一种半花菁结构的近红外染料及其制备方法和应用与流程
本发明属于有机近红外荧光染料技术领域,具体涉及一种半花菁结构的近红外染料及其制备方法和应用。
背景技术:
近红外荧光染料是一类功能性染料,由于其在近红外光区有很好的吸收,因此近红外技术在太阳能电池、肿瘤治疗、无线电射频识别系统、防伪印刷等中都有着广泛的应用。
花菁染料作为近红外染料中的一类,部分已得到实际应用,并不断有新的种类涌现。近红外甲川花菁染料吸收和发射光谱位于近红外区(>650nm),相对于可见荧光区(<650nm)检测而言,在近红外荧光区,生物样品基体的荧光吸收和荧光强度很小,因而背景干扰大大降低,并且由于散射光强度与波长的四次方成反比,随波长的增加,拉曼散射迅速减小,使散射干扰也大为减少。七甲川作为甲川花菁染料的一种,吸收波长更长,背景干扰更小,其应用于生物样品检测相比于常规荧光检测而言,具有更深的组织穿透深度,对生物组织的损伤更小,所以其应用前景也更为广阔。
基于七甲川吲哚菁染料合成的半花菁结构染料,在近红外区有较强的吸收和发射性能,可以用于光热治疗和光声成像领域;而且对于环境的pH值有响应效果,可以用作近红外区的荧光探针;另外该染料的光热和化学稳定性对比七甲川花菁染料也有了很大的提升。
技术实现要素:
解决的技术问题:本发明的目的在于提供一种带有苯并咪唑的半花菁结构的近红外染料及其制备方法,该染料具有近红外吸收和发射性能、良好的光热和化学稳定性。
技术方案:本发明一方面提供了一种半花菁结构的近红外染料,其结构式如式Ⅰ所示:
其中,R1为具有1至32个碳原子的直链、支链或者环状烷基;R2为氢、溴、甲氧基、N,N二甲氨基、乙氧基、羟基、羧基甲氧基或乙酰甲氧基中的一种。
上述半花菁结构的近红外染料的制备方法,包括以下步骤:
首先将2,3,3-三甲基-1-H-吲哚乙基化后的化合物与甲酰化的环己酮在100℃下反应得到Cy7结构的花菁染料;然后用邻苯二胺与2,4-二羟基苯甲醛在160℃下反应得到带有苯并咪唑的间苯二酚;最后在50℃下,以N,N-二甲基甲酰胺为溶剂,用Cy7花菁染料与带有苯并咪唑的间苯二酚反应,采用柱层析方法分离,即得到所述半花菁结构的近红外染料。
本发明还提供了具有硼配位的半花菁结构染料,其结构式如式Ⅱ所示:
其中,R1为具有1至32个碳原子的直链、支链或者环状烷基;R2为氢、溴、甲氧基、N,N二甲氨基、乙氧基、羟基、羧基甲氧基或乙酰甲氧基中的一种。
上述具有硼配位的半花菁结构染料的制备方法是将半花菁结构的近红外染料与三氟化硼乙醚反应,具体过程如下:
上述半花菁结构的近红外染料在光声成像领域的应用。
上述半花菁结构的近红外染料在光热治疗领域的应用。
上述半花菁结构的近红外染料在荧光传感领域的应用。
有益效果:本发明在半花菁结构的染料中引入了苯并咪唑基团,所得染料不仅在近红外区域有吸收和发射,并且对环境的pH值有响应。硼配位后的半花菁染料不仅在近红外区域有吸收和发射,对环境的pH值有响应,并且其光热与化学稳定性得到了提升。
附图说明
图1为实施例1的半花菁结构的近红外染料在酸性和碱性条件下的紫外-可见吸收光谱;
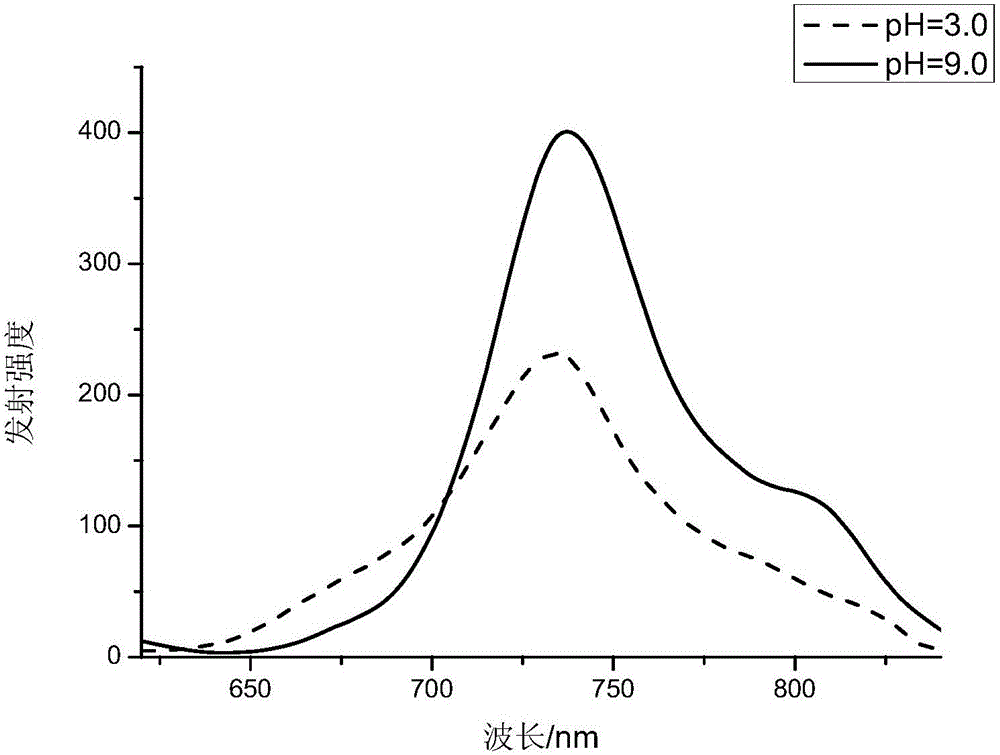
图2为实施例1的半花菁结构的近红外染料在酸性和碱性条件下的的发射光谱。
具体实施方式
为了更好地理解本发明专利的内容,下面通过具体的实例来进一步说明本发明的技术方案。具体包括合成、性质测定。但这些实施例并不限制本发明。
实施例1
带有苯并咪唑结构的半花菁结构染料的制备,R1为乙基,R2为H。
化合物C1的合成:
往双口瓶中加入固体原料2,3,3-三甲基吲哚(478mg,3mmol)后密封,抽真空,充氮气3次;然后往双口瓶中注入溶剂无水乙腈10mL和液体原料碘乙烷(515mg,3.3mmol),在70℃下搅拌反应15h。后处理:将反应后混合物冷却至室温,在搅拌下加入20mL的石油醚。搅拌2h后抽滤,得到粉红色的固体粉末567mg,产率60%。1H NMR(400MHz,DMSO)δ8.00–7.91(m,1H),7.89–7.77(m,1H),7.66–7.56(m,2H),4.47(q,J=7.3Hz,2H),2.81(s,3H),1.51(s,6H),1.42(t,J=7.3Hz,3H)。
化合物C2的合成:
将N,N-二甲基甲酰胺(1.14g,13.1mmol)与二氯甲烷(3mL)加入到双口瓶中,并在冰水浴中冷却10min。再将三氯氧磷(2g,13.1mmol)与二氯甲烷(3mL)的混合溶液逐滴加入到上述冷却好的混合溶液中,搅拌10min。往上述混合液中注入环己酮(430mg,4.38mmol)和二氯甲烷(3mL)的混合溶液,搅拌10min。在N2环境、80℃下搅拌反应6h。后处理:将反应后混合物冷却至室温后,缓慢倒入160g冰水中,有黄色固体析出,静置过夜。抽滤得到613mg黄色粉末,产率80%。不做进一步处理直接投入下一步反应。
化合物C3的合成:
将原料C1(315mg,1mmol)、C2(87.3mg,0.5mmol)、醋酸钾(45mg)加入到双口瓶中,密封,抽真空充氮气3次。注入溶剂醋酸酐8Ml,100℃下搅拌反应2h。后处理:将反应后混合物冷却至室温,抽滤,用少量乙酸酐洗涤,得到绿色带有金属光泽的粉末287.6mg,产率90%。1H NMR(400MHz,DMSO)δ8.25(d,J=14.1Hz,1H),7.62(d,J=7.4Hz,1H),7.47–7.39(m,2H),7.32–7.22(m,1H),6.30(d,J=14.2Hz,1H),4.23(q,J=7.0Hz,2H),2.69(dd,J=15.1,9.0Hz,2H),1.88–1.79(m,1H),1.68(d,J=22.1Hz,6H),1.29(t,J=7.1Hz,3H)。
化合物C4的合成:
将原料邻苯二胺(216mg,2mmol)、2,4-二羟基苯甲醛(276mg,2.2mmol)和焦亚硫酸钠(190mg,2mmol)装入双口瓶,然后注入溶剂N,N-二甲基甲酰胺15mL,在160℃下搅拌反应4小时。后处理:待反应后混合物冷却至室温后,加入50mL去离子水,抽滤得到黄色粉末状固体。将得到的固体溶在乙酸乙酯中,加入硅胶粉旋干过柱提纯,得到淡黄色粉末295mg,产率65%。1H NMR(400MHz,DMSO)δ13.17(s,1H),12.90(s,1H),9.97(s,1H),7.82(d,J=8.4Hz,1H),7.57(d,J=30.2Hz,2H),7.21(s,2H),6.42(d,J=7.7Hz,1H),6.38(s,1H)。
化合物C5的合成:
将原料C4(160mg,0.7mmol)装入双口瓶中,密封,抽真空充氮气3次。注入溶剂N,N-二甲基甲酰胺5mL和三乙胺(70.7mg,0.7mmol),在室温下搅拌10分钟。将原料C3(150mg,0.23mmol)溶解在5mL的DMF中,注入反应瓶中。50℃下搅拌反应4小时。后处理:往反应后混合物中倒入50mL二氯甲烷,用20mL去离子水将有机相洗涤3次。将得到的有机相用无水硫酸钠干燥,旋干,过柱提纯得到蓝黑色粉末62mg,产率42%。1H NMR(400MHz,DMSO)δ8.40(s,1H),8.22(d,J=13.2Hz,1H),7.77(s,1H),7.62(s,2H),7.52(d,J=6.6Hz,1H),7.35(d,J=6.7Hz,1H),7.29(s,1H),7.17(s,1H),7.13-7.17(2H),6.55(s,1H),6.00(d,J=13.0Hz,1H),4.09(q,J=8.9Hz,2H),3.58(t,J=7.0Hz,2H),2.69(t,J=7.2Hz,2H),2.59(t,J=7.0Hz,2H),1.77(s,3H),1.67(s,6H)。
化合物C6的合成:
将原料C5(100mg,0.16mmol)装入双口瓶中,密封,抽真空充氮气3次。注入溶剂四氢呋喃5mL和三氟化硼乙醚(0.1mL,过量)的混合溶液,在室温下搅拌15小时。后处理:反应后混合物直接过柱提纯得到蓝黑色粉末55mg,产率52%。1H NMR(400MHz,DMSO)δ13.20(s,1H),8.47(s,1H),8.19(d,J=13.6Hz,1H),7.89(s,1H),7.67–7.64(m,1H),7.57(d,J=6.8Hz,1H),7.49(d,J=7.6Hz,1H),7.31(d,J=7.6Hz,1H),7.19(d,J=7.9Hz,1H),7.12(dd,J=8.6,4.6Hz,3H),6.43(s,1H),5.92(d,J=13.9Hz,1H),4.03(d,J=6.8Hz,2H),2.72–2.68(m,2H),2.64(d,J=6.0Hz,2H),1.81(t,J=12.6Hz,3H),1.76(d,J=4.0Hz,2H),1.67(s,6H)。
带有苯并咪唑的半花菁结构染料C5的紫外-可见光谱测试:配制两种化合物的稀溶液(10-5M,溶剂分别为pH=3.0和pH=9.0的PBS缓冲液),移取2mL化合物溶液于荧光比色皿中,然后进行吸收和发射光谱的测试,如图1和图2。测试数据表明:该化合物在近红外荧光区具有良好的吸收和发射性质,并且在碱性条件下,该化合物的吸收和发射峰较在酸性条件下有所红移。
实施例2
带有苯并咪唑结构的半花菁结构染料的制备,R1为乙基,R2为羟基。
化合物C1、C2、C3的合成同实施例1。
化合物C4的合成:
将原料邻苯二胺(216mg,2mmol)、2,4,6-三羟基苯甲醛(311mg,2.2mmol)和焦亚硫酸钠(190mg,2mmol)装入双口瓶,然后注入溶剂N,N-二甲基甲酰胺15mL,在160℃下搅拌反应4小时。后处理:待反应后混合物冷却至室温后,加入50mL去离子水,抽滤得到黄色粉末状固体。将得到的固体溶在乙酸乙酯中,加入硅胶粉旋干过柱提纯,得到淡黄色粉末290mg,产率60%。1H NMR(400MHz,Chloroform)δ9.07(s,1H),7.57(d,J=5.6Hz,2H),7.25(d,J=2.9Hz,2H),6.36(d,J=11.4Hz,3H),6.04–5.99(m,2H)。
化合物C5的合成:
将原料C4(170mg,0.7mmol)装入双口瓶中,密封,抽真空充氮气3次。注入溶剂N,N-二甲基甲酰胺5mL和三乙胺(70.7mg,0.7mmol),在室温下搅拌10分钟。将原料C3(150mg,0.23mmol)溶解在5mL的DMF中,注入反应瓶中。50℃下搅拌反应4小时。后处理:往反应后混合物中倒入50mL二氯甲烷,用20mL去离子水将有机相洗涤3次。将得到的有机相用无水硫酸钠干燥,旋干,过柱提纯得到蓝黑色粉末46mg,产率40%。1H NMR(400MHz,Chloroform)δ9.10(s,1H),8.09(s,1H),7.69(s,1H),7.63–7.56(m,3H),7.24(d,J=2.4Hz,2H),7.16(s,1H),6.99(s,1H),6.81(s,1H),6.69(s,1H),6.43(s,1H),5.58(s,1H),5.26(s,1H),3.73(s,1H),3.47(s,1H),2.86(s,2H),2.19(s,2H),1.70(s,2H),1.47(s,3H),1.39(s,6H)。
化合物C6的合成:
将原料C5(100mg,0.15mmol)装入双口瓶中,密封,抽真空充氮气3次。注入溶剂四氢呋喃5mL和三氟化硼乙醚(0.1mL,过量)的混合溶液,在室温下搅拌15小时。后处理:反应后混合物直接过柱提纯得到蓝黑色粉末45mg,产率42%。1H NMR(400MHz,Chloroform)δ9.80(s,1H),8.12(s,1H),7.69(s,1H),7.66–7.58(m,3H),7.23(d,J=2.4Hz,2H),7.11(s,1H),6.93(s,1H),6.60(s,1H),6.41(s,1H),5.57(s,1H),5.21(s,1H),3.73(s,1H),3.41(s,1H),2.85(s,2H),2.11(s,2H),1.72(s,2H),1.50(s,3H),1.32(s,6H)。
- 还没有人留言评论。精彩留言会获得点赞!