pand片段进行清洁,再利用一步法定向克隆无缝克隆试剂盒进行重组反应,反应产物转化大肠杆菌感受态细胞后,涂布在含有卡那霉素的lb平板上。阳性克隆送测序公司测序,确定连接成功的质粒ptrc99a-pand。将构建的质粒ptrc99a-pand转化入ala1得到菌株ala2。
26.实施例3:trc启动子替换基因ppc的原始启动子构建菌株ala3
(1)ptarget-grna质粒突变以ptarget为模板,tppc-pt f和tppc-pt r为引物,pcr扩增ptarget突变质粒。pcr产物经消化处理后,转化进入dh5α感受态细胞,经测序验证,得到突变质粒ptarget-ppc-g。
27.(2)构建质粒ptarget-ppc-pdg以tppc-p1、tppc-p2和tppc-p3、tppc-p4为引物pcr扩增靶基因上下游donor dna。同时以p t+d cf com、p t+d cr com为引物,ptarget-ppc-g为模板,pcr扩增线性化ptarget-ppc-g质粒片段。dpni消化处理pcr扩增的ptarget-ppc-g线性化质粒片段后,使用dna清洁试剂盒对消化后的线性化ptarget-ppc-g质粒片段和donor dna进行clean up,再利用一步法定向克隆无缝克隆试剂盒进行重组反应,反应产物转化大肠杆菌感受态细胞后,经测序验证后得到质粒ptarget-ppc-pdg。
28.(3)ptarget-ppc-pdg的电击转化制备含pcas质粒的ala2菌株的电转感受态。取一只电转感受态,在超净台中加入2μl ptarget-ppc-pdg,冰浴1min后转移至预冷的2mm电击杯中,冰浴45s。置于电转仪中进行电击。电击后立即向杯凹槽中加入1ml预冷lb培养基,倾斜电击杯,从电击杯口吸取全部菌液,转移至2ml无菌ep管中。30℃,180rpm复苏2.5h以上;取200μl涂布于lb固体培养基(sd+kana抗性)上,30℃培养过夜。
29.后续基因编辑、验证及质粒消除同实施例1(4-7),得到菌株ala3。
30.实施例4:敲除基因pyka构建菌株ala4(1)ptarget-grna质粒突变以ptarget为模板,δpyka-pt f和δpyka-pt r为引物,pcr扩增ptarget突变质粒。pcr产物经消化处理后,转化进入dh5α感受态细胞,经测序验证,得到突变质粒ptarget-pyka-g。
31.(2)构建质粒ptarget-pyka-pdg以δpyka-p1、δpyka-p2和δpyka-p3、δpyka-p4为引物pcr扩增靶基因上下游donor dna。同时以pt+d cf com、pt+d cr com为引物,ptarget-pyka-pdg为模板,pcr扩增线性化ptarget-pyka-g质粒片段。dpni消化处理pcr扩增的ptarget-pyka-g线性化质粒片段后,使用dna清洁试剂盒对消化后的线性化ptarget-pyka-g质粒片段和donor dna进行clean up,再利用一步法定向克隆无缝克隆试剂盒进行重组反应,反应产物转化大肠杆菌感受态细胞后,经测序验证后得到质粒ptarget-pyka-pdg。
32.(3)ptarget-pyka-pdg的电击转化制备含pcas质粒的ala3菌株的电转感受态。取一只电转感受态,在超净台中加入2μl ptarget-pyka-pdg,冰浴1min后转移至预冷的2mm电击杯中,冰浴45s。置于电转仪中进行电击。电击后立即向杯凹槽中加入1ml预冷lb培养基,倾斜电击杯,从电击杯口吸取全部菌液,转移至2ml无菌ep管中。30℃,180rpm复苏2.5h以上;取200μl涂布于lb固体培养基(sd+kana抗性)上,30℃培养过夜。
33.后续基因编辑、验证及质粒消除同实施例1(4-7),得到菌株ala4。
34.实施例5:敲除基因aspa构建菌株ala5(1)ptarget-grna质粒突变以ptarget为模板,δaspa-pt f和δaspa-pt r为引物,pcr扩增ptarget突变质
粒。pcr产物经消化处理后,转化进入dh5α感受态细胞,经测序验证,得到突变质粒ptarget-aspa-g。
35.(2)构建质粒ptarget-aspa-pdg以δaspa-p1、δaspa-p2和δaspa-p3、δaspa-p4为引物pcr扩增靶基因上下游donor dna。同时以p t+d cf com、p t+d cr com为引物,ptarget-aspa-pdg为模板,pcr扩增线性化ptarget-aspa-g质粒片段。dpni消化处理pcr扩增的ptarget-aspa-g线性化质粒片段后,使用dna清洁试剂盒对消化后的线性化ptarget-aspa-g质粒片段和donor dna进行clean up,再利用一步法定向克隆无缝克隆试剂盒进行重组反应,反应产物转化大肠杆菌感受态细胞后,经测序验证后得到质粒ptarget-aspa-pdg。
36.(3)ptarget-aspa-pdg的电击转化制备含pcas质粒的ala4菌株的电转感受态。取一只电转感受态,在超净台中加入2μl ptarget-aspa-pdg,冰浴1min后转移至预冷的2mm电击杯中,冰浴45s。置于电转仪中进行电击。电击后立即向杯凹槽中加入1ml预冷lb培养基,倾斜电击杯,从电击杯口吸取全部菌液,转移至2ml无菌ep管中。30℃,180rpm复苏2.5h以上;取200μl涂布于lb固体培养基(sd+kana抗性)上,30℃培养过夜。
37.后续基因编辑、验证及质粒消除同实施例1(4-7),得到菌株ala5。
38.表1:菌株基因型表2:引物序列表
实施例6:工程菌株的摇瓶发酵和发酵罐发酵菌种活化:取-80℃保藏菌种划线接种于活化培养基(固体lb培养基)中,37℃培养过夜;种子培养:用接种环挑取活化种子接种于装有10ml种子培养基(lb培养基)的试管中,37℃,200rpm培养过夜;摇瓶发酵:按5%接种量接种种子液到装有20ml发酵培养基的500ml锥形瓶中,37℃,200rpm/min振荡培养,发酵周期48h;发酵罐发酵:取种子培养基接种于3瓶含100ml lb培养基的500ml锥形瓶中,37℃,200rpm/min振荡培养12h。按10%的接种量将摇瓶种子液接入含2l发酵培养基的5l发酵罐
中,发酵温度控制在30℃,转速500rpm/min,通过40%氨水控制发酵ph为6.8。发酵过程采用ph-stat模式控制补料,每4h取样检测生物量、残糖和β-丙氨酸的量。
39.摇瓶发酵培养基组成为:葡萄糖20g/l,硫酸铵16g/l,酵母粉2g/l,kh2po
4 1g/l,mgso
4 0.2g/l,caco
3 15g/l,微量金属盐溶液 1ml/l,溶剂为水。发酵罐发酵发酵培养基组成为:葡萄糖20 g/l,硫酸铵16 g/l,酵母粉2 g/l,kh2po
4 2 g/l,mgso
4 0.5 g/l,盐溶液 2 ml/l,溶剂为水。(微量金属盐溶液组成:10 g/l cucl2、10 g/l feso4·
7h2o、1 g/l znso4·
7h2o、0.2 g/l cuso4、0.02 g/l nicl2·
7h2o,溶剂为水)补料培养基组成如下:葡萄糖500g/l、硫酸铵10g/l、酵母浸粉2g/l、kh2po
4 14g/l、mgso
4 8g/l、微量金属盐溶液1ml/l,溶剂为水。
40.发酵结束后,使用氨基酸分析仪检测β-丙氨酸的含量。
41.表3:基因工程菌5l发酵罐补料发酵产β-丙氨酸注:nd:not detected结果显示,通过基因工程操作后,基因工程菌ala5通过摇瓶发酵β-丙氨酸产量达3.23g/l,通过5l发酵罐补料发酵,β-丙氨酸产量达到29.35g/l。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1