一种用于储氢的苯并对二吡咯分子的合成方法
1.本技术属于有机杂环芳香烃分子储氢技术领域,具体涉及一种用于储氢的苯并对二吡咯分子的合成方法。
背景技术:
2.本技术所述苯并对二吡咯分子是一种可用于储氢的有机杂环芳香烃分子。在“新型窄带隙聚方酸菁的合成与光电性质”等文章中提到所述苯并对二吡咯分子可应用于太阳能电池领域,但是该分子的合成过程表现出产率低、产量少、效率慢的不足,使其无法满足工业化生产需求,也限制了其在实际中的应用。
技术实现要素:
3.本技术提供一种用于储氢的苯并对二吡咯分子的合成方法,以解决当前的合成过程表现出产率低、产量少、效率慢的问题。
4.本技术提供一种用于储氢的苯并对二吡咯分子的合成方法,所述合成方法以n1分子为初始反应物制备得到苯并对二吡咯分子;
5.其中,所述n1分子的化学结构式为:
[0006][0007]
所述苯并对二吡咯分子的化学结构式为:
[0008][0009]
优选地,所述合成方法以n1分子为初始反应物制备得到苯并对二吡咯分子的过程包括:
[0010]
步骤1:在冰水浴中,将所述n1分子溶于浓硫酸,再滴加浓硝酸得到凝固的第一反应液,放置5-20min冷却后,进行第一后处理得到n2分子;其中,所述n2分子的化学结构式为:
[0011]
[0012]
步骤2:在氮气保护下,将所述n2分子溶于n,n-二甲基甲酰胺中,待温度升至80~100℃,加入n,n-二甲基甲酰胺二乙基缩醛,然后升温至110~130℃,反应20~30h后降至室温得到第二反应液,将所述第二反应液过滤后得到n3分子;其中,所述n3分子的化学结构式为:
[0013][0014]
步骤3:将所述n3分子溶于四氢呋喃得到第三反应液,加入贵金属质量含量为10%的pd/c催化剂,在预设条件下与氢气反应直至所述第三反应液不再吸收氢气得到第四反应液,将第四反应液进行第二后处理得到所述苯并对二吡咯分子。
[0015]
优选地,在所述步骤1中,每1.0ml的所述n1分子,需要所述浓硫酸2.0~3.0ml,所述浓硝酸0.6~1.5ml。
[0016]
优选地,在所述步骤1中,所述第一后处理包括:
[0017]
将所述凝固的第一反应液采用去离子水洗涤,除去残留的浓硫酸与浓硝酸,得到固体;
[0018]
将所述固体置于60℃的真空干燥箱中干燥3-6h后进行重结晶得到所述n2分子。
[0019]
优选地,在所述步骤2中,每1.0g的所述n2分子,需要所述n,n-二甲基甲酰胺2.0~4.0ml,所述n,n-二甲基甲酰胺二乙基缩醛3.0~4.0ml。
[0020]
优选地,在所述步骤2中,将所述第二反应液过滤后得到n3分子,所述过滤包括:自然过滤或真空抽滤。
[0021]
优选地,在所述步骤3中,每1.0g的所述n3分子,需要所述四氢呋喃1.0~3.0ml,所述贵金属质量含量为10%的pd/c催化剂0.1~0.5g。
[0022]
优选地,在所述步骤3中,所述预设条件包括:
[0023]
在完全氢气气氛下进行反应,其中,反应温度为25~30℃。
[0024]
优选地,在所述步骤3中,所述第二后处理包括:
[0025]
使用二氯甲烷和石油醚经硅胶层析柱提纯所述第四反应液得到所述苯并对二吡咯分子。
[0026]
与现有技术相比,本技术包括以下优点:
[0027]
(1)现有技术以1,3-二甲苯为初始反应底物,使得反应产物中含有大量1,3-二甲基-2,5-二硝基苯以及未反应完全的1,3-二甲基-4-硝基苯和1,3-二甲基-5-硝基苯,最终所需的反应产物选择性约为60%,且由于副产物与的结构相似性,使其难以被提纯,从而影响下一步反应的产率。本技术以代替1,3-二甲苯作为初始反应物,反应过程中不产生上述副产物,避免上述问题的产生,大幅提升了
的产率,使产率达到95%以上,并使得步骤1的反应成功率达到100%;
[0028]
(2)现有技术选择低压蒸馏的方法除去步骤2中的反应溶剂n,n-二甲基甲酰胺,该方法在应用过程中容易引起产物的喷溅,造成大量浪费且未提纯步骤2的产物,使得步骤3的反应产率较低(未超过20%)。本技术采用自然过滤或真空抽滤的方法除去步骤2中的反应溶剂n,n-二甲基甲酰胺以及溶解在n,n-二甲基甲酰胺中的少量杂质,操作过程中不会引起产物的喷溅,避免了步骤2产物的浪费且能够将步骤2的反应产物提纯,操作简单方便,大幅减少了其他不必要的杂质对步骤3反应产率的影响,可使得最终提纯后产物苯并对二吡咯分子产率稳定在70%以上;
[0029]
(3)现有技术选择室温作为步骤3的反应温度,未指出较为合适的温度范围,使得步骤3的反应产率和反应时间具有不确定性。本技术通过实验认定步骤3的反应温度保持在25~30℃范围内可以使得步骤3的反应在48h内反应完全,且使提纯前的产率达到80%以上,使得最终提纯后产物苯并对二吡咯分子产率稳定在70%以上;
[0030]
(4)步骤3中,现有技术在将步骤2产物溶解在四氢呋喃中,先通入氢气后再在密闭环境中加入pd/c催化剂,该方法操作难度大且易引入空气,会对步骤3的反应造成影响。本技术选择先加入pd/c催化剂再在完全氢气气氛下进行反应,避免了空气的引入同时降低了步骤3反应的操作难度,使得最终提纯后产物苯并对二吡咯分子产率稳定在70%以上;
[0031]
(5)最优化反应物、催化剂及溶剂加入量比,减少反应过程及提纯过程中产物的损失,使得终产物苯并对二吡咯产率稳定在70%以上;
[0032]
(6)本技术提供的合成方法可根据产物需求量进行同比例放大生产,且反应过程稳定可控。
附图说明
[0033]
图1为本技术实施例中反应终产物m1分子的合成方案;
[0034]
图2为本技术实施例中反应终产物m1分子的合成具体路径方案;
[0035]
图3为本技术实施例中步骤1反应产物n2分子的核磁共振氢谱图;
[0036]
图4为本技术实施例中步骤2反应产物n3分子的核磁共振氢谱图;
[0037]
图5为本技术实施例中步骤3反应产物m1分子的核磁共振氢谱图。
具体实施方式
[0038]
为使本技术的上述目的、特征和优点能够更加明显易懂,下面将结合实施例对本技术的优选实施方式进行详细说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0039]
申请人发现现有技术中,如下化学结构式m1所示分子苯并对二吡咯的合成过程中,其面临的出产率低、产量少、效率慢的不足尤为明显;有鉴于此,本技术针对该化学结构式的苯并对二吡咯分子的合成过程进行了优化。
[0040][0041]
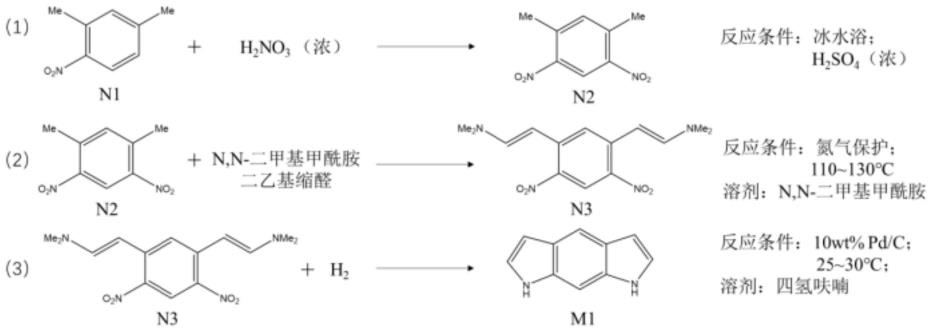
参照图1所示,示出了本技术实施例中反应终产物m1分子的合成方案;如图1所示,本技术提供一种用于储氢的苯并对二吡咯分子的合成方法,所述合成方法以n1分子为初始反应物制备得到苯并对二吡咯分子;
[0042]
其中,所述n1分子的化学结构式为:
[0043][0044]
所述苯并对二吡咯分子的化学结构式为:
[0045][0046]
在本实施例中,本技术以代替1,3-二甲苯作为初始反应物,避免反应过程中产生大量副产物,如1,3-二甲基-2,5-二硝基苯、未反应完全的1,3-二甲基-4-硝基苯和1,3-二甲基-5-硝基苯,大幅提高了反应产物的产率,使产率达到95%以上,并使得步骤1的反应成功率达到100%。且上述副产物与反应产物的结构相似性,使反应产物难以被提纯,将影响下一步反应的产率,本技术选择作为初始反应物,反应过程中不产生上述副产物,避免了上述问题的产生,进一步提高下一步反应的产率。
[0047]
示例地,在冰水浴中,将n1分子溶于浓硫酸,再滴加硝酸至反应液凝固,使用去离子水洗涤至酸液完全去除,固体干燥后重结晶得到n2分子;在氮气保护下,将n2分子溶于n,n-二甲基甲酰胺中,升温至80℃,加入n,n-二甲基甲酰胺二乙基缩醛,然后升温至120℃,反应24h,低压蒸馏除去溶剂得到n3分子混合粗产物;将n3分子粗产物溶于四氢呋喃,通入氢气后再在密闭环境中加入pd/c催化剂,设定反应温度室温,反应完全后,使用二氯甲烷和石油醚经硅胶层析柱提纯得到m1分子。
[0048]
参照图2所示,示出了本技术实施例中反应终产物m1分子的合成具体路径方案;如图2所示,所述合成具体方案包括:
[0049]
步骤1:在冰水浴中,将所述n1分子溶于浓硫酸,再滴加浓硝酸得到凝固的第一反应液,放置5-20min冷却后,进行第一后处理得到n2分子;其中,所述n2分子的化学结构式为:
[0050][0051]
参照图3所示,示出了上述n2分子的核磁共振氢谱图,1h nmr,d6-dmso:8.59(1h),7.67(1h),2.48(6h)。
[0052]
在本实施例中,每1.0ml的n1分子,需要浓硫酸2.0~3.0ml,浓硝酸0.6~1.5ml。当浓硫酸的用量小于2ml时,n1分子无法完全溶解;当浓硫酸的用量为3ml时,n1分子已经溶解完全,无需额外滴加浓硫酸。当浓硝酸的用量小于0.6ml时,反应无法完全;当浓硝酸的用量为1.5ml时,反应已经充分完全,无需额外滴加浓硝酸。
[0053]
其中,所述第一后处理包括:
[0054]
将所述凝固的第一反应液采用去离子水洗涤,除去残留的浓硫酸与浓硝酸,得到固体;
[0055]
将所述固体置于60℃的真空干燥箱中干燥3-6h后进行重结晶得到所述n2分子。
[0056]
在具体实施时,去离子水的用量可视具体情况添加。
[0057]
步骤2:在氮气保护下,将所述n2分子溶于n,n-二甲基甲酰胺中,待温度升至80~100℃,加入n,n-二甲基甲酰胺二乙基缩醛,然后升温至110~130℃,反应20~30h后降至室温得到第二反应液,将所述第二反应液过滤后得到n3分子;其中,所述n3分子的化学结构式为:
[0058][0059]
参照图4所示,示出了上述n3分子的核磁共振氢谱图,1h nmr,d6-dmso:8.52(1h),7.70(d,2h),7.56(1h),5.87(d,2h),2.96(12h)。
[0060]
在本实施例中,将n2分子溶于n,n-二甲基甲酰胺时,需将温度升至80~100℃。当温度低于80℃时,n2分子溶解速度较慢;当温度高于100℃时,由于气体膨胀导致反应器内压力太大,无法继续注入反应物。
[0061]
在加入n,n-二甲基甲酰胺二乙基缩醛后,还需要将温度升至110~130℃。当温度低于110℃时,反应效率较低;当温度高于130℃时,由于气体膨胀导致反应器内压力太大,导致安全隐患。
[0062]
在本实施例中,每1.0g的n2分子,需要n,n-二甲基甲酰胺2.0~4.0ml,所述n,n-二甲基甲酰胺二乙基缩醛3.0~4.0ml。当n,n-二甲基甲酰胺的用量小于2ml时,n2分子溶解速度较慢;当n,n-二甲基甲酰胺的用量为4ml时,n2分子已经溶解完全,无需额外加入n,n-二甲基甲酰胺。当n,n-二甲基甲酰胺二乙基缩醛的用量小于3.0ml时,反应转化率较低;当n,n-二甲基甲酰胺二乙基缩醛的用量为4.0ml时,反应已经充分完全,无需额外加入n,n-二甲
基甲酰胺二乙基缩醛。
[0063]
在本实施例中,选择自然过滤或真空抽滤将第二反应液过滤,操作简单方便。在现有技术中常选择低压蒸馏的方法除去步骤2中的反应溶剂n,n-二甲基甲酰胺以及溶解在n,n-二甲基甲酰胺中的杂质,该方法在应用过程中容易引起产物的喷溅,造成大量浪费且未提纯步骤2的产物,使得步骤3的反应产率较低(未超过20%)。本技术选择自然过滤或真空抽滤代替低压蒸馏除去步骤2中的反应溶剂n,n-二甲基甲酰胺以及溶解在n,n-二甲基甲酰胺中的少量杂质,操作过程中不会引起产物的喷溅,既能避免步骤2产物的浪费,还能将其进行提纯,大幅减少了其他不必要的杂质对步骤3反应产率的影响,可使得最终提纯后产物苯并对二吡咯分子产率稳定在70%以上。
[0064]
步骤3:将所述n3分子溶于四氢呋喃得到第三反应液,加入贵金属质量含量为10%的pd/c催化剂,在预设条件下与氢气反应直至所述第三反应液不再吸收氢气得到第四反应液,将第四反应液进行第二后处理得到所述苯并对二吡咯分子。
[0065]
参照图5所示,示出了上述苯并对二吡咯分子的核磁共振氢谱图,1h nmr,d6-dmso:10.49(2h),7.57(1h),7.28(1h),7.18(2h),6.33(2h)。
[0066]
在本实施例中,每1.0g的n3分子,需要四氢呋喃1.0~3.0ml,贵金属质量含量为10%的pd/c催化剂0.1~0.5g。当四氢呋喃的用量小于1ml时,n3分子无法完全溶解;当四氢呋喃的用量为3ml时,n3分子已经溶解完全,无需额外加入四氢呋喃。当pd/c催化剂的用量小于0.1g时,反应效率较低;当pd/c催化剂的用量为0.5g时,反应效率已满足当前反应的需求,无需额外加入pd/c催化剂。
[0067]
其中,所述预设条件包括:
[0068]
在完全氢气气氛下进行反应,其中,反应温度为25~30℃。
[0069]
在本实施例中,当反应温度低于25℃时,反应的效率太低,且经申请人通过实验认定所述反应温度保持在25~30℃范围内即可使得步骤3的反应在48h内反应完全,且使提纯前的产率达到80%以上,使得最终提纯后产物苯并对二吡咯分子产率稳定在70%以上。
[0070]
所述第二后处理包括:
[0071]
使用二氯甲烷和石油醚经硅胶层析柱提纯所述第四反应液得到所述苯并对二吡咯分子。
[0072]
在本实施例中,在得到第三反应液后,先加入pd/c催化剂再通入氢气进行反应,不同于现有技术中先通入氢气后再在密闭环境中加入pd/c催化剂,降低了操作难度,避免空气进入,影响步骤3的反应。
[0073]
在具体实施时,在通入氢气前还可以先用氩气或氮气置换空气,确保反应环境中没有空气残留。
[0074]
在符合本领域常识的基础上,上述各优选条件,可以相互组合,得到具体实施方式。
[0075]
实施例1
[0076]
步骤1:在冰水浴中,将10.0mln1分子溶于20.0ml浓硫酸,再滴加6.0ml浓硝酸得到凝固的第一反应液,放置10min冷却后,将凝固的第一反应液采用去离子水洗涤,除去残留的浓硫酸与浓硝酸,得到固体,将固体置于60℃的真空干燥箱中干燥6h后进行重结晶得到约13.8g n2分子。
[0077]
步骤2:在氮气保护下,将13.8gn2分子溶于27.6ml n,n-二甲基甲酰胺中,待温度升至80℃,加入41.4ml n,n-二甲基甲酰胺二乙基缩醛,然后升温至110℃,反应20h后降至室温得到第二反应液,将第二反应液进行自然过滤后得到约17.7gn3分子。
[0078]
步骤3:将17.7gn3分子溶于17.7ml四氢呋喃得到第三反应液,加入贵金属质量含量为10%的pd/c催化剂1.77g,设定反应温度为25℃,在完全氢气气氛下反应直至第三反应液不再吸收氢气得到第四反应液,使用二氯甲烷和石油醚经硅胶层析柱提纯第四反应液得到苯并对二吡咯分子约7.5g。
[0079]
实施例2
[0080]
步骤1:在冰水浴中,将5.0mln1分子溶于12.0ml浓硫酸,再滴加5.0ml浓硝酸得到凝固的第一反应液,放置10min冷却后,将凝固的第一反应液采用去离子水洗涤,除去残留的浓硫酸与浓硝酸,得到固体,将固体置于60℃的真空干燥箱中干燥4h后进行重结晶得到约7.1g n2分子。
[0081]
步骤2:在氮气保护下,将7.1gn2分子溶于20.0ml n,n-二甲基甲酰胺中,待温度升至85℃,加入22.0ml n,n-二甲基甲酰胺二乙基缩醛,然后升温至115℃,反应24h后降至室温得到第二反应液,将第二反应液进行自然过滤后得到约10.0gn3分子。
[0082]
步骤3:将10.0gn3分子溶于15.0ml四氢呋喃得到第三反应液,加入贵金属质量含量为10%的pd/c催化剂2.2g,设定反应温度为26℃,在完全氢气气氛下反应直至第三反应液不再吸收氢气得到第四反应液,使用二氯甲烷和石油醚经硅胶层析柱提纯第四反应液得到苯并对二吡咯分子约4.2g。
[0083]
实施例3
[0084]
步骤1:在冰水浴中,将1.2mln1分子溶于3.0ml浓硫酸,再滴加1.5ml浓硝酸得到凝固的第一反应液,放置5min冷却后,将凝固的第一反应液采用去离子水洗涤,除去残留的浓硫酸与浓硝酸,得到固体,将固体置于60℃的真空干燥箱中干燥3h后进行重结晶得到约1.67g n2分子。
[0085]
步骤2:在氮气保护下,将1.67gn2分子溶于5.0ml n,n-二甲基甲酰胺中,待温度升至90℃,加入5.8ml n,n-二甲基甲酰胺二乙基缩醛,然后升温至120℃,反应20h后降至室温得到第二反应液,将第二反应液进行自然过滤后得到约2.43gn3分子。
[0086]
步骤3:将2.43gn3分子溶于5.0ml四氢呋喃得到第三反应液,加入贵金属质量含量为10%的pd/c催化剂0.6g,设定反应温度为25℃,在完全氢气气氛下反应直至第三反应液不再吸收氢气得到第四反应液,使用二氯甲烷和石油醚经硅胶层析柱提纯第四反应液得到苯并对二吡咯分子约1.3g。
[0087]
实施例4
[0088]
步骤1:在冰水浴中,将25.0mln1分子溶于3.0ml浓硫酸,再滴加55.0ml浓硝酸得到凝固的第一反应液,放置20min冷却后,将凝固的第一反应液采用去离子水洗涤,除去残留的浓硫酸与浓硝酸,得到固体,将固体置于60℃的真空干燥箱中干燥6h后进行重结晶得到约35.51g n2分子。
[0089]
步骤2:在氮气保护下,将35.51gn2分子溶于90.0ml n,n-二甲基甲酰胺中,待温度升至95℃,加入120.0ml n,n-二甲基甲酰胺二乙基缩醛,然后升温至125℃,反应30h后降至室温得到第二反应液,将第二反应液进行真空抽滤后得到约49.9gn3分子。
[0090]
步骤3:将49.9gn3分子溶于80.0ml四氢呋喃得到第三反应液,加入贵金属质量含量为10%的pd/c催化剂15.5g,设定反应温度为28℃,在完全氢气气氛下反应直至第三反应液不再吸收氢气得到第四反应液,使用二氯甲烷和石油醚经硅胶层析柱提纯第四反应液得到苯并对二吡咯分子约20.6g。
[0091]
实施例5
[0092]
步骤1:在冰水浴中,将8.0mln1分子溶于18.0ml浓硫酸,再滴加10.0ml浓硝酸得到凝固的第一反应液,放置10min冷却后,将凝固的第一反应液采用去离子水洗涤,除去残留的浓硫酸与浓硝酸,得到固体,将固体置于60℃的真空干燥箱中干燥4h后进行重结晶得到约11.41g n2分子。
[0093]
步骤2:在氮气保护下,将11.4gn2分子溶于35.0ml n,n-二甲基甲酰胺中,待温度升至85℃,加入40.0ml n,n-二甲基甲酰胺二乙基缩醛,然后升温至125℃,反应28h后降至室温得到第二反应液,将第二反应液进行真空抽滤后得到约16.4gn3分子。
[0094]
步骤3:将16.4gn3分子溶于26.0ml四氢呋喃得到第三反应液,加入贵金属质量含量为10%的pd/c催化剂2.5g,设定反应温度为30℃,在完全氢气气氛下反应直至第三反应液不再吸收氢气得到第四反应液,使用二氯甲烷和石油醚经硅胶层析柱提纯第四反应液得到苯并对二吡咯分子约7.0g。
[0095]
实施例6
[0096]
步骤1:在冰水浴中,将13.0mln1分子溶于32.0ml浓硫酸,再滴加15.0ml浓硝酸得到凝固的第一反应液,放置10min冷却后,将凝固的第一反应液采用去离子水洗涤,除去残留的浓硫酸与浓硝酸,得到固体,将固体置于60℃的真空干燥箱中干燥4h后进行重结晶得到约18.4g n2分子。
[0097]
步骤2:在氮气保护下,将18.4gn2分子溶于65.0ml n,n-二甲基甲酰胺中,待温度升至90℃,加入70.0ml n,n-二甲基甲酰胺二乙基缩醛,然后升温至115℃,反应24h后降至室温得到第二反应液,将第二反应液进行真空抽滤后得到约25.5gn3分子。
[0098]
步骤3:将25.5gn3分子溶于55.0ml四氢呋喃得到第三反应液,加入贵金属质量含量为10%的pd/c催化剂10.0g,设定反应温度为27℃,在完全氢气气氛下反应直至第三反应液不再吸收氢气得到第四反应液,使用二氯甲烷和石油醚经硅胶层析柱提纯第四反应液得到苯并对二吡咯分子约10.4g。
[0099]
实施例7
[0100]
步骤1:在冰水浴中,将10.0mln1分子溶于30.0ml浓硫酸,再滴加15.0ml浓硝酸得到凝固的第一反应液,放置10min冷却后,将凝固的第一反应液采用去离子水洗涤,除去残留的浓硫酸与浓硝酸,得到固体,将固体置于60℃的真空干燥箱中干燥6h后进行重结晶得到约14.4g n2分子。
[0101]
步骤2:在氮气保护下,将14.4gn2分子溶于57.6ml n,n-二甲基甲酰胺中,待温度升至100℃,加入57.6ml n,n-二甲基甲酰胺二乙基缩醛,然后升温至130℃,反应30h后降至室温得到第二反应液,将第二反应液进行真空抽滤后得到约20.7gn3分子。
[0102]
步骤3:将20.7gn3分子溶于62.1ml四氢呋喃得到第三反应液,加入贵金属质量含量为10%的pd/c催化剂10.35g,设定反应温度为30℃,在完全氢气气氛下反应直至第三反应液不再吸收氢气得到第四反应液,使用二氯甲烷和石油醚经硅胶层析柱提纯第四反应液
得到苯并对二吡咯分子约9.3g。
[0103]
对于方法实施例,为了简单描述,故将其都表述为一系列的动作组合,但是本领域技术人员应该知悉,本技术并不受所描述的动作顺序的限制,因为依据本技术,某些步骤可以采用其他顺序或者同时进行。其次,本领域技术人员也应该知悉,说明书中所描述的实施例均属于优选实施例,所涉及的动作和部件并不一定是本技术所必须的。
[0104]
以上对本技术所提供的一种用于储氢的苯并对二吡咯分子的合成方法,进行了详细介绍,本文中应用了具体个例对本技术的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本技术的方法及其核心思想;同时,对于本领域的一般技术人员,依据本技术的思想,在具体实施方式及应用范围上均会有改变之处,综上所述,本说明书内容不应理解为对本技术的限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1