长余辉组合物、长余辉元件及波长控制方法与流程
本发明涉及一种容易控制发光波长的长余辉组合物及长余辉元件。又,本发明也涉及一种长余辉材料的波长控制方法。
背景技术:
长余辉材料是在激发光照射期间蓄积能量,在激发光的照射中断后,也通过所蓄积的能量而发光的发光材料。长余辉材料是用于暗处或夜间发光的时钟的文字盘、标识或引导板等的文字、图形等用途的夜光涂料,最近,在即便不供给电力也可照明的长余辉照明中的利用也不断推进。
此种长余辉材料之中,特别是作为发光时间长的长余辉材料,已知有包含eu、ce、tb等稀土类元素的无机盐(例如,参照专利文献1)。
[现有技术文献]
[专利文献]
专利文献1:日本专利特开2006-206618号公报
技术实现要素:
[发明所要解决的问题]
然而,包含这些无机盐的长余辉材料(无机长余辉材料)存在包含稀土类元素,需要高温工艺,不溶于溶剂的缺点。因此,本发明人等人以提供一种可使用有机材料控制发光波长的长余辉材料是为目的而进行了努力研究。
[解决问题的技术手段]
进行了努力研究,结果本发明人等人发现通过在包含第一有机化合物及第二有机化合物的长余辉材料中添加满足规定条件的第三有机化合物,可控制长余辉发光色。本发明是基于此种见解而提出,具有以下的构成。
[1]一种有机长余辉组合物,至少包含第一有机化合物、第二有机化合物及第三有机化合物,且满足下述式(1),
式(1)es1(b)-es1(a)≦0.15ev
(上式中,es1(a)表示包含所述第一有机化合物及所述第二有机化合物的发光的最低激发单重态能级,es1(b)表示所述第三有机化合物的最低激发单重态能级;es1(a)与es1(b)的单位为ev)。
[2]根据[1]所述的长余辉组合物,满足下述式(2),
式(2)0ev<es1(b)-es1(a)≦0.15ev。
[3]根据[1]所述的长余辉组合物,满足下述式(3),
式(3)es1(b)-es1(a)<0ev。
[4]根据[1]至[3]中任一项所述的长余辉组合物,其中包含所述第一有机化合物及所述第二有机化合物的组合物为长余辉材料。
[5]根据[1]至[4]中任一项所述的长余辉组合物,其中通过对所述长余辉组合物的光照射,而使所述第一有机化合物与所述第二有机化合物形成激基复合物。
[6]根据[1]至[5]中任一项所述的长余辉组合物,其中所述第一有机化合物为电子给体,所述第二有机化合物为电子受体。
[7]根据[1]至[6]中任一项所述的长余辉组合物,其中所述第三有机化合物形成凝聚体。
[8]根据[7]所述的长余辉组合物,其中所述凝聚体为准分子。
[9]根据[1]至[8]中任一项所述的长余辉组合物,其中所述第三有机化合物为荧光材料。
[10]一种长余辉元件,在支持体上具有根据[1]至[9]中任一项所述的长余辉组合物。
[11]一种波长控制方法,是控制包含第一有机化合物及第二有机化合物的长余辉材料的发光波长的方法,其特征在于,
在所述长余辉材料中添加满足下述式(1)的第三有机化合物,
式(1)es1(b)-es1(a)≦0.15ev
(上式中,es1(a)表示包含所述第一有机化合物及所述第二有机化合物的发光的最低激发单重态能级,es1(b)表示所述第三有机化合物的最低激发单重态能级;es1(a)与es1(b)的单位为ev)。
[12]根据[11]所述的波长控制方法,其中所述第三有机化合物满足下述式(2),
式(2)0ev<es1(b)-es1(a)≦0.15ev。
[13]根据[12]所述的波长控制方法,其中通过添加所述第三化合物,而将长余辉材料的发光波长控制在短波长侧。
[14]根据[11]所述的波长控制方法,其中所述第三有机化合物满足下述式(3),
式(3)es1(b)-es1(a)<0ev。
[15]根据[14]所述的波长控制方法,其中通过添加所述第三化合物,而将长余辉材料的发光波长控制在长波长侧。
[发明的效果]
根据本发明,可容易地控制长余辉材料的发光波长。又,可将现有的无机长余辉材料难以实现的红色光以较高的色纯度进行发光,又,也可实现白色光。
附图说明
图1是表示本发明的长余辉组合物的发光机制的示意图。
图2是表示tmb/ppt膜的余辉光谱及余辉持续时间特性的图表。
图3是表示m-mtdata/ppt膜的余辉光谱及余辉持续时间特性的图表。
图4是表示cv/ppt膜的余辉光谱及余辉持续时间特性的图表。
图5是表示tmb/ppt/ttpa膜、tmb/ppt/tbrb膜、tmb/ppt/tbrb膜的余辉持续时间特性的图表。
图6是tmb/ppt/ttpa膜、tmb/ppt/tbrb膜、tmb/ppt/tbrb膜、tmb/ppt/tbpe膜的余辉光谱。
图7是tmb/ppt/ttpa膜、tmb/ppt膜、及ttpa甲苯溶液的发光光谱、以及ttpa甲苯溶液的光吸收光谱。
图8是表示tmb/ppt/ttpa膜、tmb/ppt膜、及含有ttp与ttpa的膜的余辉持续时间特性的图表。
图9是tmb/ppt/tbrb膜、tmb/ppt膜、及tbrb甲苯溶液的发光光谱、以及tbrb甲苯溶液的光吸收光谱。
图10是表示tmb/ppt/tbrb膜、tmb/ppt膜的余辉持续时间特性的图表。
图11是tmb/ppt/dcm2膜、tmb/ppt膜、及dcm2甲苯溶液的发光光谱、以及dcm2甲苯溶液光吸收光谱。
图12是表示tmb/ppt/dcm2膜、tmb/ppt膜、及ppt/dcm2膜的余辉持续时间特性的图表。
图13是tmb/ppt/tbpe/dbp膜的发光光谱。
图14是表示tmb/ppt/tbpe/dbp膜的余辉持续时间特性的图表。
具体实施方式
以下,对本发明的内容进行详细说明。以下所记载的构成要件的说明存在基于本发明的代表性的实施形态或具体例而进行的情况,但本发明并不限定于此种实施形态或具体例。再者,在本说明书中使用“~”所表示的数值范围意指包含“~”的前后所记载的数值作为下限值及上限值的范围。又,本发明中所使用的化合物的分子内所存在的氢原子的同位素种类并无特别限定,例如分子内的氢原子可全部为1h,也可一部分或全部为2h(氘d)。
本说明书中的“室温”意指20℃。
本说明书中的“激发光”是激发测定对象物产生发光的光,可使用与所述测定对象物的吸收波长一致的波长的光。
本说明书中的“拉电子基”意指哈密特(hammett)的σp值为正的取代基,“推电子基”意指哈密特的σp值为负的取代基。对于关于哈密特的σp值的说明及各取代基的数值,可参照汉斯,c等人(hansch,c.et.al.,)的《化学评论(chem.rev.)》,91,165–195(1991)中的关于σp值的记载。
[长余辉组合物]
本发明的长余辉组合物至少含有第一有机化合物、第二有机化合物及第三有机化合物。又,本发明的长余辉组合物满足下述式(1)。
式(1)es1(b)-es1(a)≦0.15ev
式(1)中,es1(a)表示包含所述第一有机化合物及所述第二有机化合物的发光的最低激发单重态能级,es1(b)表示所述第三有机化合物的最低激发单重态能级。es1(a)与es1(b)的单位为ev。
根据本发明,通过在由第一有机化合物与第二有机化合物所形成的长余辉材料中使用满足式(1)的关系的第三有机化合物,可容易地控制长余辉材料的发光波长。例如,若使用如满足下述式(2)的第三有机化合物,则可将长余辉材料的发光波长较容易地控制在短波长侧。
式(2)0ev<es1(b)-es1(a)≦0.15ev
又,例如若使用如满足下述式(3)的第三有机化合物,则可将长余辉材料的发光波长较容易地控制在长波长侧。由此,可实现例如色纯度高的红色发光。
式(3)es1(b)-es1(a)<0ev
因此,通过在包含第一有机化合物及第二有机化合物的长余辉材料中使用适当的第三有机化合物,可提供控制为所需的发光波长的长余辉组合物。又,通过适当地选择第三有机化合物,可提供具有多种色调的长余辉组合物。
通过在包含第一有机化合物及第二有机化合物的长余辉材料中进而使用第三有机化合物所控制的发光波长偏移幅度可设为例如5nm以上、10nm以上、20nm以上、40nm以上、70nm以上、100nm以上、130nm以上。又,通过在包含第一有机化合物及第二有机化合物的长余辉材料中进而使用第三有机化合物所控制的发光波长偏移幅度可设为例如300nm以下、200nm以下、150nm以下。此种波长控制通过自包含第一有机化合物及第二有机化合物的长余辉材料向第三有机化合物产生能量转移而实现。再者,此处所谓的波长控制幅度意指峰值波长的偏移幅度。
再者,es1(b)-es1(a)的值也可选自例如-0.1ev以下、-0.2ev以下、-0.3ev以下、-0.4ev以下、-0.5ev以下、-0.6ev以下的区域。
本发明中的“第一有机化合物”优选为伴随着对长余辉组合物的光照射释出电子而成为自由基阳离子状态的分子,本发明中的“第二有机化合物”优选为接受第一有机化合物所释出的电子而成为自由基阴离子状态的分子。“自由基阳离子”或“自由基阴离子”等自由基的存在可通过电子自旋共振(electronspinresonance,esr)测定等进行确认。
本发明中的“长余辉材料”是包含此种第一有机化合物及第二有机化合物的材料,且通过光照射而发光,在停止所述光照射后也继续发光。此处,在本说明书中,有时将自停止光照射的时间点起的发光称为“余辉”,将自停止光照射的时间点起至变得无法检测出发光强度的时间称为“余辉时间”。本申请所谓的长余辉材料意指余辉时间为0.1秒以上的长余辉材料,本发明的长余辉材料的余辉时间优选为1秒以上,更优选为5秒以上,进而优选为5分钟以上,进而更优选为20分钟以上。
发光强度可使用例如分光测定装置(滨松光子(hamamatsuphotonics)公司制造:pma-50)进行测定。未达0.01mcd/m2的发光可视为无法检测出发光强度。
包含第一有机化合物及第二有机化合物的长余辉材料可通过添加第三有机化合物而控制其发光波长。本发明中,将此种包含第一有机化合物、第二有机化合物及第三有机化合物的组合物称为“长余辉组合物”。
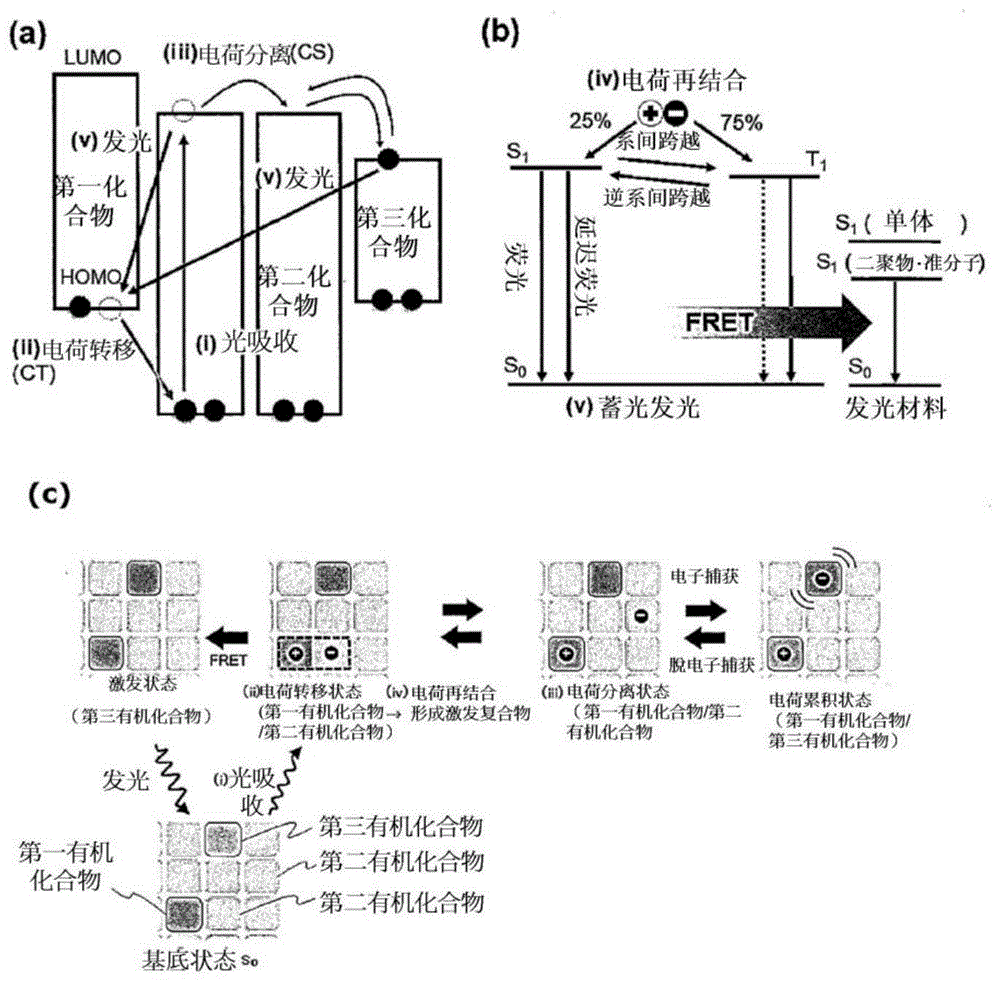
推测本发明的长余辉组合物的发光是通过图1的示意图中所示的发光机制所产生。以下,对所述长余辉组合物的发光机制进行说明。在图1中,(a)表示长余辉组合物中的电子的转移过程,(b)表示长余辉组合物中的能量状态的跃迁过程。图1(c)是表示俯视下第三有机化合物具有捕获功能的情形时的电子的转移过程的图。在图1(c)的各图中,左侧列的黑框所包围的区域表示第一有机化合物,中央列的黑框所包围的区域表示第三有机化合物,其他区域表示第二有机化合物。再者,图1(c)所示的利用第三有机化合物的电子捕获的电荷累积状态在本发明中并非必需的过程。又,以下的说明中的带圆括弧的罗马数字对应图1中的带圆括弧的罗马数字,表示各发光过程的顺序。但,本发明的长余辉组合物的发光机制并不应通过以下所说明的发光机制而限定性地进行解释。
首先,若光照射在长余辉组合物,则如图1(a)所示,第二有机化合物吸收光,电子自其最高占有分子轨道(highestoccupiedmolecularorbital,homo)向最低未占分子轨道(lowestunoccupiedmolecularorbital,lumo)跃迁(i),电子自第一有机化合物的homo向所述第二有机化合物的homo转移(ii)。或者,发生自第一有机化合物的homo向第二有机化合物的lumo的由光吸收所引起的直接电子跃迁。其结果为,生成包含在homo产生有空穴的第一有机化合物(自由基阳离子状态的第一有机化合物)与在lumo进入有多余电子的第二有机化合物(自由基阴离子状态的第二有机化合物)的电荷转移状态。进入第二有机化合物的lumo的电子自所述第二有机化合物分离,并依次向接近的第二有机化合物的lumo转移而扩散(iii)。并且,若电子进入接近第三有机化合物的第二有机化合物的lumo,则所述电子转移至第三有机化合物的lumo后,与第一有机化合物的空穴再结合。通过所述再结合能量,第一有机化合物与第二有机化合物缔合而形成激基复合物(激发状态)。
如图1(b)所示,所述激发状态中的激发单重态s1与激发三重态t1的产生概率为25%:75%,在这些激发状态恢复至基底状态时发射光(v)。作为所述光的发射路径,考虑有以下的(a)~(e)的路径。即,在长余辉材料中,(a)在激发单重态s1恢复至基底状态时发射荧光,(b)在激发三重态t1恢复至基底状态时发射磷光。又,(c)产生自激发三重态t1向激发单重态s1的逆系间跨越,在所述激发单重态s1恢复至基底状态时发射荧光(延迟荧光)。进而,又,(d)产生自激发单重态s1向激发三重态t1的系间跨越,在所述激发三重态t1恢复至基底状态时发射磷光。并且,(e)在第三有机化合物为荧光材料的情形时,长余辉材料的激发单重态s1的能量通过福斯特
在以上的发光机制中,长余辉材料形成激基复合物而发光者可通过所述长余辉材料的发光光谱与以第一有机化合物单独观测的发光光谱及以第二有机化合物单独观测的发光光谱为不同的图案而确认。作为发光光谱的图案不同的例,可列举:发光极大波长不同的情形、发光峰值的半峰宽或上升的斜率不同的情形、发光峰值的数量不同的情形。在利用此种激基复合物的发光中,起因于第一有机化合物与第二有机化合物空间分离,最低激发单重态能级与最低激发三重态能级的差δest极小,容易产生自激发三重态t1向激发单重态s1的逆系间跨越。由此,激发三重态t1的能量也可有效地利用于荧光发光,可获得高的发光效率。此处,经过所述逆系间跨越所发射的荧光是迟于来自由基底状态直接跃迁的激发单重态s1的荧光所观测到的荧光,本说明书中称为“延迟荧光”。
又,如上所述,长余辉材料中的发光路径可列举(a)~(e)的路径,长余辉材料的发光利用(a)~(e)中的至少一个路径进行即可,可利用(a)~(e)中的任一个路径发光,也可利用(a)~(e)中的2个以上的组合发光。
又,第三有机化合物可以各个分子独立的单量体的状态(不凝聚的状态)发光,也可所激发的第三有机化合物的分子彼此凝聚而形成准分子,以所述准分子的状态发光。以准分子的状态发光的光相较于以不凝聚的状态发光的光,存在波长长的倾向。因此,通过使用此种来自准分子的发光,而变得可将长余辉组合物的发光色更容易地向长波长侧控制发光色。
又,此处,以第三有机化合物为荧光材料的情形为例说明发光机制,在使用磷光材料或延迟荧光材料作为第三有机化合物的情形时,也以相同的机制产生发光。此处,作为代替(e)的路径,在第三有机化合物为磷光材料的情形时,通过自长余辉材料利用戴克特(dexter)转移机制转移而来的激发三重态能量,第三有机化合物跃迁至激发三重态t1,在所述激发三重态t1恢复至基底状态时发射磷光。又,在第三有机化合物为延迟荧光材料的情形时,通过自长余辉材料利用戴克特(dexter)转移机制转移而来的激发三重态能量,第三有机化合物跃迁至激发三重态t1后,产生自激发三重态t1向激发单重态t1的逆系间跨越。在所述激发单重态s1恢复至基底状态时发射荧光。
又,如图1(c)所示,经过(i)光吸收、(ii)电荷转移状态及(iii)电荷分离状态自第二有机化合物交接电子的第三有机化合物也可发挥暂时捕捉所述电子并累积后,通过热活化等释出电子而使所述电子返回至第二有机化合物的捕获功能(电子捕获-脱电子捕获功能)。在所述情形时,自第三有机化合物向第二有机化合物交接的电子在第二有机化合物彼此之间转移而与第一有机化合物的空穴再结合(iv)。通过所述电荷再结合,在第一有机化合物与第二有机化合物之间形成激基复合物,通过所述(a)~(d)的路径产生发光。进而,又,(e)激基复合物的激发能量通过福斯特
其次,对所述长余辉组合物所含有的长余辉材料(第一有机化合物及第二有机化合物)及第三有机化合物、根据需要添加的其他成分进行说明。
[长余辉材料]
本发明的长余辉组合物所含有的长余辉材料包含第一有机化合物及第二有机化合物,也可仅包含第一有机化合物及第二有机化合物。
长余辉材料的发光波长并无特别限制,优选为200nm~2000nm。例如,可选自400nm以上、600nm以上、800nm以上、1000nm以上、1200nm以上的波长区域,且可选自1500nm以下、1100nm以下、900nm以下、700nm以下、500nm以下的区域。
以下,对于构成长余辉材料的第二有机化合物及第一有机化合物进行说明。
(第一有机化合物)
构成长余辉材料的第一有机化合物是可与第二有机化合物协动而实现余辉发射的化合物,优选为电子给体。此处所谓的电子给体意指对第二有机化合物容易释出电子的化合物。并且,第一有机化合物优选为与第二有机化合物形成激基复合物而发光的化合物。又,第一有机化合物的自由基阳离子状态稳定,在与第二有机化合物组合而构成长余辉材料时,也优选为以10k(及优选为在20℃下)发射余辉的化合物。由此,可获得余辉持续时间更长的长余辉组合物。
优选为第一有机化合物的homo高于第二有机化合物的homo,其lumo高于第二有机化合物的lumo。由此,变得容易产生自第一有机化合物的homo向第二有机化合物的homo或lumo的电子转移,可高效率地生成电荷分离状态。具体而言,第一有机化合物的homo优选为-3.5ev~-8.0ev,更优选为-4.0ev~-7.0ev,进而优选为-4.5ev~-6.0ev。
第一有机化合物的homo可通过光电子光谱法或循环伏安法测定,又,lumo可通过循环伏安法或吸收光谱求得。
作为第一有机化合物,就自由基阳离子的稳定性的方面而言,优选为使用具有推电子基的化合物,更优选为使用与推电子基具有共轭系的化合物,进而优选为使用具有二烷基氨基与芳香环的化合物。又,第一有机化合物优选为不包含稀土类原子及金属原子,更优选为仅包含选自碳、氢、氮、氧、硫、磷的原子。
在第一有机化合物为具有二烷基氨基与芳香环的化合物的情形时,所述芳香环可为芳香族烃,也可为芳香族杂环,优选为芳香族烃。关于芳香族烃的说明及优选的范围,可参照关于所述的ar15及ar16为经取代或未经取代的亚芳基时的构成亚芳基的芳香环的说明及优选的范围。又,关于芳香族杂环的说明及优选的范围,可参照关于所述的ar15及ar16为经取代或未经取代的亚杂芳基时的构成亚杂芳基的杂环的说明及优选的范围。这些之中,芳香环优选为苯环、联苯环,更优选为联苯环。芳香环也可经取代基取代。关于可取代在芳香环上的取代基的具体例及优选的范围,可参照所述的ar15及ar16中的可取代在亚芳基等上的取代基的具体例及优选的范围。另一方面,二烷基氨基优选为取代在芳香环上。第一有机化合物所包含的二烷基氨基的数量可为1个,也可为2个,优选为1~4个,更优选为2个或4个,进而优选为2个。关于二烷基氨基的烷基的说明及优选的范围、具体例,可参照关于下述的r21等中的烷基的说明及优选的范围、具体例。二烷基氨基的烷基也可经取代基取代。关于所述取代基的说明及优选的范围,可参照关于r21等中的可取代在烷基上的取代基的说明及优选的范围。
第一有机化合物优选为下述通式(1)所表示的化合物。
[化1]
通式(1)
通式(1)中,ar21表示经取代或未经取代的亚芳基。关于构成ar21的亚芳基的芳香环的说明及优选的范围、亚芳基的具体例,可参照关于所述的ar15及ar16为经取代或未经取代的亚芳基时的构成亚芳基的芳香环的说明及优选的范围、亚芳基的具体例。ar21优选为经取代或未经取代的亚苯基、经取代或未经取代的联苯二基,更优选为经取代或未经取代的联苯二基。关于可取代在亚芳基上的取代基的具体例及优选的范围,可参照所述的ar15及ar16中的可取代在亚芳基等上的取代基的具体例及优选的范围。
r21~r24分别独立地表示经取代或未经取代的烷基。r21~r24可相互相同也可不同。r21~r24中的烷基可为直链状、支链状、环状中的任一者。优选的碳数为1~20,更优选为1~10,进而优选为1~6。例如可例示:甲基、乙基、正丙基、异丙基等。作为可取代在烷基上的取代基,可列举:碳数6~40的芳基、碳数3~40的杂芳基、碳数2~10的烯基、碳数2~10的炔基等。这些取代基也可进而经取代基取代。
以下,列举可用作第一有机化合物的优选的化合物。但,本发明中可使用的第一有机化合物并不应通过这些具体例而限定性地进行解释。
[化2]
又,本发明中所使用的第一有机化合物也可为在作为单体的第一有机化合物中导入聚合性基,将其作为单体进行聚合而成的聚合物。作为可用作第一有机化合物的聚合物的具体例,可列举具有下述结构的聚合物。在下述式中,n为1以上的整数。但,在本发明中可用作第一有机化合物的聚合物并不应通过所述具体例而限定性地进行解释。
[化3]
(第一有机化合物的含量)
长余辉材料中的第一有机化合物的含量相对于第一有机化合物与第二有机化合物的合计摩尔数优选为未达60mol%,更优选为未达30mol%,进而优选为未达20mol%,进而更优选为未达10mol%。又,长余辉材料中的第一有机化合物的含量相对于第一有机化合物与第二有机化合物的合计摩尔数优选为超过0.001mol%,更优选为超过0.01mol%,进而优选为超过1mol%。通过改变第一有机化合物的含量,可控制长余辉组合物发光的发光色。例如,可见第一有机化合物的含量越多,激基复合物形成越强,成为越长波长的发光的现象。通过利用此种现象,可控制长余辉组合物的发光色或发光时间。例如,包含下述的第二有机化合物(左侧)及第一有机化合物(右侧)的长余辉材料中,在第一有机化合物的含量未达30mol%的情形时,可观测到蓝色光,在第一有机化合物的含量超过30mol%的情形时,观测到黄色光。
[化4]
(第二有机化合物)
构成长余辉材料的第二有机化合物是可与第一有机化合物协动而实现余辉发射的化合物,优选为电子受体。此处所谓的电子受体意指自第一有机化合物容易接受电子的化合物。并且,第二有机化合物优选为与第一有机化合物形成激基复合物而发光的化合物。又,第二有机化合物的自由基阴离子状态稳定,在与第一有机化合物组合而构成长余辉材料时,也优选为以10k(及优选为在20℃下)发射余辉的化合物。由此,可获得余辉持续时间更长的长余辉组合物。
第二有机化合物的homo与lumo的能隙优选为1.0ev~3.5ev,更优选为1.5ev~3.4ev,进而优选为2.0ev~3.3ev。由此,伴随着对长余辉组合物的光照射,可高效率地产生自其homo向lumo的电子跃迁。又,第二有机化合物的lumo优选为6.0ev以下,更优选为5.5ev以下,进而优选为5.0ev以下。由此,电荷分离状态生成后,电子容易自第二有机化合物的lumo向lumo转移,可高概率地发生与空穴的再结合。
第二有机化合物的homo可通过光电子光谱法或循环伏安法测定,又,lumo可通过循环伏安法或吸收光谱求得。
又,关于第二有机化合物,为了在室温中可以玻璃状态存在,优选为具有高的玻璃化温度tg的化合物,又,优选为在成膜时可获得高的膜密度的化合物。通过膜中的第二有机化合物的密度高,电荷分离状态生成后,电子容易自第二有机化合物的lumo向lumo扩散,可高概率地发生电子与空穴的再结合。
作为第二有机化合物,就自由基阴离子的稳定性的方面而言,优选为使用具有阴电性高的原子或拉电子基的化合物,更优选为使用与阴电性高的原子或拉电子基具有共轭系的化合物。又,第二有机化合物优选为不包含稀土类原子及金属原子,更优选为仅包含选自碳、氢、氮、氧、硫、磷中的原子。
作为第二有机化合物的优选的例子,可列举包含1个以上的氧化膦结构r3p(=o)(r表示取代基,3个r彼此可相互相同也可不同)的化合物,作为进而优选的例子,可列举包含1个以上的氧化膦结构r3p(=o)且除此以外包含1个以上的杂原子的化合物。作为杂原子,可列举n、o、s、p等,可仅包含其中1种,也可包含2种以上。第二有机化合物所包含的氧化膦结构的数量优选为2个以上,在所述情形时,多个氧化膦结构可相互相同也可不同。又,多个氧化膦结构优选为其取代基r的至少一个经由杂原子连结于其他氧化膦结构的取代基r,更优选为其取代基r的至少一个经由杂原子连结于其他氧化膦结构的取代基r,并且其所连结的取代基r中的与键结杂原子的原子不同的其他原子彼此相互以单键连结。
氧化膦结构的取代基r优选为经取代或未经取代的芳基、经取代或未经取代的杂芳基。关于构成芳基的芳香环的说明及优选的范围、芳基的具体例,可参照关于下述的ar11等为经取代或未经取代的芳基时的构成芳基的芳香环的说明及优选的范围、芳基的具体例。关于构成杂芳基的杂环的说明及优选的范围、杂芳基的具体例,可参照关于下述的ar11等为经取代或未经取代的杂芳基时的构成杂芳基的杂环的说明及优选的范围、杂芳基的具体例。关于可取代在芳基及杂芳基上的取代基的具体例及优选的范围,可参照ar11等中的可取代在芳基及杂芳基上的取代基的具体例及优选的范围。
第二有机化合物优选为下述通式(2)所表示的化合物。
[化5]
通式(2)
在通式(2)中,ar11~ar14分别独立地表示经取代或未经取代的芳基或者经取代或未经取代的杂芳基,优选为经取代或未经取代的芳基。ar11~ar14可相互相同也可不同。ar15及ar16分别独立地表示经取代或未经取代的亚芳基或者经取代或未经取代的亚杂芳基,ar15与ar16也可相互以单键连结而形成缩环结构。ar15及ar16可相互相同也可不同。ar15及ar16优选为经取代或未经取代的亚芳基,更优选为所述亚芳基彼此相互以单键连结而形成缩环结构。
ar11~ar14为经取代或未经取代的芳基时的构成芳基的芳香环、以及ar15及ar16为经取代或未经取代的亚芳基时的构成亚芳基的芳香环可为单环,也可为2个以上的芳香环缩合而成的稠环,还可为2个以上的芳香环连结而成的连结环。在2个以上的芳香环连结的情形时,可为连结成直链状,也可为连结成支链状。构成芳基及亚芳基的芳香环的碳数优选为6~40,更优选为6~22,进而优选为6~18,进而更优选为6~14,特别优选为6~10。作为芳基的具体例,可列举苯基、萘基、联苯基。作为亚芳基的具体例,可列举亚苯基、萘二基、联苯二基。这些之中,作为ar11~ar14特别优选者为经取代或未经取代的苯基。又,作为ar15及ar16特别优选者为经取代或未经取代的亚苯基,尤其更优选为所述亚苯基彼此相互以单键连结而形成3环结构(苯环与包含x11的五员环与苯环的3环结构)。
ar11~ar14为经取代或未经取代的杂芳基时的构成杂芳基的杂环、以及ar15及ar16为经取代或未经取代的亚杂芳基时的构成亚杂芳基的杂环可为单环,也可为1个以上的杂环与芳香环或杂环缩合而成的稠环,还可为1个以上的杂环与芳香环或杂环连结而成的连结环。构成杂芳基的杂环的碳数优选为3~40,更优选为5~22,进而优选为5~18,进而更优选为5~14,特别优选为5~10。构成杂环的杂原子优选为氮原子。作为杂环的具体例,可列举吡啶环、哒嗪环、嘧啶环、三唑环、苯并三唑环。
作为ar11~ar14中的可取代在芳基及杂芳基上的取代基、ar15及ar16中的可取代在亚芳基及亚杂芳基上的取代基,例如可列举:羟基、卤素原子、碳数1~20的烷基、碳数1~20的烷氧基、碳数1~20的烷硫基、碳数1~20的烷基取代氨基、碳数1~20的芳基取代氨基、碳数6~40的芳基、碳数3~40的杂芳基、碳数2~10的烯基、碳数2~10的炔基、碳数2~20的烷基酰胺基、碳数7~21的芳基酰胺基、碳数3~20的三烷基硅烷基等。这些具体例之中,可经取代基取代者也可进而经取代。更优选的取代基为碳数1~20的烷基、碳数1~20的烷氧基、碳数1~20的烷硫基、碳数1~20的烷基取代氨基、碳数1~20的芳基取代氨基、碳数6~40的芳基、碳数3~40的杂芳基。
x11表示nr11、o或s,r11表示氢原子或取代基。作为r11可采取的取代基,例如可列举:碳数1~20的烷基、碳数6~40的芳基、碳数3~40的杂芳基、碳数2~10的烯基、碳数2~10的炔基等。这些取代基也可进而经取代基取代。r11优选为氢原子、经取代或未经取代的芳基,更优选为经取代或未经取代的芳基,进而优选为经取代或未经取代的苯基。
以下,列举可用作第二有机化合物的优选的化合物。但,本发明中可使用的第二有机化合物并不应通过这些具体例而限定性地进行解释。
[化6]
又,本发明中所使用的第二有机化合物也可为在作为单体的第二有机化合物中导入聚合性基,将其作为单体进行聚合而成的聚合物。可用作第二有机化合物的聚合物的具体例,可列举具有下述结构的聚合物。在下述式中,n为1以上的整数。但,本发明中可用作第二有机化合物的聚合物并不应通过这些具体例而限定性地进行解释。
[化7]
[第三有机化合物]
本发明的长余辉组合物中所使用的第三有机化合物优选为发光材料。第三有机化合物的发光波长例如可选自可见光区域或近红外线区域。具体而言,第三有机化合物的发光波长优选为200nm~2000nm。例如,可选自400nm以上、600nm以上、800nm以上、1000nm以上、1200nm以上的波长区域,且可选自1500nm以下、1100nm以下、900nm以下、700nm以下、500nm以下的区域。又,第三有机化合物也优选为具有载流子捕获功能。
第三有机化合物可为荧光材料、磷光材料、延迟荧光材料中的任一者,根据目标的发光色,可自公知者中选择而使用。此处,“荧光材料”是在室温下荧光的发光强度比磷光的发光强度更高的发光材料,“磷光材料”是在室温下磷光的发光强度比荧光的发光强度更高的发光材料,“延迟荧光材料”是在室温下观测到余辉持续时间短的荧光及余辉持续时间长的荧光(延迟荧光)的两者的发光材料。通常的荧光(非延迟荧光的荧光)的余辉持续时间为ns等级,磷光通常余辉持续时间为ms等级,故而荧光与磷光可通过余辉持续时间区别。又,有机金属络合物以外的发光性的有机化合物通常为荧光材料或延迟荧光材料。
例如,作为荧光材料,可优选地使用实施例中所使用的ttpa(绿色发光材料)、tbrb(橙色发光材料)、dcm2(红色发光材料)。又,作为磷光材料,可优选地使用实施例中所使用的ir(ppy)3、ir(piq)3、ir(btp)2(acac)、ptoep。
长余辉组合物中的第三有机化合物的含量相对于第一有机化合物、第二有机化合物及第三有机化合物的合计摩尔数优选为未达50mol%,更优选为未达25mol%,进而优选为0.001~20mol%,进而更优选为0.001~10mol%。通过改变长余辉组合物中的第三有机化合物的浓度,可控制长余辉组合物的发光波长。例如,使用es1(b)-es1(a)成为负值的第三有机化合物的长余辉组合物中,若提高第三有机化合物的浓度,则存在发光波长发生长波长化的倾向,也可获得近红外线区域的发光。
[其他成分]
长余辉组合物可仅包含第一有机化合物、第二有机化合物及第三有机化合物,也可添加其他成分,还可包含溶解长余辉组合物的成分的溶剂。作为其他成分,可列举载流子捕获材料。通过添加载流子捕获材料,而产生通过电荷分离所产生的自第二有机化合物的自由基阳离子向载流子捕获材料的电子转移,可在载流子捕获材料中更稳定地累积电子。载流子捕获材料中所累积的电子通过热等能量再次返回至第二有机化合物并向第三有机化合物转移,与第一有机化合物的空穴再结合,由此获得长余辉发光。
作为载流子捕获材料,优选为其lumo能级与第二有机化合物的lumo能级相近的材料。载流子捕获材料的lumo能级优选为比第二有机化合物的lumo能级低0.001ev以上,更优选为低0.01ev以上,进而优选为低0.1ev以上。又,载流子捕获材料的lumo能级与第二有机化合物的lumo能级的差优选为0.5ev以下,更优选为0.4ev以下,进而优选为0.3ev以下。
[发光的形态]
本发明的长余辉组合物通过照射光,而在停止光照射后,也长时间继续发光(余辉)。
长余辉组合物的发光至少包含来自长余辉材料的发光及来自第三有机化合物的发光。来自长余辉材料的发光优选为所激发的第一有机化合物与第二有机化合物缔合(凝聚)而形成激基复合物,由来自所述激基复合物的发射缓和所引起的发光(激基复合物发光)。在长余辉材料形成激基复合物而发光的情形时,来自长余辉材料的发光可仅为激基复合物发光,也可包含来自未与第二有机化合物缔合的第一有机化合物的发光、或来自未与第一有机化合物缔合的第二有机化合物的发光。发光的光可为荧光及磷光中的任一者,也可为荧光与磷光的两者,进而也可包含延迟荧光。
用于自长余辉组合物获得余辉的激发光可为太阳光,也可为来自出射特定的波长范围的光的人工光源的光。
为了自长余辉组合物获得余辉而进行的光照射的时间优选为1μ秒以上,更优选为1m秒以上,进而优选为1秒以上,进而更优选为10秒以上。由此,可充分地生成自由基阴离子与自由基阳离子,在停止光照射后,可长时间继续发光。
长余辉组合物的发光波长可通过第一有机化合物~第三有机化合物的组合而广泛控制,并无特别限制,优选为200nm~2500nm。又,长余辉组合物的发光波长也优选为380nm~750nm的可见光区域,也优选为700nm~2500nm的近红外线区域。近红外线区域的光由于对活体的透过性高,故而可用于活体深部的可视化或诊断,可用于生物学或医疗技术、光损耗较少的通信技术等各种领域。
[长余辉组合物的形态]
本发明的长余辉组合物只要含有第一有机化合物、第二有机化合物及第三有机化合物即可,其形态并无特别限定。因此,可为混合有第一有机化合物、第二有机化合物及第三有机化合物的混合物,也可为构成长余辉材料的第二有机化合物及第一有机化合物与第三有机化合物分别存在于不同区域者。作为混合有长余辉材料与第三有机化合物的混合物,例如可列举将长余辉材料与第三有机化合物溶解于溶剂而获得的溶液、或包含长余辉材料及第三有机化合物的薄膜(长余辉膜)。又,作为第二有机化合物、第一有机化合物及第三有机化合物分别存在于不同区域的例子,可列举:具有相对于第二有机化合物及第三有机化合物的各者包含100质量倍以上的量的第一有机化合物的区域、相对于第一有机化合物及第三有机化合物的各者包含100质量倍以上的量的第二有机化合物的区域、及相对于第一有机化合物及第二有机化合物的各者包含100重量倍以上的量的第三有机化合物的区域者,具有不包含第二有机化合物及第三有机化合物而包含第一有机化合物的区域、不包含第一有机化合物及第三有机化合物而包含第二有机化合物的区域、及不包含第一有机化合物及第二有机化合物而包含第三有机化合物的区域者,这些3个区域相互相接者,这些区域为层状(包含薄膜)者。
使用长余辉材料与第三有机化合物的薄膜可为通过干式工艺、湿式工艺的任一者而成膜的薄膜。例如,可为在经加热熔融的第二有机化合物的熔融液中添加第一有机化合物及第三有机化合物进行混合,并进行冷却而获得的玻璃状的薄膜。通过湿式工艺成膜时所使用的溶剂只要为与成为溶质的第一有机化合物或第二有机化合物、第三有机化合物具有相溶性的有机溶剂即可。使用有机溶剂,例如可制备第二有机化合物、第一有机化合物及第三有机化合物的混合溶液,或制备仅溶解有第二有机化合物的溶液,或制备仅溶解有第一有机化合物的溶液,或制备仅溶解有第三有机化合物的溶液。若将混合溶液涂布于支持体上并进行干燥,则可形成第一有机化合物、第二有机化合物及第三有机化合物的混合薄膜,又,通过在支持体上依次涂布第一有机化合物的溶液、第二有机化合物的溶液及第三有机化合物的溶液并进行干燥,也可将第一有机化合物的薄膜、第二有机化合物的薄膜及第三有机化合物的薄膜以相互相接的方式形成(第一有机化合物的溶液、第二有机化合物的溶液及第三有机化合物的溶液的涂布顺序不同)。
薄膜的平面形状可根据用途适当选择,例如可为正方形状、长方形状等多边形状、正圆状、椭圆状、长圆状、半圆状之类的连续形状,也可为对应几何学花纹或文字、图形等的特定图案。
[长余辉元件]
本发明的长余辉元件在支持体上具有本发明的长余辉组合物。长余辉组合物通常是制成膜状而形成于支持体上。形成于支持体上的膜可为单层膜,也可为包含多层膜。单层膜或多层膜中的一部分膜可制成包含第一有机化合物、第二有机化合物及第三有机化合物中的两者以上的膜。又,也可将多层膜中的一部分膜制成包含第二有机化合物且不包含第一有机化合物及第三有机化合物的膜,将一部分膜制成包含第一有机化合物且不包含第二有机化合物及第三有机化合物的膜,进而将一部分膜制成包含第三有机化合物且不包含第一有机化合物及第二有机化合物的膜。此时,这些3种膜也可以相互相接的方式构成。
关于长余辉组合物,可参照长余辉组合物的栏的对应的记载。再者,关于长余辉组合物的形态,可参照长余辉组合物的形态的栏的关于薄膜的记载。
关于支持体,并无特别限制,只要为长余辉材料所惯用的支持体即可。作为支持体的材料,例如可列举:纸、金属、塑料、玻璃、石英、硅等。由于也可形成于具有可挠性的支持体,故而也可根据用途制成各种形状。
长余辉膜优选为整体通过密封材所覆盖。密封材可使用玻璃、环氧树脂等水或氧的透过率低的透明材料。
根据本发明,可提供透明的组合物。因此,与现有的无机材料不同,可用于多种用途加以应用。例如,通过利用由玻璃等透明材料制成的2片支持体夹入本发明的透明的长余辉组合物,可形成透明的长余辉板等。若调节支持体的透明性,则也可制成半透明的长余辉板。又,根据本发明,通过层叠发光色不同的透明的长余辉膜,也可调整向外部释出的光的色调。
[长余辉组合物的用途]
本发明的长余辉组合物如上所述,具有容易控制发光波长的特征,并且例如,仅通过将作为有机化合物的第一有机化合物、第二有机化合物及第三有机化合物在溶剂中混合而进行涂布,便可构成长余辉品。因此,本发明的长余辉组合物与使用包含稀有元素的无机材料的高温焙烧、微粒子化及分散步骤而构成长余辉品的无机长余辉材料相比,具有如下优点:容易供应材料,并且将长余辉品的制造成本抑制得低,又,可使长余辉品具有透明性、可挠性、柔软性。因此,本发明的长余辉组合物除可用于通常的长余辉品以外,也可有效利用所述的特征,实现至今没有的新颖的用途。
例如,本发明的长余辉组合物通过与第一有机化合物及第二有机化合物组合的第三有机化合物的选择,可在遍及蓝色光~近红外线区域的广的波长范围内控制发光波长。其中,发出绿色光的长余辉组合物由于绿色的视感强度强,故而可有效地用作标识用的长余辉涂料。又,发出红色~近红外区域的光的长余辉组合物由于所述波长区域的光容易透过活体,故而作为用于生物学成像的标记材料有用。进而,通过组合使用各种发光色的长余辉组合物,可提供设计性优异的物品,又,也可应用于护照等的公文防伪系统等。
又,通过将本发明的长余辉组合物溶解于溶剂,可构成涂布性优异的长余辉涂料。通过将此种长余辉涂料整面涂布于道路或建筑物内装面,可实现不需要电源的大规模长余辉照明。又,在通过所述长余辉涂料划出车道外侧线的情形时,变得即便在暗处也可识别车道外侧线,从而可显著提高车辆通行的安全性。
进而,若使用通过所述长余辉涂料所绘制的安全引导标识,则除实现灾害时长时间的安全的避难引导以外,可将所述长余辉涂料涂布于节能照明、住宅建筑材料、铁道、移动设备等而构筑灾害时避难系统。
此外,含有本发明的长余辉组合物的长余辉涂料也可用作印刷用墨水。由此,可获得设计性优异,也可用于暗处或灾害时的引导的印刷物。此种长余辉印刷用墨水可优选地用于例如封皮、封装、海报、叠合式封装(packageonpackage,pop)、贴纸(sticker)、引导看板、避难引导信号、安全用品、防盗用品的用途的印刷。
另外,通过使用第一有机化合物、第二有机化合物及第三有机化合物的至少任一者为聚合物的长余辉组合物(长余辉聚合物)、或在本发明的长余辉组合物中添加有市售的半导体性聚合物的组合物,可获得长余辉成形品。
作为此种长余辉成形品,例如可列举:发光广告牌、商品显示器、液晶背光源、照明显示器、照明器具罩、交通标识、安全标识、夜间视认性提升构件、信号板、屏幕、反射板或仪表零件等汽车零件、娱乐设施的游戏道具或玩具、笔记型计算机、移动电话等移动设备、以及汽车室内或建筑物内的标示按钮、时钟的文字盘、配件类、文具类、运动用品、各种电气/电子/办公自动化(officeautomation,oa)设备等领域中的外壳或开关、按钮类等。
此外,由于本发明的长余辉组合物的透明性优异,故而通过将所述长余辉组合物涂布于玻璃的表面,或将长余辉组合物与树脂的混合物成形为薄板状,可实现具有长余辉功能的调光窗。进而,在将包含长余辉组合物的薄板与反射板层叠的情形时,可获得高亮度的长余辉板。此种长余辉板可作为发光引导磁砖而用于伴随着各种灾害的避难路径道路构件、楼梯段板、竖板、框材、槽盖材、室外停车场构件、港口配备构件、道路设施安全构件、高空作业支架构件、海上浮动设施支架构件、山路步行道相关构件、耐盐害性耐候招牌或广告牌等。
并且,通过将本发明的长余辉组合物涂布于纤维,可获得长余辉纤维或使用其的布类或长余辉衣类。作为此种长余辉纤维品,可列举:夜间用作业服、帽子、紧急通道用地毯、婚礼服装、壁挂、车辆用内装材等。
此外,本发明的长余辉组合物可构成长余辉膜、长余辉带、长余辉封条、长余辉建筑材料、长余辉喷雾器等各种原材料。在任一者中,均可容易地控制发光波长,及可由有机化合物构成各成分,由此可对各原材料赋予颜色的选择范围广,透明且柔软的性状,从而可制成设计性或标识性、操作性优异者。例如,长余辉膜可广泛地用作避难引导或防灾物品的包装材。
进而,本发明的长余辉组合物有长余辉材料的电荷分离状态的寿命相对较长的倾向。因此,可在广泛领域中用于各种用途。例如,本发明的长余辉组合物可应用于通过光能形成电荷分离状态而导入至物质生产的人工光合成的领域。而且,本发明的长余辉组合物也可有效地用作应答热能或力学能的元件。例如,作为应答热能的元件,可列举通过激发光的照射而使长余辉材料成为电荷分离状态后,对长余辉组合物施加热而使其瞬间发光的热开关。此外,作为应答力学能的元件,可列举通过对使长余辉材料成为电荷分离状态的长余辉组合物施加压力等力学能而发光的元件、或通过对使长余辉材料成为电荷分离状态的长余辉组合物施加压力等力学能而发光状态发生变化的元件。另外,作为这些的应用例,可列举应答热等外部刺激的互动发光技术。
[实施例]
以下列举实施例更具体地说明本发明的特征。以下所示的材料、处理内容、处理程序等只要不脱离本发明的主旨,则可进行适当变更。因此,本发明的范围并不应通过以下所示的具体例而限定性地进行解释。再者,激发光使用340nm的发光二极管(lightemittingdiode,led)灯(光机械(thorlabs)公司制造:m340l4)。又,发光光谱、余辉光谱及余辉持续时间、光致发光量子产率的测定使用分光测定装置(堀场若比伊冯(horibajobinyvon)公司制造:弗洛麦克斯(fluoromax)、滨松光子(hamamatsuphotonics)公司制造:pma-12)、多通道光谱仪(海洋光学(oceanoptics)公司制造:qe-pro)、万用表(是德科技(keysight)公司制造:34461a)、光致发光(photoluminescence,pl)量子产率测定装置(滨松光子(hamamatsuphotonics)公司制造:库安陶若斯-qy(quantaurus-qy))进行。
实施例或比较例所使用的化合物的最低激发单重态能级(es1)与最低激发三重态能级(et1)通过以下的程序而求得。
(1)最低激发单重态能级(es1)
制备包含测定对象化合物的10-5mol/l浓度的甲苯溶液或氯仿溶液。在常温(300k)下对所述试样照射激发光,测定荧光光谱,累计激发光刚入射后至入射后100纳秒的发光,由此获得纵轴为发光强度、横轴为波长的荧光光谱。荧光光谱将纵轴设为发光、将横轴设为波长。针对所述发光光谱的短波长侧的上升引出切线,求得所述切线与横轴的交点的波长值λedge[nm]。将所述波长值通过如下所示的换算式换算成能量值所得的值设为es1。
换算式:es1[ev]=1239.85/λedge
(2)最低激发三重态能级(et1)
将与最低激发单重态能级(es1)相同的试样冷却至77[k],对磷光测定用试样照射激发光(340nm),使用pma-12,测定磷光强度。累计激发光入射后1毫秒至入射后10毫秒的发光,由此获得纵轴为发光强度、横轴为波长的磷光光谱。针对所述磷光光谱的短波长侧的上升引出切线,求得所述切线与横轴的交点的波长值λedge[nm]。将所述波长值通过如下所示的换算式换算成能量值所得的值设为et1。
换算式:et1[ev]=1239.85/λedge
针对磷光光谱的短波长侧的上升的切线是以如下方式引出。自磷光光谱的短波长侧至光谱的极大值中最短波长侧的极大值在光谱曲线上移动时,朝向长波长侧考虑曲线上的各点处的切线。所述切线随着曲线上升(即随着纵轴增加),斜率增加。将所述斜率的值取极大值的点处所引出的切线设为针对所述磷光光谱的短波长侧的上升的切线。
[实施例及比较例中所使用的化合物]
实施例及比较例中,使用tmb作为第一有机化合物,使用ppt、m-mtdata、cv作为第二有机化合物,使用4czbn、2czpn、flrpic、tbpe、4czipn、5czpn、ttpa、tbrb、dcm2、nilered、dbp、ir(ppy)3、ir(piq)3、ir(btp)2(acac)、ptoep作为第三有机化合物。这些化合物的结构如下。
[化8-1]
第一有机化合物
第二有机化合物
[化8-2]
第三有机化合物
[化8-3]
[化8-4]
[1]第一有机化合物与第二有机化合物的组合的研究
在氮气氛围的手套箱中,将石英基板加热至ppt的熔点以上(250℃以上)的温度,在所述石英基板上使ppt熔解。在所述ppt的熔融液中以1mol%的浓度添加tmb进行混合,并进行急冷,由此形成玻璃状态的长余辉材料膜,通过玻璃基板与紫外线硬化树脂密封。对所形成的膜照射340nm的激发光,结果确认到发光,在停止照射后,也确认到余辉。图2表示余辉光谱及余辉持续时间特性。余辉光谱的峰值波长为526nm。
变更如下方面,即,使用m-mtdata或cv代替tmb,同样地形成玻璃状态的膜。使用m-mtdata及cv的膜在停止照射后也确认到余辉。针对使用m-mtdata的膜,调查余辉光谱与余辉持续时间特性,所得的结果示于图3,针对使用cv的膜,调查余辉光谱与余辉持续时间特性,所得的结果示于图4。使用m-mtdata的膜的余辉光谱的峰值波长为523nm,使用cv的膜的余辉光谱的峰值波长为513nm。
[2]溶液及膜状的长余辉组合物的制备与特性的评价
(实施例1~实施例7、比较例1~比较例4)
相对于ppt99mol%将tmb以成为1mol%的量进行混合,在ppt的熔点以上(250℃以上)的温度下加热熔解后,进行急冷而获得长余辉材料膜。测定来自所述膜的发光的最低激发单重态能级,设为es1(a)。
又,针对4czbn、2czpn、5czpn、flrpic、tbpe、4czipn、ttpa、tbrb、dcm2、nilered、dbp的各者,制备10-5mol/l浓度的甲苯溶液,测定最低激发单重态能级,由此决定各化合物的es1(b)。结果示于表1。
相对于ppt98mol%将tmb以成为1mol%的量进行混合,将作为第三化合物的4czbn以成为1mol%的量进行混合,加热熔解后,进行急冷而获得长余辉膜。进而,使用2czpn、5czpn、flrpic、tbpe、4czipn、ttpa、tbrb、dcm2、nilered、dbp代替4czbn作为第三化合物,同样地获得长余辉膜。
针对所制备的各长余辉膜,照射340nm激发光,结果确认到发光,在停止照射后,也自所有的长余辉膜确认到余辉。调查长余辉膜的余辉光谱及余辉持续时间特性,决定峰值波长与半峰宽(fullwidthathalfmaximum,fwhm)。结果示于表1。作为代表性的余辉持续时间特性,包含ttpa、tbrb、dcm2的长余辉膜的余辉持续时间特性示于图5。图中“olpl”表示tmb/ppt的组合。又,作为代表性的余辉光谱,包含ttpa、tbrb、dcm2、tbpe的长余辉膜的余辉光谱示于图6。这些余辉光谱与tmb/ppt的余辉光谱明显不同,峰值波长也偏移,可控制发光波长。特别是,若为包含式(1)的es1(b)-es1(a)为负的ttpa、tbrb、dcm2的长余辉膜,则可达成向长波长侧的控制,若为包含es1(b)-es1(a)取超过0且0.15ev以下的正值的tbpe的长余辉膜,则可达成向短波长侧的控制。另一方面,对于包含不满足式(1)的关系的4czbn、2czpn、5czpn、flrpic的长余辉膜,可确认余辉光谱的峰值波长与tmb/ppt溶液相同,未发生自tmb/ppt向这些第三有机化合物的能量转移。
[表1]
又,针对实施例1、实施例3、实施例4、实施例5中所制作的长余辉膜、及无第三有机化合物的长余辉膜,测定利用340nm激发光的光致发光量子产率(pl量子产率)与余辉时间。结果示于表2。此处,余辉时间是自停止激发光的照射的时间点起至发光强度变为未达3pw为止的时间。
[表2]
如表2所示,在长余辉膜中添加有第三有机化合物的各实施例的长余辉膜的光致发光量子产率均比无第三有机化合物的长余辉膜高,且余辉长时间持续。特别是,添加有tbrb作为第三有机化合物的实施例4的长余辉膜与无第三有机化合物的长余辉膜相比,可达成3.5倍的光致发光量子产率及接近6倍的余辉时间。由此可知,通过在长余辉膜中添加第三有机化合物,发光效率与余辉时间显著改善。
(实施例8)
相对于ppt83mol%将tmb以成为1mol%的量进行混合,将作为第三化合物的dcm2以成为16mol%的量进行混合,加热熔解后,进行急冷而获得长余辉膜。
针对所述长余辉膜,照射340nm激发光,结果确认到峰值波长为711nm的发光,在停止照射后,也自长余辉膜确认到余辉。又,其发光峰值的半峰宽(fwhm)为134nm。由此可知,通过在长余辉膜中以高浓度添加dcm2,而发光波长发生长波长化,可实现近红外发光。
(比较例5)ppt与ttpa的膜的制作
在氮气氛围的手套箱中,将石英基板加热至ppt的熔点以上(250℃以上)的温度,在所述石英基板上使ppt熔解。在所述ppt的熔融液中以1重量%的浓度添加ttpa进行混合,并进行急冷,由此形成玻璃状态的长余辉材料膜,通过玻璃基板与紫外线硬化树脂密封。
(比较例6)ppt与dcm2的膜的制作
使用dcm2代替ttpa,除此以外,与比较例1同样地制作膜。
包含tmb与ppt的长余辉材料膜、实施例3中所制作的包含tmb/ppt/ttpa的长余辉膜及ttpa的甲苯溶液(10-5mol/l)的由340nm激发光所得的发光光谱、及ttpa的甲苯溶液的光吸收光谱示于图7,调查所述长余辉膜、长余辉材料膜、及比较例5中所制作的ppt与ttpa的膜的余辉持续时间,所得的结果示于图8。
包含tmb与ppt的长余辉材料膜、实施例4中所制作的包含tmb/ppt/tbrb的长余辉膜及tbrb的甲苯溶液(10-5mol/l)的由340nm激发光所得的发光光谱、及tbrb的甲苯溶液的光吸收光谱示于图9,调查所述长余辉膜及长余辉材料膜的余辉持续时间,所得的结果示于图10。
包含tmb及ppt的长余辉材料膜、实施例5中所制作的包含tmb/ppt/dcm2的长余辉膜及dcm2的甲苯溶液(10-5mol/l)的由340nm激发光所得的发光光谱、及dcm2的甲苯溶液的光吸收光谱示于图11,调查所述长余辉膜、长余辉材料膜、及比较例6中所制作的ppt与dcm2的膜的余辉持续时间,所得的结果示于图12。
图7、图9、图11中,虚线表示光吸收光谱,其他波形表示发光光谱。
观察图7、图9、图11可知,含有长余辉材料与第三有机化合物的长余辉膜的发光峰值相较于长余辉材料膜或第三有机化合物的甲苯溶液的发光峰值,向长波长侧偏移。由此可知,通过组合使用长余辉材料与第三有机化合物,可获得仅为长余辉材料或第三有机化合物无法获得的波长更长的发光色,广的波长范围内的发光色控制成为可能。特别是,极为划时代的是可以高的色纯度观测到无机长余辉材料难以实现的红色发光(参照图11)。
此外,根据图8、图10、图12可知,各实施例中所制作的长余辉膜相较于长余辉材料膜,寿命更长,发光强度也提高。
(实施例9)白色发光膜的制作
相对于tmb1当量,将ppt以成为99当量、将tbpe以成为1当量、将dbp以成为0.01当量的量进行混合,加热熔解后,进行急冷而获得长余辉膜。对所制作的长余辉膜照射340nm激发光,结果确认到国际照明委员会(internationalcommissiononillumination,cie)坐标(x=0.37,y=0.42)的白色发光。图13表示发光光谱,图14表示调查余辉持续时间的结果。
实施例9中,通过并用作为蓝绿色发光体的tbpe与作为红色发光体的dbp作为第三化合物,可实现白色发光。
(实施例10~实施例13)
实施例10~实施例13中,使用磷光材料作为第三有机化合物。对于磷光材料,由于无法测定最低激发单重态能级es1(b),故而将最低激发三重态能级et1(b)设为es1(b)的指标。对于通常的磷光材料,由于最低激发单重态能级与最低激发三重态能级无法极端地分离,故而只要et1(b)-es1(a)未达-0.1ev、优选为未达-0.3ev,则可视为es1(b)-es1(a)≦0.15ev。
具体而言,首先,针对ir(ppy)3、ir(piq)3、ir(btp)2(acac)、ptoep的各者,制备10-5mol/l浓度的氯仿溶液,照射激发光380nm,测定最低激发三重态能级et1(b)。结果示于表3。
相对于ppt98mol%,将tmb以成为1mol%的量进行混合,将作为第三化合物之ir(ppy)3以成为1mol%的量进行混合,加热熔解后,进行急冷而获得长余辉膜。进而,使用ir(piq)3、ir(btp)2(acac)、ptoep代替ir(ppy)3作为第三化合物,同样地获得长余辉膜。
针对所制备的各长余辉膜,照射340nm激发光,结果确认到发光,在停止照射后,也自所有的长余辉膜确认到余辉。调查长余辉膜的余辉光谱,决定峰值波长与半峰宽(fwhm)。结果示于表3。如表3所示,可确认这些峰值波长自tmb/ppt(表3中的第三有机化合物“无”的例子)的峰值波长向长波长侧偏移,可控制发光波长。
[表3]
[产业上的可利用性]
本发明的长余辉组合物容易控制发光色,可将现有的无机长余辉材料难以实现的红色光也以高的色纯度进行发光,此外,也可实现白色光。因此,根据本发明,可实现具有多种色调的长余辉材料。因此,本发明的长余辉组合物的产业上的可利用性高。
- 还没有人留言评论。精彩留言会获得点赞!
- 光交叉连接装置的制造方法
- 一种激光雷达用3341nm、850nm双波长光纤输出激光器的制造方法
- 一种激光雷达用3196nm、862nm双波长光纤输出激光器的制造方法
- 一种激光雷达用2043nm、780nm双波长光纤输出激光器的制造方法
- 一种激光雷达用3196nm、1208nm双波长光纤输出激光器的制造方法
- 一种激光雷达用808nm、872nm双波长光纤输出激光器的制造方法
- 一种激光雷达用872nm、1500nm双波长光纤输出激光器的制造方法
- 一种海洋探测用2404nm波长光纤输出激光器的制造方法
- 一种海洋探测用2230nm波长光纤输出激光器的制造方法
- 一种激光雷达用2970nm、880nm双波长光纤输出激光器的制造方法