一种卡利拉嗪中间体的制备方法与流程
1.本发明涉及一种卡利拉嗪中间体的制备方法,属于有机合成技术领域。
背景技术:
2.卡利拉嗪(cariprazine)由gedeon richter ltd和forest laboratories公司联合开发,首次报道其具有多巴胺d3/d2部分激动剂,兼具优先结合d3r和da部分激动剂的特点,用于治疗精神分裂症、躁狂症、重度抑郁症,其盐酸盐于2015年作为抗精神分裂症药于美国上市,化学名为反式-1-{4-[2-[4-(2,3-二氯苯基)-哌嗪-1-基]乙基]环已基}-3,3-二甲基脲盐酸盐。
[0003]
而反式n-叔丁氧羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环已胺(int.3)是卡利拉嗪的前一步重要原料,在此将有基因毒性杂质完原去除,从而更加提高最重原料药卡利拉嗪的安全性。
[0004]
然而经典合成工艺存在如下问题:其使用了对甲苯磺酰氯而产生基因毒性杂质(对甲苯磺酸酯);中间体smo1对哌嗪对接温度需要80℃以上,否则反应不完全,而且smo1对温度不稳定,在60℃就开始有降解,使得smo1和哌嗪对接的收率始终只有75-80%左右,反应时间需要10-12h左右;存在工艺杂质的产生;因此本领域亟待一种解决上述技术问题的新的卡利拉嗪的制备方法。
技术实现要素:
[0005]
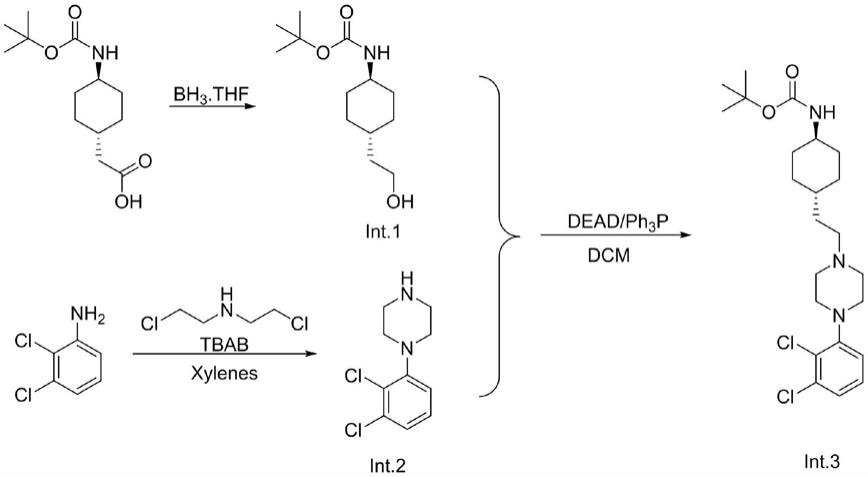
为了克服上述技术不足,本发明提供了一种卡利拉嗪中间体的制备方法。首先用原料反式-(n-boc-4-氨基环已基)乙酸还原得到反式-(n-boc-4-氨基环已基)乙醇(int.1),随后用原料2,3-二氯苯胺和双(2-氯乙基)乙胺在相转移催化作用下分子内关环得到1-(2,3-二氯苯基)哌嗪(int.2),随后int.1与int.2发生光延反应,纯化除掉三苯基氧磷得到卡利拉嗪中间体(化学式所式int.3)。本发明原料易得,步骤简洁,反应条件温和且连续,对设备要求相对较低,产品纯度大于99.0%。
[0006]
本发明所述一种卡利拉嗪中间体的制备方法,反应方程式如下:
[0007]
包括如下步骤:
[0008]
环原反应:将反式-(n-boc-4-氨基环已基)乙酸在有机溶剂中混合,随后滴加硼烷四氢呋喃溶液,反应得到反式-(n-boc-4-氨基环已基)乙醇(int.1);
[0009]
胺化反应:将2,3-二氯苯胺、双(2-氯乙基)乙胺、催化剂和相转移催化剂在二甲苯中混合,升温回流得到1-(2,3-二氯苯基)哌嗪(int.2);
[0010]
光延反应:将int.1、int.2和三苯基磷混合在有机溶剂中,滴加光延反应试剂发生光延反应,纯化除掉三苯基氧磷得到卡利拉嗪中间体(化学式所式int.3);
[0011]
进一步地,在上述技术方案中,环原反应中,所述有机溶剂选自四氢呋喃或甲基四氢呋喃。
[0012]
进一步地,在上述技术方案中,环原反应中,所述反式-(n-boc-4-氨基环已基)乙酸与硼烷四氢呋喃溶液摩尔比为1:1.80-2.30。
[0013]
进一步地,在上述技术方案中,胺化反应中,所述催化剂选自对甲苯磺酸,相转移催化剂选自四丁基溴化胺或18-冠醚-6。
[0014]
进一步地,在上述技术方案中,胺化反应中,所述2,3-二氯苯胺、双(2-氯乙基)乙胺、催化剂和相转移催化剂摩尔比为1:1.0-1.05:0.05-0.1:0.05-0.1。
[0015]
进一步地,在上述技术方案中,光延反应中,所述光延反应试剂选自偶氮二甲酸二乙酯(dead)或偶氮二甲酸二异丙酯(diad)。
[0016]
进一步地,在上述技术方案中,光延反应中,所述int.1、int.2、三苯基磷与光延反应试剂摩尔比为1:1.00-1.05:1.2-1.3:1.25-1.35。
[0017]
发明有益效果
[0018]
步骤少,纯度高,收率高,原料易得。
具体实施例
[0019]
下面通过具体实例对本发明进行进一步说明。这些实施例应理解为仅用于说明本发明而不用于限制本发明的保护范围。在阅读了本发明记载的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等效变化和修改同样落入本发明权利要求所限定的
范围。
[0020]
卡利拉嗪中间体的制备
[0021]
实施例1
[0022][0023]
向反应瓶内加入反式-(n-boc-4-氨基环已基)乙酸(25.7g,0.1mol)溶于300ml四氢呋喃中,温度降低为0℃,滴加1.0m/l硼烷四氢呋喃溶液150ml(0.15mol),该温度下反应3小时。加水淬灭,加入mtbe,用饱和碳酸氢钠水溶液洗涤两次,有机相浓缩至不流液,得到反式-(n-boc-4-氨基环已基)乙醇22.5g,hplc96%,收率92%。
[0024]
实施例2
[0025][0026]
向反应瓶内加入2,3-二氯苯胺1(10g,61.72mmol,1eq.)和双(2-氯乙基)乙胺1(8.7g,1eq,61.72mmol)的搅拌溶液中加入对甲苯磺酸(1.17g,0.1eq,6.17mmol)和四丁基溴化铵(1.5g,0.1eq,6.17mmol)在二甲苯(150ml)中的溶液。将所得反应混合物130-135℃加热48小时。反应完成后。将反应混合物冷却至室温,用氨水将溶液的ph值调节至ph 6-7。有机化合物用乙酸乙酯萃取,用硫酸钠干燥,减压干燥。浓缩后得到1-(2,3-二氯苯基)哌嗪12.5g,hplc91.6%,收率88%。无需纯化直接下一步。
[0027]
实施例3
[0028][0029]
氮气保护下向反应瓶内加入反式-(n-boc-4-氨基环已基)乙醇(int.1)24.3g
(0.1mol)、1-(2,3-二氯苯基)哌嗪(int.2)25.4g(0.11mol)、三苯基膦31.5(0.12mol)和干燥后的二氯甲烷240ml,氮气氛围下冰浴控温0-5℃滴加偶氮二甲酸二乙酯22.6g(0.123mol)的二氯甲烷210ml,滴毕保持温度搅拌反应3小时,hplc监控反应完毕,加入饱和碳酸氢钠水溶液,有机相浓缩,减压抽滤,滤饼用20ml二氯甲烷洗涤一次,滤饼大量三苯基氧磷,滤液加入氯化锌,过滤,滤液浓缩,加入正庚烷打浆得反式n-叔丁氧炭基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺(iii)化合物32.8g,hplc99.7%,收率71.44%。
[0030]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
技术特征:
1.一种卡利拉嗪中间体的制备方法,其特征在于,包括如下步骤:环原反应:将反式-(n-boc-4-氨基环已基)乙酸在有机溶剂中混合,随后滴加硼烷四氢呋喃溶液,反应得到反式-(n-boc-4-氨基环已基)乙醇(int.1);胺化反应:将2,3-二氯苯胺、双(2-氯乙基)乙胺、催化剂和相转移催化剂在二甲苯中混合,升温回流得到1-(2,3-二氯苯基)哌嗪(int.2);光延反应:将int.1、int.2和三苯基磷混合在有机溶剂中,滴加光延反应试剂发生光延反应,纯化除掉三苯基氧磷得到卡利拉嗪中间体(化学式所式int.3)。2.根据权利要求1所述卡利拉嗪中间体的制备方法,其特征在于:环原反应中,所述有机溶剂选自四氢呋喃或甲基四氢呋喃。3.根据权利要求1所述卡利拉嗪中间体的制备方法,其特征在于:环原反应中,所述反式-(n-boc-4-氨基环已基)乙酸与硼烷四氢呋喃溶液摩尔比为1:1.80-2.30。4.根据权利要求1所述卡利拉嗪中间体的制备方法,其特征在于:胺化反应中,所述催化剂选自对甲苯磺酸,相转移催化剂选自四丁基溴化胺或18-冠醚-6。5.根据权利要求1所述卡利拉嗪中间体的制备方法,其特征在于:胺化反应中,所述2,3-二氯苯胺、双(2-氯乙基)乙胺、催化剂和相转移催化剂摩尔比为1:1.0-1.05:0.05-0.1:0.05-0.1。6.根据权利要求1所述卡利拉嗪中间体的制备方法,其特征在于:光延反应中,所述光延反应试剂选自偶氮二甲酸二乙酯(dead)或偶氮二甲酸二异丙酯(diad)。7.根据权利要求1所述卡利拉嗪中间体的制备方法,其特征在于:光延反应中,所述int.1、int.2、三苯基磷与光延反应试剂摩尔比为1:1.00-1.05:1.2-1.3:1.25-1.35。
技术总结
本发明公开了一种卡利拉嗪中间体的制备方法,属于有机合成技术领域。首先用原料反式-(N-Boc-4-氨基环已基)乙酸还原得到反式-(N-Boc-4-氨基环已基)乙醇(Int.1),随后用原料2,3-二氯苯胺和双(2-氯乙基)乙胺在相转移催化作用下分子内关环得到1-(2,3-二氯苯基)哌嗪(Int.2),随后Int.1与Int.2发生光延反应,纯化除掉三苯基氧磷得到卡利拉嗪中间体(化学式所式Int.3)。本发明原料易得,步骤简洁,反应条件温和且连续,对设备要求相对较低,产品纯度大于99.0%。于99.0%。
技术研发人员:李国伟 孙桂淦 徐清雨
受保护的技术使用者:泰州精英化成医药科技有限公司
技术研发日:2022.01.27
技术公布日:2022/5/31
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1