发光材料、有机发光元件及化合物的制作方法
本发明涉及一种发光效率高的有机发光元件。另外,本发明涉及一种能够有效地用于该有机发光元件的发光材料和化合物。
背景技术:
业界正积极地进行提高有机电致发光元件(有机EL(Electroluminescence,电致发光)元件)等有机发光元件的发光效率的研究。尤其正进行通过新开发出构成有机电致发光元件的发光材料或主体材料、电子传输材料、电洞传输材料等并加以组合而提高发光效率的各种钻研。其中,也可见和利用如下化合物的有机电致发光元件相关的研究,该化合物具有(N,N-二芳基氨基)芳基在吡口井环上进行取代而成的结构。
例如,在专利文献1中,记载了使用下述通式所表示的聚氮杂并苯化合物作为有机发光元件的发光材料。下述通式中的R1~R4表示氢原子或经取代或者未经取代的(N,N-二芳基氨基)芳基,R1~R4的至少1个为经取代或者未经取代的(N,N-二芳基氨基)芳基,W1、W2、X1、X2、Y1、Y2、Z1及Z2表示碳原子或氮原子。然而,在专利文献1中,未记载R1~R4为(N,N-二芳基氨基)芳基以外的取代基的化合物。
[化1]
另外,在专利文献2中,记载了使用下述通式所表示的吡口井衍生物作为构成发光元件的发光层的主体材料或发光性材料。下述通式中的R1~R3表示氢原子、烷基、或芳基的任一种,A表示通式(a-1)~(a-4)所表示的取代基的任一种,R4表示烷基、或芳基,R5~R7表示氢原子、烷基、或芳基的任一种,Ar1~Ar7表示芳基,α表示亚芳基。另外,作为具体例,记载了使用A为N,N-二苯基氨基、R3为4-(N,N-二苯基氨基)苯基、R1、R2为苯基的吡口井衍生物作为磷光材料的主体材料的例子。然而,专利文献2中所记载的化合物均为中心骨架包含单环的吡口井环的吡口井衍生物,在同一文献中,未记载具有多个吡口井环进行融合而成的结构的中心骨架(聚氮杂并苯骨架)的化合物。
[化2]
[背景技术文献]
[专利文献]
[专利文献1]日本专利特开2014-9352号公报
[专利文献2]日本专利第5227510号公报
技术实现要素:
[发明要解决的问题]
如上所述,在专利文献1中记载了经(N,N-二芳基氨基)芳基取代而成的聚氮杂并苯化合物能够用作发光材料。然而,专利文献1中所记载的化合物在聚氮杂并苯骨架上进行取代的取代基均为(N,N-二芳基氨基)芳基,在同一文献中,未对其它取代基在聚氮杂并苯骨架上进行取代而成的化合物进行记载。
相对于此,本发明者等人对具有(N,N-二芳基氨基)芳基在聚氮杂并苯骨架上进行取代而成的结构的化合物群开始各种研究,结果首次发现,具有(N,N-二芳基氨基)芳基和不具有N,N-二芳基氨基的芳基在聚氮杂并苯骨架上进行取代而成的结构的化合物群作为发光材料有高有用性,并进一步进行研究。如上所述,关于(N,N-二芳基氨基)芳基在聚氮杂并苯骨架上进行取代而成的化合物,在专利文献1中记载了能够用作有机发光元件的发光材料。然而,在同一文献中,对于(N,N-二芳基氨基)芳基和不具有N,N-二芳基氨基的芳基在聚氮杂并苯骨架上进行取代而成的化合物未进行研究。另一方面,在专利文献2中,记载了4-(N,N-二苯基氨基)苯基和不具有取代基的苯基在单环的吡口井环上进行取代而成的化合物。然而,在同一文献中,未记载具有多个吡口井环进行融合而成的聚氮杂并苯骨架的化合物。因此,完全未预测(N,N-二芳基氨基)芳基和不具有N,N-二芳基氨基的芳基在聚氮杂并苯骨架上进行取代而成的化合物作为发光材料的有用性。
在这种情况下,本发明者等人针对(N,N-二芳基氨基)芳基和不具有N,N-二芳基氨基的芳基进行取代而成的聚氮杂并苯化合物,为了评价其作为有机发光元件的发光材料的有用性而进行研究。另外,也为了导出能够用作发光材料的化合物的通式,使发光效率高的有机发光元件的构成概括化而进行努力研究。
[解决问题的技术手段]
本发明者等人为了达成所述目的而进行努力研究,结果明确,具有特定的结构的聚氮杂并苯化合物作为有机发光元件的发光材料而极有用。并且,发现这种聚氮杂并苯化合物中有能够用作延迟荧光材料的化合物,且明确能够提供一种发光效率高的有机发光元件。本发明者等人基于这些见解,提供以下的本发明作为解决所述课题的方法。
[1]一种发光材料,其包含下述通式(1)所表示的化合物。
[化3]
通式(1)
[在通式(1)中,R1~R5分别独立表示氢原子或哈米特的σp值为0以上的取代基;R6~R20分别独立表示氢原子或者取代基,R6~R20的至少一个为经取代或者未经取代的N,N-二芳基氨基;m表示1或2]
[2]根据[1]所述的发光材料,其特征在于:所述哈米特的σp值为0以上的取代基为卤素原子、酰氧基、烷氧基羰基、芳氧基羰基、苯基或氰基。
[3]根据[1]或[2]所述的发光材料,其特征在于:所述经取代或者未经取代的N,N-二芳基氨基为下述通式(2)所表示的基。
[化4]
通式(2)
[在通式(2)中,Ar1及Ar2分别独立表示碳数6~10的经取代或者未经取代的芳香族基;﹡表示键结部位;在通式(1)所表示的化合物中存在多个通式(2)所表示的基的情况下,Ar1及Ar2所表示的基可以相同也可以不同]
[4]根据[3]所述的发光材料,其特征在于:所述Ar1及Ar2直接或者间接连结而形成环。
[5]根据[4]所述的发光材料,其特征在于:所述通式(2)所表示的基是由下述通式(3)所表示。
[化5]
通式(3)
[在通式(3)中,Ra及Rb分别独立表示氢原子、经取代或者未经取代的碳数1~5的烷基、或经取代或者未经取代的碳数6~10的芳香族基;﹡表示键结部位;另外,在通式(1)所表示的化合物中存在多个通式(3)所表示的基的情况下,Ra及Rb所表示的基可以相同也可以不同]
[6]根据[1]至[5]中任一项所述的发光材料,其特征在于:在所述通式(1)中,m为1。
[7]根据[1]至[6]中任一项所述的发光材料,其特征在于:其发射延迟荧光。
[8]一种化合物,其是由下述通式(11)所表示。
[化6]
通式(11)
[在通式(11)中,R1'~R5'分别独立表示氢原子或哈米特的σp值为0以上的取代基;R6'~R20'分别独立表示氢原子或者取代基,R6'~R20'的至少一个为经取代或者未经取代的N,N-二芳基氨基;m'表示1或2]
[9]根据[8]所述的化合物,其特征在于:通式(11)所表示的化合物是由下述通式(12)所表示。
[化7]
通式(12)
[在通式(12)中,R1a~R5a及R16a~R19a分别独立表示氢原子或哈米特的σp值为0以上的取代基;R6a~R15a分别独立表示氢原子、哈米特的σp值为0以上的取代基或经取代或者未经取代的N,N-二芳基氨基;n表示1或2;Z表示用以形成6员环或者7员环的包含碳链的连结基、或用以形成6员环的氧原子]
[10]根据[9]所述的化合物,其特征在于:所述R1a~R5a分别独立为氢原子或氟原子。
[11]一种有机发光元件,其特征在于:在基板上具有包含根据所述[1]至[7]中任一项所述的发光材料的发光层。
[12]根据[11]所述的有机发光元件,其特征在于:其发射延迟荧光。
[13]根据[11]或[12]所述的有机发光元件,其特征在于:其为有机电致发光元件。
[发明效果]
本发明的有机发光元件具有发光效率高的特征。另外,本发明的化合物或发光材料具有在用作有机发光元件的发光层时能够提高发光效率的特征。其中,如果使用发射延迟荧光的本发明的化合物或发光材料,那么能够飞跃性地提高发光效率。
附图说明
图1是表示有机电致发光元件的层构成例的概略剖面图。
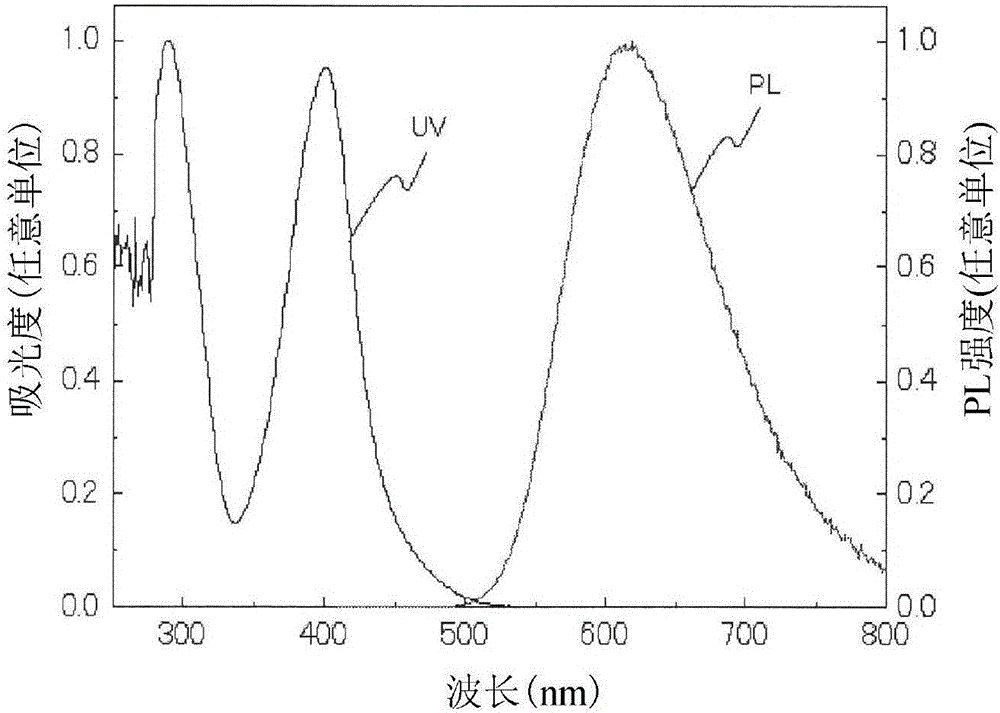
图2是实施例1的例示化合物(1)的甲苯溶液的发光光谱和吸收光谱。
图3是实施例1的例示化合物(1)的甲苯溶液的暂态衰减曲线。
图4是实施例2的例示化合物(2)的甲苯溶液的发光光谱和吸收光谱。
图5是实施例2的例示化合物(2)的甲苯溶液的暂态衰减曲线。
图6是实施例3的例示化合物(2)的有机光致发光元件的发光光谱和吸收光谱。
图7是实施例3的例示化合物(2)的有机光致发光元件的暂态衰减曲线。
图8是实施例4的例示化合物(2)的有机电致发光元件的发光光谱。
图9是表示实施例4的例示化合物(2)的有机电致发光元件的电压-电流密度-亮度特性的曲线图。
图10是表示实施例4的例示化合物(2)的有机电致发光元件的电流密度-电流效率-功率效率特性的曲线图。
图11是表示实施例4的例示化合物(2)的有机电致发光元件的电流密度-外部量子效率特性的曲线图。
具体实施方式
以下,对本发明的内容详细地进行说明。以下所记载的构成要素的说明存在基于本发明的代表性实施方式或具体例而进行的情况,但本发明并不限定于这种实施方式或具体例。另外,在本说明书中,使用“~”表示的数值范围意指包含“~”的前后所记载的数值作为下限值及上限值的范围。另外,本发明中所使用的化合物的分子内所存在的氢原子的同位素种类并无特别限定,例如,分子内的氢原子可以全部为1H,也可以一部分或全部为2H(氘D)。
[通式(1)所表示的化合物]
本发明的发光材料的特征在于包含下述通式(1)所表示的化合物。
[化8]
通式(1)
在通式(1)中,R1~R5分别独立表示氢原子或哈米特的σp值为0以上的取代基。所谓哈米特的σp值,是依据下述式(I)而算出的值。所谓该σp值为0以上,是指其取代基为吸电子性的取代基。
[数1]
式(I)
σp=logKX-logKH
[在式(I)中,KH为苯甲酸在25℃的水中的离子化常数,KX是取代基在p位进行取代而成的苯甲酸在25℃的水中的离子化常数]
作为哈米特的σp值为0以上的取代基,例如可以列举:卤素原子、酰氧基、烷氧基羰基、芳氧基羰基、苯基或氰基,其中优选卤素原子、氰基,更优选卤素原子。R1~R5中的取代基的数量并无特别限制,也可以全部未经取代(即氢原子)。另外,在R1~R5中的2个以上为取代基的情况下,多个取代基可以相互相同也可以不同。
R6~R20分别独立表示氢原子或者取代基,R6~R20的至少一个为经取代或者未经取代的N,N-二芳基氨基。表示经取代或者未经取代的N,N-二芳基氨基的只要为R6~R20中的1个以上即可,也可以为多个,R6~R10、R11~R15、R16~R20各者中的经取代或者未经取代的N,N-二芳基氨基的数量的上限优选2个,更优选1个。R6~R20中的表示经取代或者未经取代的N,N-二芳基氨基的优选R7~R9、R12~R14、R17~R19中的1~3个,更优选R7、R8、R12、R13、R17、R18中的1~3个,进而优选R8、R13、R18中的1~3个,特别优选R13及R18的至少任一个,但并无特别限制。
优选R6~R20能够采用的经取代或者未经取代的N,N-二芳基氨基为下述通式(2)所表示的基。
[化9]
通式(2)
在通式(2)中,﹡表示键结部位。Ar1及Ar2分别独立表示碳数6~10的经取代或者未经取代的芳香族基。这里所述的芳香族基可以包含单环,也可以包含稠环。例如可以列举苯基、萘基作为优选例,更优选苯基。作为具体例,可以列举:苯基、1-萘基、2-萘基。Ar1和Ar2可以相同也可以不同。另外,在分子内存在多个通式(2)所表示的经取代或者未经取代的N,N-二芳基氨基的情况下,这些经取代或者未经取代的N,N-二芳基氨基的Ar1彼此可以相互相同也可以不同,另外,这些经取代或者未经取代的N,N-二芳基氨基的Ar2彼此可以相互相同也可以不同。
在通式(2)中,Ar1及Ar2也可以相互连结而形成环状结构。在相互连结而形成环状结构时,Ar1和Ar2也可以直接或者间接进行连结而形成环。也就是说,构成Ar1的芳香族基和构成Ar2的芳香族基相互可以利用单键进行连结,也可以利用连结基进行连结。在利用连结基进行连结时,连结基的连结原子数优选1~3个,更优选1或2个。在连结原子数为2个以上时,在连结原子之间可以存在不饱和键也可以不存在不饱和键,优选存在不饱和键。例如可以列举下述式(4)所表示的连结基作为优选例。在下述式(4)中,R21及R22分别独立表示氢原子或取代基,R21及R22也可以相互键结而形成环状结构。作为环状结构,可以列举:苯环或萘环等芳基环、吡啶环或吡口井环等杂芳基环、环戊二烯环或环己烯环等不饱和脂肪环。
[化10]
通式(4)
作为通式(2)所表示的N,N-二芳基氨基的具体例,可以列举:N,N-二苯基氨基、N-苯基-N-(1-萘基)氨基、N-苯基-N-(2-萘基)氨基、N,N-二(1-萘基)氨基、N,N-二(2-萘基)氨基、咔唑-9-基、5H-二苯并[b,f]氮呯-5-基。其中,优选N,N-二苯基氨基、N-苯基-N-(1-萘基)氨基、咔唑-9-基、5H-二苯并[b,f]氮呯-5-基。这里所例示的N,N-二芳基氨基也可以经取代。
另外,通式(2)所表示的N,N-二芳基氨基也优选下述通式(3)所表示的基。
[化11]
通式(3)
在通式(3)中,﹡表示键结部位。Ra及Rb分别独立表示氢原子、经取代或者未经取代的碳数1~5的烷基、或经取代或者未经取代的碳数6~10的芳香族基。关于Ra及Rb能够采用的烷基,只要碳数为1~5,那么并无特别限制,优选甲基。关于Ra及Rb能够采用的芳香族基的说明和优选的范围,可以参照Ar1及Ar2能够采用的芳香族基的说明和优选的范围。另外,Ra和Rb可以相互相同也可以不同。另外,在通式(1)所表示的化合物中存在多个通式(3)所表示的基的情况下,这些Ra彼此可以相互相同也可以不同,另外,这些Rb彼此可以相互相同也可以不同。
作为通式(2)的Ar1及Ar2可以采用的取代芳香族基的取代基、通式(3)的Ra及Rb可以采用的取代烷基及取代芳香族基的取代基、通式(4)的R21及R22可以采用的取代基,例如可以列举:羟基、卤素原子、氰基、碳数1~20的烷基、碳数1~20的烷氧基、碳数6~40的经取代或者未经取代的芳氧基、碳数1~20的烷基硫基、碳数1~20的烷基取代氨基、碳数2~20的酰基、碳数3~40的杂芳基、碳数12~40的二芳基氨基、碳数12~40的经取代或者未经取代的咔唑基、碳数2~10的烯基、碳数2~10的炔基、碳数2~10的烷氧基羰基、碳数1~10的烷基磺酰基、碳数1~10的卤烷基、酰胺基、碳数2~10的烷基酰胺基、碳数3~20的三烷基硅烷基、碳数4~20的三烷基硅烷基烷基、碳数5~20的三烷基硅烷基烯基、碳数5~20的三烷基硅烷基炔基及硝基等。这些具体例中,可以进一步经取代基取代的取代基也可以经取代。更优选的取代基为卤素原子、氰基、碳数1~20的经取代或者未经取代的烷基、碳数1~20的烷氧基、碳数6~40的经取代或者未经取代的芳氧基、碳数3~40的经取代或者未经取代的杂芳基、碳数12~40的经取代或者未经取代的二芳基氨基、碳数12~40的经取代或者未经取代的咔唑基。进而优选的取代基为碳数1~10的经取代或者未经取代的烷基、碳数6~40的经取代或者未经取代的芳氧基、碳数12~40的经取代或者未经取代的二芳基氨基。
本说明书中所述的烷基可以为直链状、支链状、环状的任一种,更优选碳数1~6,作为具体例,可以列举:甲基、乙基、丙基、丁基、叔丁基、戊基、己基、异丙基。烷氧基可以为直链状、支链状、环状的任一种,更优选碳数1~6,作为具体例,可以列举:甲氧基、乙氧基、丙氧基、丁氧基、叔丁氧基、戊氧基、己氧基、异丙氧基。二烷基氨基的2个烷基可以相互相同也可以不同,优选相同。二烷基氨基的2个烷基可以分别独立为直链状、支链状、环状的任一种,更优选碳数1~6,作为具体例,可以列举:甲基、乙基、丙基、丁基、戊基、己基、异丙基。杂芳基可以仅包含单环,也可以包含稠环,作为具体例,可以列举:吡啶基、嗒口井基、嘧啶基、三口井基、三唑基、苯并三唑基。这些杂芳基可以为经由杂原子而键结的基,也可以为经由构成杂芳基环的碳原子而键结的基。
另外,在通式(1)中,m表示1或2,优选1。
以下,例示通式(1)所表示的化合物的具体例,但本发明中能够使用的通式(1)所表示的化合物不应受到这些具体例的限定性解释。
[化12]
[化13]
[化14]
[化15]
[化16]
关于通式(1)所表示的化合物的分子量,例如在意图利用蒸镀法将包含通式(1)所表示的化合物的有机层制膜并加以利用的情况下,优选1500以下,更优选1200以下,进而优选1000以下,进而更优选800以下。分子量的下限值为通式(1)所表示的分子量最小的化合物的分子量。
通式(1)所表示的化合物无论分子量如何,均能够利用涂布法进行成膜。如果使用涂布法,那么即便为分子量相对大的化合物也能够进行成膜。
也可以想到应用本发明而将分子内包含多个通式(1)所表示的结构的化合物用于有机发光元件的发光层中。
例如可以想到将使具有通式(1)所表示的结构的聚合性单体进行聚合而成的聚合物用于有机发光元件的发光层中。具体而言,可以想到准备在通式(1)的R1~R20的任一个中具有聚合性官能基的单体,使其均聚合、或和其它单体一起共聚合,由此获得具有重复单元的聚合物,并将该聚合物用于有机发光元件的发光层中。或者,也可以想到通过使具有通式(1)所表示的结构的化合物彼此反应而获得二聚物或三聚物,并将这些用于有机发光元件的发光层中。
作为构成包含通式(1)所表示的结构的聚合物的重复单元的结构例,可以列举通式(1)的R1~R20的任一个为下述通式(21)或(22)所表示的结构的重复单元。
[化17]
在通式(21)及(22)中,L1及L2表示连结基。连结基的碳数优选0~20,更优选1~15,进而优选2~10。连结基优选具有-X11-L11-所表示的结构。这里,X11表示氧原子或硫原子,优选氧原子。L11表示连结基,优选经取代或者未经取代的亚烷基、或经取代或者未经取代的亚芳基,更优选碳数1~10的经取代或者未经取代的亚烷基、或经取代或者未经取代的亚苯基。
在通式(21)及(22)中,R101、R102、R103及R104分别独立表示取代基。优选碳数1~6的经取代或者未经取代的烷基、碳数1~6的经取代或者未经取代的烷氧基、卤素原子,更优选碳数1~3的未经取代的烷基、碳数1~3的未经取代的烷氧基、氟原子、氯原子,进而优选碳数1~3的未经取代的烷基、碳数1~3的未经取代的烷氧基。
作为重复单元的具体的结构例,可以列举在通式(1)的R1~R20的任一个中包含下述式(23)~(26)的任一种结构的重复单元。也可以R1~R20中的2个以上包含下述式(23)~(26)的任一种结构,优选的情况为R1~R20中的1个包含下述式(23)~(26)的任一种结构。
[化18]
具有包含这些式(23)~(26)的重复单元的聚合物能够通过预先将通式(1)的R1~R20的至少1个设为包含羟基的基,将其作为连结子使下述化合物反应而导入聚合性基,并使该聚合性基进行聚合而合成。
[化19]
分子内包含通式(1)所表示的结构的聚合物可以为仅包含具有通式(1)所表示的结构的重复单元的聚合物,也可以为包含具有其以外的结构的重复单元的聚合物。另外,聚合物中所含的具有通式(1)所表示的结构的重复单元可以为单独一种,也可以为两种以上。
作为不具有通式(1)所表示的结构的重复单元,可以列举由通常的共聚合中所使用的单体衍生的重复单元。例如可以列举由乙烯、苯乙烯等具有乙烯性不饱和键的单体衍生的重复单元。
[通式(11)所表示的化合物]
在通式(1)所表示的化合物中,下述通式(11)所表示的化合物为新颖化合物。
[化20]
通式(11)
在通式(11)中,R1'~R5'分别独立表示氢原子或哈米特的σp值为0以上的取代基。R6'~R20'分别独立表示氢原子或者取代基,R6'~R20'的至少一个为经取代或者未经取代的N,N-二芳基氨基。m'表示1或2。
关于通式(11)中的R1'~R5'、R6'~R20'、m'的说明和优选的范围,可以参照通式(1)所表示的化合物的说明。
另外,通式(11)所表示的化合物优选下述通式(12)所表示的化合物。
[化21]
通式(12)
在通式(12)中,R1a~R5a及R16a~R19a分别独立表示氢原子或哈米特的σp值为0以上的取代基。R6a~R15a分别独立表示氢原子、哈米特的σp值为0以上的取代基或经取代或者未经取代的N,N-二芳基氨基。n表示1或2。Z表示用以形成6员环或者7员环的包含碳链的连结基、或用以形成6员环的氧原子。
关于R1a~R5a及R16a~R19a可以采用的取代基的说明和优选例,可以参照通式(1)的R1~R5可以采用的取代基的说明和优选例,其中R1a~R5a所采用的取代基优选氟原子。关于R6a~R15a可以采用的N,N-二芳基氨基的说明和优选的范围,可以参照通式(1)的R6~R20可以采用的N,N-二芳基氨基的说明和优选的范围。n为1或2,优选1。在Z为碳链时,碳链的碳数为1或2。在碳数为2时,在碳原子之间可以存在不饱和键。另外,碳链也可以经取代基取代。关于该取代基的说明和优选的范围,可以参照通式(2)的Ar1及Ar2可以采用的取代芳香族基等取代基的说明和优选的范围。
[通式(11)所表示的化合物的合成法]
通式(11)所表示的化合物的合成法并无特别限制。通式(11)所表示的化合物的合成可以通过将已知的合成法或条件适当组合而进行。
例如,R18'为通式(3)所表示的基、且m为1的通式(11)所表示的化合物可以根据下述流程进行合成。
[化22]
流程中的R1'~R17'、R19'、R20'是和所述通式(11)含义相同的基。Ra及Rb是和所述通式(3)含义相同的基。X表示卤素原子。作为卤素原子,可以列举:氟原子、氯原子、溴原子、碘原子,优选氟原子、氯原子、溴原子。关于各步骤的反应条件或程序,可以适当选择采用在类似的合成反应中所采用的公知的反应条件或程序。
R18'为通式(3)所表示的基、且m为1以外的通式(11)所表示的化合物能够通过设置所述流程而进行合成。另外,关于通式(11)所表示的化合物的合成反应的详细情况,可以将下述合成例作为参考。另外,通式(11)所表示的化合物也能够通过将其它公知的合成反应组合而进行合成。
[有机发光元件]
本发明的通式(1)所表示的化合物能够用作有机发光元件的发光材料。因此,本发明的通式(1)所表示的化合物在有机发光元件的发光层中能够有效地用作发光材料。在通式(1)所表示的化合物中,含有发射延迟荧光的延迟荧光材料(延迟荧光体)。也就是说,本发明也提供具有通式(1)所表示的结构的延迟荧光体的发明、使用通式(1)所表示的化合物作为延迟荧光体的发明、及使用通式(1)所表示的化合物使延迟荧光发光的方法的发明。使用这种化合物作为发光材料的有机发光元件具有发射延迟荧光、且发光效率高的特征。如果以有机电致发光元件为例对其原理进行说明,那么如下所述。
在有机电致发光元件中,从正负两电极在发光材料中注入载子,产生激发状态的发光材料,使其发光。通常,在载子注入型的有机电致发光元件的情况下,所产生的激子中,被激发成激发单重态的激子为25%,剩余75%被激发成激发三重态。因此,利用作为来自激发三重态的发光的磷光的有机电致发光元件的能量利用效率高。然而,激发三重态由于寿命长,所以产生激发状态的饱和或因和激发三重态的激子的相互作用所引起的能量失活,通常大多情况下磷光的量子产率不高。另一方面,延迟荧光材料通过系间窜越等而使能量向激发三重态跃迁后,通过三重态-三重态湮灭或热能的吸收,反向系间窜越(inverse intersystem crossing)至激发单重态而发射荧光。在有机电致发光元件中,认为其中利用热能的吸收的热活化型的延迟荧光材料尤其有用。在有机电致发光元件利用延迟荧光材料的情况下,激发单重态的激子像通常那样发射荧光。另一方面,激发三重态的激子吸收器件所发出的热,向激发单重态进行系间窜越而发射荧光。这时,由于为来自激发单重态的发光,所以为和荧光相同波长的发光,但因通过从激发三重态向激发单重态的反向系间窜越,所产生的光的寿命(发光寿命)较通常的荧光或磷光长,所以作为较这些延迟的荧光而被观察到。能够将其定义为延迟荧光。如果使用这种热活化型的激子移动机制,那么通过在载子注入后经过热能的吸收,能够将通常仅产生25%的激发单重态的化合物的比率提高至25%以上。如果使用在未达100℃的低温下也发出强荧光及延迟荧光的化合物,那么利用器件的热而充分地产生从激发三重态向激发单重态的系间窜越而发射延迟荧光,所以能够飞跃性地提高发光效率。
通过使用本发明的通式(1)所表示的化合物作为发光层的发光材料,能够提供有机光致发光元件(有机PL(Photoluminescence,光致发光)元件)或有机电致发光元件(有机EL元件)等优异的有机发光元件。这时,本发明的通式(1)所表示的化合物也可以作为所谓辅助掺杂剂而具有辅助发光层中所含的其它发光材料的发光的功能。也就是说,发光层中所含的本发明的通式(1)所表示的化合物也可以具有发光层中所含的主体材料的最低激发单重态能阶和发光层中所含的其它发光材料的最低激发单重态能阶之间的最低激发单重态能阶。
有机光致发光元件具有在基板上至少形成发光层的构造。另外,有机电致发光元件具有至少形成阳极、阴极、及阳极和阴极之间的有机层的构造。有机层至少包含发光层,可以仅包含发光层,也可以除了发光层以外还具有1层以上的有机层。作为这种其它有机层,可以列举:电洞传输层、电洞注入层、电子阻挡层、电洞阻挡层、电子注入层、电子传输层、激子阻挡层等。电洞传输层也可以为具有电洞注入功能的电洞注入传输层,电子传输层也可以为具有电子注入功能的电子注入传输层。将具体的有机电致发光元件的结构例示于图1。在图1中,1表示基板,2表示阳极,3表示电洞注入层,4表示电洞传输层,5表示发光层,6表示电子传输层,7表示阴极。
以下,对有机电致发光元件的各构件及各层进行说明。另外,基板和发光层的说明也适于有机光致发光元件的基板和发光层。
(基板)
本发明的有机电致发光元件优选被基板支撑。关于该基板,并无特别限制,只要一直以来在有机电致发光元件中所惯用即可,例如可以使用包含玻璃、透明塑胶、石英、硅等的基板。
(阳极)
作为有机电致发光元件中的阳极,优选使用将功函数大(4eV以上)的金属、合金、导电性化合物及这些的混合物作为电极材料的阳极。作为这种电极材料的具体例,可以列举:Au等金属、CuI、铟锡氧化物(ITO,Indium Tin Oxide)、SnO2、ZnO等导电性透明材料。另外,也可以使用IDIXO(In2O3-ZnO)等非晶质且能够制作透明导电膜的材料。阳极可以利用蒸镀或溅镀等方法使这些电极材料形成薄膜,并利用光微影法而形成所需形状的图案,或在不太需要图案精度的情况下(100μm以上左右),也可以在所述电极材料的蒸镀或溅镀时介隔所需形状的遮罩形成图案。或者,在使用像有机导电性化合物那样能够涂布的材料的情况下,也可以使用印刷方式、涂布方式等湿式成膜法。在从该阳极提取发光的情况下,理想为使透过率大于10%,另外,阳极的薄片电阻优选几百Ω/□以下。进而,膜厚通常是在10~1000nm、优选10~200nm的范围内选择,但也取决于材料。
(阴极)
另一方面,作为阴极,可以使用将功函数小(4eV以下)的金属(称为电子注入性金属)、合金、导电性化合物及这些的混合物作为电极材料的阴极。作为这种电极材料的具体例,可以列举:钠、钠-钾合金、镁、锂、镁/铜混合物、镁/银混合物、镁/铝混合物、镁/铟混合物、铝/氧化铝(Al2O3)混合物、铟、锂/铝混合物、稀土金属等。这些中,就电子注入性及对氧化等的耐久性的观点而言,优选电子注入性金属和作为功函数的值更大而稳定的金属的第二金属的混合物、例如镁/银混合物、镁/铝混合物、镁/铟混合物、铝/氧化铝(Al2O3)混合物、锂/铝混合物、铝等。阴极能够通过利用蒸镀或溅镀等方法使这些电极材料形成薄膜而制作。另外,阴极的薄片电阻优选几百Ω/□以下,膜厚通常是在10nm~5μm、优选50~200nm的范围内选择。另外,为了使所发出的光透过,只要有机电致发光元件的阳极或阴极的任一个为透明或半透明,那么发光亮度提高而适合。
另外,通过将阳极的说明中所列举的导电性透明材料用于阴极,能够制作透明或半透明的阴极,通过应用该阴极,能够制作阳极和阴极两者具有透过性的元件。
(发光层)
发光层是在通过从阳极及阴极分别注入的电洞及电子再结合而产生激子后,进行发光的层,可以将发光材料单独用在发光层中,优选包含发光材料和主体材料。作为发光材料,可以使用选自通式(1)所表示的本发明的化合物群中的一种或两种以上。为了使本发明的有机电致发光元件及有机光致发光元件表现出高发光效率,重要的是将发光材料中所产生的单重态激子及三重态激子封闭在发光材料中。因此,优选在发光层中除了发光材料以外还使用主体材料。作为主体材料,可以使用激发单重态能量、激发三重态能量的至少任一个具有高于本发明的发光材料的值的有机化合物。其结果为,能够将本发明的发光材料中所产生的单重态激子及三重态激子封闭在本发明的发光材料的分子中,从而能够充分地提升其发光效率。但是,由于也存在即便无法充分地封闭单重态激子及三重态激子也能够获得高发光效率的情况,所以只要为能够实现高发光效率的主体材料,那么可以无特别限制地用于本发明中。在本发明的有机发光元件或有机电致发光元件中,发光是由发光层中所含的本发明的发光材料所产生。该发光包括荧光发光及延迟荧光发光两者。但也可以使发光的一部分或局部地存在来自主体材料的发光。
在使用主体材料的情况下,作为发光材料的本发明的化合物在发光层中所含有的量优选0.1重量%以上,更优选1重量%以上,另外,优选50重量%以下,更优选20重量%以下,进而优选10重量%以下。
作为发光层中的主体材料,优选具有电洞传输能力、电子传输能力,且防止发光的长波长化,并且具有高玻璃转移温度的有机化合物。
(注入层)
所谓注入层,是为了降低驱动电压或提高发光亮度而设置于电极和有机层间的层,有电洞注入层和电子注入层,也可以使其存在于阳极和发光层或电洞传输层之间、及阴极和发光层或电子传输层之间。注入层可以视需要而设置。
(阻挡层)
阻挡层是能够阻挡存在于发光层中的电荷(电子或者电洞)及/或激子向发光层外扩散的层。电子阻挡层可以配置在发光层及电洞传输层之间,阻挡电子朝向电洞传输层而通过发光层。同样地,电洞阻挡层可以配置在发光层及电子传输层之间,阻挡电洞朝向电子传输层而通过发光层。另外,阻挡层可以用于阻挡激子扩散至发光层的外侧。也就是说,电子阻挡层、电洞阻挡层也可以分别兼具作为激子阻挡层的功能。本说明书中所述的电子阻挡层或激子阻挡层是以包含以一层而具有电子阻挡层及激子阻挡层的功能的层的含义使用。
(电洞阻挡层)
所谓电洞阻挡层,广义上具有电子传输层的功能。电洞阻挡层具有一面传输电子一面阻挡电洞到达电子传输层的作用,由此能够提高发光层中的电子和电洞的再结合机率。作为电洞阻挡层的材料,视需要能够使用下述电子传输层的材料。
(电子阻挡层)
所谓电子阻挡层,广义上具有传输电洞的功能。电子阻挡层具有一面传输电洞一面阻挡电子到达电洞传输层的作用,由此能够提高发光层中的电子和电洞进行再结合的机率。
(激子阻挡层)
所谓激子阻挡层,是用以阻挡通过电洞和电子在发光层内再结合而产生的激子扩散至电荷传输层的层,能够通过插入本层而将激子有效率地封闭在发光层内,从而能够提高元件的发光效率。激子阻挡层能够和发光层相邻而插入至阳极侧、阴极侧的任一侧,也可以在两侧同时插入。也就是说,于在阳极侧具有激子阻挡层的情况下,能够在电洞传输层和发光层之间和发光层相邻而插入该层,在插入至阴极侧的情况下,能够在发光层和阴极之间和发光层相邻而插入该层。另外,在阳极、和与发光层的阳极侧相邻的激子阻挡层之间可以具有电洞注入层或电子阻挡层等,在阴极、和与发光层的阴极侧相邻的激子阻挡层之间可以具有电子注入层、电子传输层、电洞阻挡层等。在配置阻挡层的情况下,优选用作阻挡层的材料的激发单重态能量及激发三重态能量的至少任一个高于发光材料的激发单重态能量及激发三重态能量。
(电洞传输层)
所谓电洞传输层,包含具有传输电洞的功能的电洞传输材料,电洞传输层能够设置单层或多层。
作为电洞传输材料,具有电洞的注入或传输、电子的障壁性的任一种,可以为有机物、无机物的任一种。作为能够使用的公知的电洞传输材料,例如可以列举:三唑衍生物、口咢二唑衍生物、咪唑衍生物、咔唑衍生物、吲哚咔唑衍生物、多芳基烷烃衍生物、吡唑啉衍生物及吡唑啉酮衍生物、苯二胺衍生物、芳基胺衍生物、氨基取代查耳酮衍生物、口咢唑衍生物、苯乙烯基蒽衍生物、茀酮衍生物、腙衍生物、茋衍生物、硅氮烷衍生物、苯胺系共聚物、以及导电性高分子低聚物、尤其是噻吩低聚物等,优选使用卟啉化合物、芳香族三级胺化合物及苯乙烯基胺化合物,更优选使用芳香族三级胺化合物。
(电子传输层)
所谓电子传输层,包含具有传输电子的功能的材料,电子传输层可以设置单层或多层。
作为电子传输材料(也存在兼作电洞阻挡材料的情况),只要具有将从阴极注入的电子传导至发光层的功能即可。作为能够使用的电子传输层,例如可以列举:硝基取代茀衍生物、二苯基醌衍生物、噻喃二氧化物衍生物、碳二酰亚胺、亚茀基甲烷衍生物、蒽醌二甲烷及蒽酮衍生物、口咢二唑衍生物等。进而,在所述口咢二唑衍生物中,将口咢二唑环的氧原子取代为硫原子而成的噻二唑衍生物、具有作为拉电子基而已知的喹口咢啉环的喹口咢啉衍生物也能够用作电子传输材料。进而也可以使用将这些材料导入至高分子链中、或将这些材料作为高分子的主链的高分子材料。
在制作有机电致发光元件时,不仅可以将通式(1)所表示的化合物用在发光层中,也可以用在发光层以外的层中。这时,发光层中所使用的通式(1)所表示的化合物、和发光层以外的层中所使用的通式(1)所表示的化合物可以相同也可以不同。例如,也可以在所述注入层、阻挡层、电洞阻挡层、电子阻挡层、激子阻挡层、电洞传输层、电子传输层等中使用通式(1)所表示的化合物。这些层的制膜方法并无特别限定,可以利用干式工艺、湿式工艺的任一种而制作。
以下,具体地例示有机电致发光元件中能够使用的优选材料。但,本发明中能够使用的材料不受以下的例示化合物的限定性解释。另外,即便为作为具有特定功能的材料而例示的化合物,也能够转用作具有其它功能的材料。另外,以下的例示化合物的结构式中的R、R2~R7分别独立表示氢原子或取代基。n表示3~5的整数。
首先,列举也能够用作发光层的主体材料的优选化合物。
[化23]
[化24]
[化25]
[化26]
[化27]
其次,列举能够用作电洞注入材料的优选化合物例。
[化28]
其次,列举能够用作电洞传输材料的优选化合物例。
[化29]
[化30]
[化31]
[化32]
[化33]
[化34]
其次,列举能够用作电子阻挡材料的优选化合物例。
[化35]
其次,列举能够用作电洞阻挡材料的优选化合物例。
[化36]
其次,列举能够用作电子传输材料的优选化合物例。
[化37]
[化38]
[化39]
其次,列举能够用作电子注入材料的优选化合物例。
[化40]
列举作为能够进一步添加的材料而优选的化合物例。例如能够想到作为稳定材料而添加等。
[化41]
利用所述方法制作的有机电致发光元件是通过在所获得的元件的阳极和阴极之间施加电场而发光。这时,如果为利用激发单重态能量的发光,那么和其能阶对应的波长的光是作为荧光发光及延迟荧光发光而被确认到。另外,如果为利用激发三重态能量的发光,那么和其能阶对应的波长是作为磷光而被确认到。通常的荧光由于荧光寿命短于延迟荧光发光,所以发光寿命能够由荧光和延迟荧光加以区别。
另一方面,关于磷光,对于像本发明的化合物那样的通常的有机化合物而言,由于激发三重态能量不稳定而被转换为热等,寿命短而立即失活,所以在室温下几乎无法观测到。为了测定通常的有机化合物的激发三重态能量,能够通过观测极低温的条件下的发光而进行测定。
本发明的有机电致发光元件在单一元件、包含以阵列状配置的结构的元件、将阳极和阴极以X-Y矩阵状配置的结构的任一个中均能够应用。根据本发明,通过使发光层中含有通式(1)所表示的化合物,能够获得发光效率经大幅度地改善的有机发光元件。本发明的有机电致发光元件等有机发光元件能够进而应用于各种用途中。例如能够使用本发明的有机电致发光元件而制造有机电致发光显示装置,关于详细情况,可以参照时任静士、安达千波矢、村田英幸共著的“有机EL显示器”(Ohm公司)。另外,尤其本发明的有机电致发光元件也能够应用于需求大的有机电致发光照明或背光。
实施例
以下列举合成例及实施例而更具体地说明本发明的特征。如下所示的材料、处理内容、处理程序等只要不脱离本发明的主旨,那么能够适当进行变更。因此,本发明的范围不应受到如下所示的具体例的限定性解释。另外,发光特性的评价是使用电源电表(Keithley公司制造:2400系列)、半导体参数分析器(Agilent Technology公司制造:E5273A)、光功率计测定装置(Newport公司制造:1930C)、光学分光器(Ocean Optics公司制造:USB2000)、分光发射计(TOPCON公司制造:SR-3)及快速照相机(streak camera)(Hamamatsu Photonics股份有限公司制造的C4334型)而进行。
另外,各材料的单重态能量(ES1)和三重态能量(ET1)的差(ΔEST)是利用以下的方法算出单重态能量(ES1)和三重态能量,并利用ΔEST=ES1-ET1而求出。
(1)单重态能量ES1
通过将测定对象化合物和mCBP(3,3'-di(9H-carbazol-9-yl)biphenyl,3,3'-二(9H-咔唑-9-基)联苯)以使测定对象化合物的浓度成为6重量%的方式共蒸镀,而在Si基板上制作厚度100nm的试样。在常温(300K)下测定该试样的荧光光谱。将从激发光刚入射后至入射后100纳秒为止的发光累计,由此获得将纵轴设为发光强度、横轴设为波长的荧光光谱。荧光光谱中,将纵轴设为发光、横轴设为波长。对该发光光谱的短波侧的上升作切线,求出该切线和横轴的交点的波长值λedge[nm]。将利用如下所示的换算式将该波长值换算成能量值而得的值设为ES1。
换算式:ES1[eV]=1239.85/λedge
在发光光谱的测定中,激发光源是使用氮气雷射(Lasertechnik Berlin公司制造,MNL200),检测器是使用快速照相机(Hamamatsu Photonics公司制造,C4334)。
(2)三重态能量ET1
将和单重态能量ES1相同的试样冷却至5[K],对磷光测定用试样照射激发光(337nm),使用快速照相机测定磷光强度。将从激发光入射后1毫秒起至入射后10毫秒为止的发光累计,由此获得将纵轴设为发光强度、横轴设为波长的磷光光谱。对该磷光光谱的短波长侧的上升作切线,求出该切线和横轴的交点的波长值λedge[nm]。将利用如下所示的换算式将该波长值换算成能量值而得的值设为ET1。
换算式:ET1[eV]=1239.85/λedge
对磷光光谱的短波长侧的上升的切线是以如下方式作出。在从磷光光谱的短波长侧起,在光谱曲线上移动至光谱的极大值中的最为短波长侧的极大值时,朝向长波长侧考虑曲线上的各点的切线。该切线随着曲线上升(也就是说,随着纵轴增加)而增加斜率。将在该斜率的值取极大值的点所作出的切线设为对该磷光光谱的短波长侧的上升的切线。
另外,具有光谱的最大波峰强度的10%以下的波峰强度的极大点不包括在所述最为短波长侧的极大值中,将在最接近最为短波长侧的极大值且斜率的值取极大值的点所作出的切线设为对该磷光光谱的短波长侧的上升的切线。
(合成例1)例示化合物(1)的合成
在本合成例中,依据以下的流程,对例示化合物(1)进行合成。
[化42]
(1)2,3-二氨基-5,6-二苯基吡口井的合成步骤
基于J.Chem.Research(s),1997,250-251中所记载的方法,利用以下的方法合成2,3-二氨基-5,6-二苯基吡口井。
将3,4-二氨基-1,2,5-噻二唑(7.9g,68.2mmol)、苯偶酰(15.8g,75.0mmol)、乙酸230ml投入至500ml三口烧瓶中,通过回流进行5小时搅拌。将反应溶液放置冷却至室温后,将溶剂蒸馏去除,使用管柱层析法进行精制,由此以产量8.0g、产率41%获得5,6-二苯基[1,2,5]噻二唑并[3,4-b]吡口井。
其次,将5,6-二苯基[1,2,5]噻二唑并[3,4-b]吡口井(8.0g,27.7mmol)、氯化锡(II)二水合物(31.2g,138.5mmol)、甲醇160ml投入至1000ml三口烧瓶中后,缓慢添加浓盐酸160ml,在氮气气流下,在60℃下进行2小时搅拌。使用饱和碳酸钠水溶液将放置冷却至室温的溶液中和至pH值8~9后,将溶液内的甲醇某程度蒸馏去除。过滤取出沉淀物,使用乙酸乙酯反复进行3次固体萃取后,利用管柱层析法进行精制,由此以产量4.8g、产率66%获得2,3-二氨基-5,6-二苯基吡口井。
(2)中间物B的合成步骤
将2,3-二氨基-5,6-二苯基吡口井(1.0g,3.8mmol)、利用Eur.J.Org.Chem.,2012,320-328中所记载的方法合成的1-(4-溴苯基)-2-苯基乙烷-1,2-二酮(1.2g,4.2mmol)、乙酸30ml投入至100ml三口烧瓶中,进行4小时加热回流搅拌。将溶液放置冷却至室温后,将溶剂蒸馏去除,使用管柱层析法进行精制,由此以产量1.6g、产率88%获得中间物B。
(3)例示化合物(1)的合成步骤
基于Eur.J.Inorg.Chem.,2006,3676-3683中所记载的方法,利用以下的方法对例示化合物(1)进行合成。
将中间物B(0.39mmol)、利用国际公开WO2012/039561号中所记载的方法合成的9,9-二甲基-9,10-二氢吖啶(89mg,0.43mmol)、三(二亚苄基丙酮)二钯(0)(36mg,0.039mmol)、叔丁醇钠(41mg,0.43mmol)、甲苯20ml、三(叔丁基)膦(10mg,0.050mmol)投入至100ml三口烧瓶中,一面搅拌,一面迅速反复3次脱气和氮气置换。将该混合物在氮气气流下,通过回流进行8小时搅拌。反应后,在放置冷却至室温的反应溶液中添加水,利用二氯甲烷进行萃取,并利用饱和盐水进行洗净。利用硫酸钠对有机层进行干燥后,将溶剂蒸馏去除,并使用管柱层析法进行精制,由此以产量173mg、产率69%获得例示化合物(1)。
熔点为300℃以上。1H-NMR(δppm,CDCl3)1.69(6H,s),6.31(2H,dd,J=8.4,1.2),6.96(2H,td,J=7.2,0.12),7.33~7.48(13H,m),7.71(4H,d,J=7.6),7.77(2H,d,J=8.0),7.91(2H,d,J=8.0)
(合成例2)例示化合物(2)的合成
在本合成例中,依据以下的流程,对例示化合物(2)进行合成。
[化43]
(1)中间物C的合成步骤
将3,4-二氨基-1,2,5-噻二唑(9.9g,85.2mmol)、4,4'-二溴苯偶酰(24.4g,66.4mmol)、乙酸300ml投入至1000ml三口烧瓶中,通过回流进行24小时搅拌。将反应溶液放置冷却至室温后,将溶剂蒸馏去除,使用管柱层析法进行精制,由此以产量19.0g、产率64%获得中间物C。
(2)中间物D的合成步骤
将中间物C(21.4g,47.8mmol)、氯化锡(II)二水合物(50.3g,222.9mmol)、甲醇400ml投入至1000ml三口烧瓶中后,缓慢添加浓盐酸400ml,在氮气气流下,在60℃下进行3小时搅拌。使用饱和碳酸钠水溶液将放置冷却至室温的溶液中和至pH值8~9后,将溶液内的甲醇某程度蒸馏去除。过滤取出沉淀物,使用乙酸乙酯反复3次固体萃取后,利用管柱层析法进行精制,由此以产量12.0g、产率60%获得中间物D。
(3)中间物E的合成步骤
将中间物D(0.6g,1.4mmol)、苯偶酰(0.33g,1.6mmol)、乙酸10ml投入至50ml三口烧瓶中,通过回流进行4小时搅拌。将溶液放置冷却至室温后,将溶剂蒸馏去除,使用管柱层析法进行精制,由此以产量0.62g、产率73%获得中间物E。
(4)例示化合物(2)的合成步骤
使用中间物E(0.39mmol)代替中间物B(0.39mmol),使用9,9-二甲基-9,10-二氢吖啶2倍摩尔量,除此以外,通过进行和合成例1的步骤(3)相同的步骤,以产量0.24g、产率50%获得例示化合物(2)。
熔点为300℃以上。1H-NMR(δppm,CDCl3)1.69(12H),6.33(4H,m),6.90(8H,m),7.37~7.53(14H,m),7.74(4H,m),8.01(4H,d,J=8.0)
(合成例3)例示化合物(4)的合成
在本合成例中,依据以下的流程,对例示化合物(4)进行合成。
[化44]
(1)中间物F的合成步骤
将3,4-二氨基-1,2,5-噻二唑(9.9g,85.2mmol)、利用Bioorgnic&Medicinal Chem.2007,15,3801-3817中所记载的方法合成的3,3'-二氟苯偶酰(16.3g,66.4mmol)、乙酸300ml投入至1000ml三口烧瓶中,通过回流进行24小时搅拌。将溶液放置冷却至室温后,将溶剂蒸馏去除,使用管柱层析法进行精制,由此以产量18.1g、产率65%获得中间物F。
(2)中间物G的合成步骤
将中间物F(15.6g,47.8mmol)、氯化锡(II)二水合物(50.3g,222.9mmol)、甲醇400ml投入至1000ml三口烧瓶中后,缓慢添加浓盐酸400ml,在氮气气流下,在60℃下进行3小时搅拌。使用饱和碳酸钠水溶液将放置冷却至室温的溶液中和至pH值8~9后,将溶液内的甲醇某程度蒸馏去除。过滤取出沉淀物,使用乙酸乙酯反复3次固体萃取后,利用管柱层析法进行精制,由此以产量8.6g、产率60%获得中间物G。
(3)中间物H的合成步骤
将中间物G(0.60g,1.4mmol)、4-溴苯偶酰(0.46g,1.6mmol)、乙酸10ml投入至50ml三口烧瓶中,通过回流进行4小时搅拌。将溶液放置冷却至室温后,将溶剂蒸馏去除,使用管柱层析法进行精制,由此以产量0.56g、产率73%获得中间物H。
(4)例示化合物(4)的合成步骤
使用中间物H(0.39mmol)代替中间物B(0.39mmol),使用9,9-二甲基-9,10-二氢吖啶2倍摩尔量,除此以外,进行和合成例1的步骤(3)相同的步骤,由此以产量0.15g、产率55%获得例示化合物(4)。
熔点为300℃以上。
(合成例4)例示化合物(7)的合成
在本合成例中,依据以下的流程,对例示化合物(7)进行合成。
[化45]
(1)中间物I的合成步骤
将3,4-二氨基-1,2,5-噻二唑(10.0g,86.1mmol)、利用Bioorgnic&Medicinal Chem.2007,15,3801-3817中所记载的方法合成的3,3'-二氟苯偶酰(16.5g,67.2mmol)、乙酸300ml投入至1000ml三口烧瓶中,通过回流进行24小时搅拌。将溶液放置冷却至室温后,将溶剂蒸馏去除,使用管柱层析法进行精制,由此以产量14.6g、产率65%获得中间物I。
(2)中间物J的合成步骤
将中间物I(21.4g,47.8mmol)、氯化锡(II)二水合物(50.3g,222.9mmol)、甲醇400ml投入至1000ml三口烧瓶中后,缓慢添加浓盐酸400ml,在氮气气流下,在60℃下进行3小时搅拌。使用饱和碳酸钠水溶液将放置冷却至室温的溶液中和至pH值8~9后,将溶液内的甲醇某程度蒸馏去除。过滤取出沉淀物,使用乙酸乙酯反复3次固体萃取后,利用管柱层析法进行精制,由此以产量9.6g、产率60%获得中间物J。
(3)中间物K的合成
将中间物J(0.6g,1.8mmol)、4-溴苯偶酰(0.58g,2.0mmol)、乙酸10ml投入至50ml三口烧瓶中,通过回流进行4小时搅拌。将溶液放置冷却至室温后,将溶剂蒸馏去除,使用管柱层析法进行精制,由此以产量0.78g、产率73%获得中间物K。
(4)例示化合物(7)的合成步骤
使用中间物K(0.39mmol)代替中间物B(0.39mmol),除此以外,进行和合成例1的步骤(3)相同的步骤,由此以产量0.14g、产率50%获得例示化合物(7)。
熔点为300℃以上。
(合成例5)例示化合物(11)的合成
将在合成例1中所合成的中间物B(0.2g,0.39mmol)、啡口咢口井(78mg,0.43mmol)、乙酸钯(II)(5mg,0.023mmol)、碳酸钾(0.16g,1.16mmol)、甲苯20ml、三(叔丁基)膦(17mg,0.085mmol)投入至100ml三口烧瓶中,一面搅拌,一面迅速反复3次脱气和氮气置换。将该混合物在氮气气流下,通过回流进行10小时搅拌。反应后,在放置冷却至室温的反应溶液中添加水,利用二氯甲烷进行萃取,并利用饱和盐水进行洗净。利用硫酸钠对有机层进行干燥后,将溶剂蒸馏去除,并使用管柱层析法进行精制,由此以产量0.14g、产率59%获得例示化合物(11)。
熔点为300℃以上。1H-NMR(δppm,CDCl3)5.96(2H,dd,J=8.0,1.6),6.60~6.78(6H,m),7.33~7.53(12H,m),7.67~7.80(6H,m),7.87(2H,d,J=8.0)
(合成例6)例示化合物(12)的合成
将在合成例2中所合成的中间物E(0.2g,0.34mmol)、啡口咢口井(0.14g,0.74mmol)、乙酸钯(II)(5mg,0.020mmol)、碳酸钾(0.28g,2.02mmol)、甲苯20ml、三(叔丁基)膦(15mg,0.074mmol)投入至100ml三口烧瓶中,一面搅拌,一面迅速反复3次脱气和氮气置换。将混合物在氮气气流下,通过回流进行9小时搅拌。反应后,在放置冷却至室温的反应溶液中添加水,利用二氯甲烷进行萃取,并利用饱和盐水进行洗净。利用硫酸钠对有机层进行干燥后,将溶剂蒸馏去除,并使用管柱层析法进行精制,由此以产量0.19g、产率71%获得例示化合物(12)。
熔点为300℃以上。
1H-NMR(δppm,CDCl3)5.96(4H,dd,J=8.0,1.6),6.53(4H,td,J-=8.0,1.6),6.66(4H,td,J=8.0,1.6),6.71(4H,dd,J=8.0,1.6),7.37~7.48(10H,m),7.72(4H,d,J=7.2),7.95(4H,d,J=8.8)
(合成例7)例示化合物(25)的合成
在本合成例中,依据以下的流程,对例示化合物(25)进行合成。
[化46]
(1)中间物L的合成步骤
将4-溴苯偶酰(7.9g,27.2mmol)、二氨基顺丁烯二腈(3.5g,32.6mmol)、乙酸120ml投入至500ml三口烧瓶中,通过回流进行4小时搅拌。反应结束后,将反应液放置冷却,将溶剂蒸馏去除后,利用管柱层析法进行精制,由此以产量9.8g、产率93%获得中间物L。
(2)中间物M的合成步骤
将中间物L(1.9g,5.2mmol)、2,3-二氨基-5,6-二苯基吡口井(1.0g,5.8mmol)、碳酸钾(1.3g,9.5mmol)、二甲基亚砜30ml投入至100ml三口烧瓶中后,反复3次脱气和氮气置换。将混合溶液在氮气气流下,在120℃下进行7小时搅拌。将反应液放置冷却后,利用二氯甲烷、蒸馏水进行萃取。将有机层和亚二硫磺酸钠水溶液注入至分液漏斗中而进行分液,利用硫酸钠对该有机层进行干燥后,利用管柱层析法进行精制,由此以产量0.89g、产率30%获得中间物M。
(3)中间物N的合成步骤
将中间物M(0.63g,1.1mmol)、二碳酸二叔丁酯(0.52g,2.4mmol)、三乙胺(0.24g,2.37mmol)、N,N-二甲基-4-氨基吡啶(26mg,0.22mmol)、四氢呋喃30ml投入至100ml烧瓶中,在室温下进行7小时搅拌。反应后,将溶剂从反应液中蒸馏去除,在其残留物中添加20~30ml左右的乙酸乙酯,进行过滤,由此将杂质去除至滤液侧。利用管柱层析法对所获得的残留物进行精制,由此以产量0.56g、产率66%获得中间物N。
(4)例示化合物(25)的合成步骤
将中间物N(0.31g,0.40mmol)、亚氨基茋(0.12g,0.60mmol)、三(二亚苄基丙酮)二钯(0)(36mg,0.040mmol)、叔丁醇钠(0.57g,5.96mmol)、甲苯20ml、三(叔丁基)膦(10mg,0.052mmol)投入至100ml三口烧瓶中,一面搅拌,一面迅速反复3次脱气和氮气置换。将混合物在氮气气流下,通过回流进行26小时搅拌。反应后,在放置冷却至室温的反应溶液中添加水,利用二氯甲烷进行萃取,并利用饱和盐水进行洗净。利用硫酸钠对有机层进行干燥后,将溶剂蒸馏去除,并使用管柱层析法进行精制,由此以产量0.13g、产率46%获得例示化合物(25)。
熔点为300℃以上。
(实施例1)例示化合物(1)的溶液的制作和评价
制备在合成例1中所合成的例示化合物(1)的甲苯溶液(浓度10-5M)。
对该甲苯溶液测定由380nm激发光所产生的光致发光量子效率,结果光致发光量子效率在未通入氮气的甲苯溶液中为12.5%,在通入了氮气的甲苯溶液中为33.4%。
另外,将对该甲苯溶液测定利用400nm激发光所得的发光光谱和紫外线吸收光谱的结果示于图2。最大发光波长λmax为651nm。
将测定利用340nm激发光所得的暂态衰减曲线的结果示于图3。暂态衰减曲线表示对化合物照射激发光而测定发光强度逐渐失活的过程所得的发光寿命测定结果。如果为通常的单一成分的发光(荧光或者磷光),那么发光强度以单一指数函数的形式衰减。这意味着在曲线图的纵轴为半log的情况下,线性衰减。在图3所示的例示化合物(1)的暂态衰减曲线中,虽然在观测初期观测到这种线性成分(荧光),但数μ秒后出现偏离线性的成分。其为延迟成分的发光,和初期的成分相加的信号成为向长时间侧延伸的平缓曲线。通过像这样测定发光寿命,确认到例示化合物(1)是除了包含荧光成分以外也包含延迟成分的发光体。
关于发光寿命,在未通入氮气的甲苯溶液中为0.37μs,在通入了氮气的甲苯溶液中为36.1μs。
该甲苯溶液的激发单重态和激发三重态的能量差ΔEst为0.002eV,作为S0→S1跃迁的频度因数的f值为0.0002。ΔEst值用作热活化的容易产生程度的标准,认为该值越小越有可能容易产生热活化延迟荧光。另外,f值为从S0(基态)向S1(激发单重态)的跃迁的容易产生程度,此次的计算中使用f值作为发光的容易产生程度(荧光强度)的标准,认为该值越大越有可能显示出强荧光。
(实施例2)例示化合物(2)的溶液的制作和评价
制备在合成例2中所合成的例示化合物(2)的甲苯溶液(浓度10-5M)。
对该甲苯溶液测定由440nm激发光所产生的光致发光量子效率,结果光致发光量子效率在未通入氮气的甲苯溶液中为12.0%,在通入了氮气的甲苯溶液中为32.4%。
另外,将对该甲苯溶液测定利用444nm激发光所得的发光光谱和紫外线吸收光谱的结果示于图4,将对利用340nm激发光所得的暂态衰减曲线进行测定的结果示于图5。根据图4,最大发光波长λmax为621nm。根据图5,能够观测到发光寿命短的成分(即时荧光成分)和发光寿命长的成分(延迟荧光成分),关于延迟荧光成分的发光寿命,在未通入氮气的甲苯溶液中为0.40μs,在通入了氮气的甲苯溶液中为23.5μs。另外,该甲苯溶液的激发单重态和激发三重态的能量差ΔEst为0.002eV,f值为0。
(比较例1)比较化合物(A)的溶液的制作和评价
制备比较化合物(A)的甲苯溶液(浓度10-5M)。
对该甲苯溶液评价特性,结果由320nm激发光所产生的光致发光量子效率在未通入氮气的甲苯溶液中为1.2%,在通入了氮气的甲苯溶液中为2.3%,最大发光波长为706nm。另外,激发单重态和激发三重态的能量差ΔEst为0.047eV,f值为0.031。该比较化合物(A)的甲苯溶液的发光弱,未能确认到延迟荧光。
[化47]
比较化合物(A)
(比较例2)比较化合物(B)的溶液的制作和评价
制备比较化合物(B)的甲苯溶液(浓度10-5M)。
对该甲苯溶液评价特性,结果由360nm激发光所产生的光致发光量子效率在未通入氮气的甲苯溶液中为1.1%,在通入了氮气的甲苯溶液中为2.3%,最大发光波长为697nm。另外,激发单重态和激发三重态的能量差ΔEst为0.006eV,f为0.041。该比较化合物(B)的甲苯溶液的发光弱,未能确认到延迟荧光。
[化48]
比较化合物(B)
(比较例3)比较化合物(C)的溶液的制作和评价
制备比较化合物(C)的甲苯溶液(浓度10-5M)。
使用该甲苯溶液对由360nm激发光所产生的光致发光进行观测,结果未能明确地确认到延迟荧光。
[化49]
比较化合物(C)
(实施例3)例示化合物(2)的薄膜型有机光致发光元件的制作和评价
在硅基板上利用真空蒸镀法,在真空度3.0×10-4Pa以下的条件下从不同的蒸镀源对mCBP(3,3'-Bis(N-carbazolyl)-1,1'-biphenyl,3,3'-二(N-咔唑基)-1,1'-联苯)和例示化合物(2)进行蒸镀,以50nm的厚度形成例示化合物(2)的浓度为6.0重量%的薄膜而制成薄膜型有机光致发光元件。
对该薄膜型有机光致发光元件,将测定由400nm激发光所发出的发光光谱和紫外线吸收光谱的结果示于图6,将在大气下测定暂态衰减曲线的结果示于图7。光致发光量子效率在大气下为48.9%,在含氮环境下为50.1%,发光波长为586nm。另外,根据图7能够观测到发光寿命不同的3个成分,各成分的发光寿命为7.81ns、969.0ns、5.73μs。
(实施例4)使用例示化合物(2)的有机电致发光元件的制作和评价
在形成膜厚100nm的包含铟锡氧化物(ITO)的阳极的玻璃基板上,利用真空蒸镀法在真空度3.0×10-4Pa下积层各薄膜。首先,在ITO上以35nm的厚度形成α-NPD(4,4'-bis[N-(1-naphthyl)-N-phenyl-amino]-biphenyl,4,4'-双[N-(1-萘基)-N-苯基-氨基]-联苯)。其次,从不同的蒸镀源将例示化合物(2)和mCBP共蒸镀,形成15nm的厚度的层作为发光层。这时,例示化合物(2)的浓度是设为6.0重量%。其次,以65nm的厚度形成TPBi(1,3,5-Tris(1-phenyl-1H-benzimidazol-2-yl)benzene,1,3,5-三(1-苯基-1H-苯并咪唑-2-基)苯),进而将氟化锂(LiF)真空蒸镀0.8nm,继而以100nm的厚度蒸镀铝(Al),由此形成阴极,制成有机电致发光元件。
对所制造的有机电致发光元件,将在1mA/cm2、10mA/cm2、100mA/cm2的各条件下测得的发光光谱示于图8,将电压-电流密度-亮度特性示于图9,将电流密度-电流效率-功率效率特性示于图10,将电流密度-外部量子效率特性示于图11。该有机电致发光元件的接通电压为3.2V,最大亮度为14820cd/m2,最大电流效率为27.7cd/A,最大功率效率为25.1lm/W。另外,该有机电致发光元件达成10.5%的高外部量子产率。如果使用发光量子效率为100%的荧光材料尝试制作取得了平衡的理想的有机电致发光元件,那么只要光提取效率为20~30%,荧光发光的外部量子效率便达到5~7.5%。该值通常被视为使用荧光材料的有机电致发光元件的外部量子效率的理论极限值。就实现超出理论极限值的高外部量子效率的观点而言,本发明的有机电致发光元件极优异。
[化50]
[工业上的可利用性]
本发明的有机发光元件可实现高发光效率。另外,本发明的化合物能够用作这种有机发光元件用的发光材料。因此,本发明的产业上的可利用性高。
[符号的说明]
1 基板
2 阳极
3 电洞注入层
4 电洞传输层
5 发光层
6 电子传输层
7 阴极
- 还没有人留言评论。精彩留言会获得点赞!