一种结晶诱导橙红色发光固态碳点的制备方法
本发明涉及纳米材料制备技术领域,尤其涉及一种结晶诱导橙红色发光固态碳点的制备方法。
背景技术:
碳点(cds)是近年来发现的一种新型碳材料,由于其独特的光学、化学和物理特性而得到了广泛的研究。作为传统的半导体量子点和有机染料最有前途的替代,碳点不仅保持了碳材料毒性小、生产成本低、生物相容性好等优点,还拥有发光范围可调、光稳定性好、易于功能化等优势。故在传感器、光催化、led、指纹识别等领域有着良好的应用前景。
然而,多数制备的碳点仅在溶液态发光,干燥溶液后因聚集诱导猝灭导致在固态下不发光或者发出微弱的荧光,即光减弱。为了拓展此类材料在led和显示等领域的实际应用,固态时优异的发光性能成为这些材料的瓶颈之一。
目前,复合制备(多数采用二步法)是多数研究者采用的制备固态发光碳点的方法,借助pva/pvp、无机化合物、硅烷类材料甚至mof等多孔材料进行发光碳点的包覆,实现“限域”作用。尽管碳点发光性能得以保护,同时又带来了新的问题,如发光性能改变,二次复合增加了工艺的复杂程度,复合引入新的机理问题等。另外,现有多数碳点发光为蓝绿色,长波长区的红色发光碳点材料较少,即使已经发现了红色发光的碳点,它们的荧光量子产率(flqys)也远远低于前期蓝绿光区的碳点。所以,高效红色发光碳点及碳点复合材料的制备也是目前碳点发光材料领域急需突破的课题之一,这也限制了这些材料在生物成像、wled以及显示等方面的应用推广。
显然,一步合成固态高效发光碳点将节约制备该类材料的时间成本,缩短工艺流程,减少二次复合带来的不确定因素。而直接获得固态碳点材料的高产率橙红色发光更能为该类材料的应用推广提供现实的可能性。目前,尚未有高产率橙红色发光碳点材料制备的相关报导。
技术实现要素:
本发明为了弥补现有技术的不足,提供了一种结晶诱导橙红色发光固态碳点的制备方法,固态发光量子产率达17%,具有溶剂用量少、制备工艺简单、可重复性高的优点,弥补了现有固态发光碳点大多只发蓝绿光的局限,解决了现有技术中存在的问题。
本发明是通过如下技术方案实现的:
一种结晶诱导橙红色发光固态碳点的制备方法,包括如下操作步骤:
(1)将一定摩尔配比的邻苯二甲酸和硫脲药品充分溶解到n,n-二甲基甲酰胺溶剂或乙醇溶剂中,得溶液;
(2)将步骤(1)得到的溶液进行微波加热反应;
(3)将步骤(2)得到的反应溶液使用去离子水进行提纯;
(4)将步骤(3)提纯得到的沉积物烘干、研磨,得固态碳点粉末。
进一步地,步骤(1)所述邻苯二甲酸和硫脲药品的摩尔比为(2-10):5;溶剂优选n,n-二甲基甲酰胺溶剂;步骤(1)采用超声的方式进行充分溶解。
进一步地,步骤(1)溶剂用量为10-20ml。
进一步地,步骤(2)微波加热反应功率为640-800w,反应时间2min-3min。
进一步地,步骤(3)提纯方式为超声搅拌后离心、去除上清液;步骤(3)提纯操作至少重复三次。
进一步地,离心操作条件为10000rpm-12000rpm离心3min-5min。
进一步地,步骤(4)烘干温度为70-85℃,恒温烘干时间为2-4h。
如上所述的制备方法制得的固态碳点在指纹显现、指纹检测及光电器件中的应用。
本发明的固态碳点可较好的应用于指纹潜影及led器件中。经过简单研磨并用于指纹的潜影探测时,该碳点能够清晰显示指纹细节且在紫外光照射下,没有出现明显的光衰。考虑到稀缺的橙红色发光,固态碳点作为荧光粉用于led器件时,首先制备出了单色led。混合传统蓝色和绿色的稀土荧光粉,基于三基色原理也制备了发暖白光的wled器件(i=20ma时,cri=82,cct=3100k),其光谱在蓝光区相对较弱,在照明中的防蓝光危害方面有潜在的应用价值。
本发明的有益效果:
本发明原料选用将特定配合比例的邻苯二甲酸和硫脲药品溶解至溶剂中,硫脲药品含有氮和硫两种元素,相较于单一的氮掺杂,更易实现发光红移;且相较现有元素掺杂实现红光发射的方法得到的产品均为液态发光、固态不发光或发光效果不好、容易氧化变质,本发明碳点固态发红光,且对碳点形成了很好的结晶保护,碳点稳定,溶剂用量少、制备工艺简单、可重复性高,发光性能可稳定维持、放置数月不变,相较于现有包覆隔离,在化学性能上更有益于产品发光性能的调控,效果优异;固态发光量子产率可达17%,这在现有碳点固态橙红色发光中优势明显。
本发明制备方法加热采用微波反应,微波反应条件对本发明制得的橙红色发光固态碳点的发光状态及发光颜色具有明显影响,获得的结晶橙红色固态碳点稳定,品质优异;制备过程中,提纯采用去离子水洗,相较于现有碳点制备提纯采用透析等工艺,显著提升了工作效率,更简洁、环保,降低了成本。
附图说明
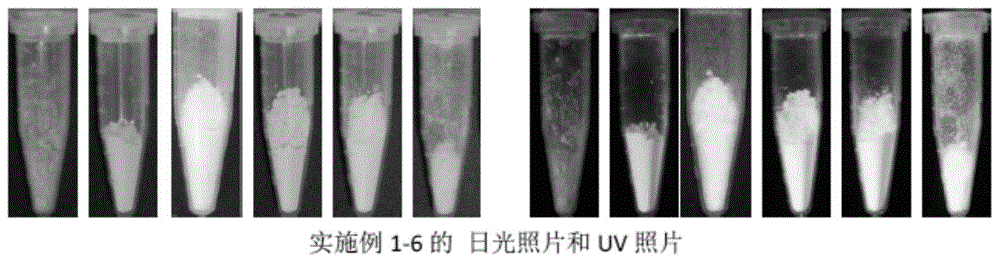
图1为本发明实施例1-5制备的产物粉末日光照片和uv照片;
图2为本发明对比例1-6制备的产物粉末日光照片和uv照片;
图3为本发明实施例3中产物对应的粉末衍射图及结构解析;
图4为本发明实施例3中产物固体碳点复合物的tem(hrtem)和sem形貌结构;
图5为本发明实施例3中产物固体碳点复合物的xps和ft-ir光谱;
图6为本发明实施例3中产物固体碳点粉末的日光下和紫外光365nm激发下的实物图;其中,图6(a1)、图6(a2)为日光下的实物图;图6(b1)、图6(b2)为紫外光激发下的实物图;图6(a1)、图6(b1)为9月前制备的产品,图6(a2),图6(b2)为现制备的产品;
图7为图6实物a2对应的光致发光谱;
图8为本发明制备工艺流程示意图;
其中,图1中各实施例依次排列;图2中,上层为日光照片,下层为uv照片,对比例依次排列。
具体实施方式
为能清楚说明本方案的技术特点,下面通过具体实施方式,结合附图,对本发明进行详细阐述。
实施例1
一种结晶诱导橙红色发光固态碳点的制备方法,包括以下步骤:
(1)称量0.1398g邻苯二甲酸和0.1602g硫脲(邻苯二甲酸:硫脲=2:5摩尔比),倒入20ml的烧杯内,向其加入10ml的n,n-二甲基甲酰胺(dmf)溶剂,超声5min,使其充分溶解,将溶解后的溶液转移到50ml的圆底烧瓶内;
(2)将圆底烧瓶放置微波化学反应仪内,在700w的功率下反应3min,待冷却至室温后,将反应好的溶液移至20ml的烧杯内;
(3)向步骤(2)得到的溶液中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;
(4)在步骤(3)的沉积物中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;反复提纯3次;
(5)将提纯后的沉积物放入70℃真空烘箱内进行烘干4h后,进行研磨得到固态碳点粉末样品。
实施例2
一种结晶诱导橙红色发光固态碳点的制备方法,包括以下步骤:
(1)称量0.2330g邻苯二甲酸和0.2670g硫脲(邻苯二甲酸:硫脲=2:5摩尔比),倒入20ml的烧杯内,向其加入10ml的n,n-二甲基甲酰胺(dmf)溶剂,超声5min,使其充分溶解,将溶解后的溶液转移到50ml的圆底烧瓶内;
(2)将圆底烧瓶放置微波化学反应仪内,在700w的功率下反应3min,待冷却至室温后,将反应好的溶液移至20ml的烧杯内;
(3)向步骤(2)得到的溶液中加入15ml的去离子水超声搅拌均匀,将所得悬浊液以12000rpm离心3min,移除上清液后,得到沉积物;
(4)在步骤(3)的沉积物中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以12000rpm离心3min,移除上清液后,得到沉积物;反复提纯3次;
(5)将提纯后的沉积物放入75℃真空烘箱内进行烘干4h后,进行研磨得到固态碳点粉末样品。
实施例3
一种结晶诱导橙红色发光固态碳点的制备方法,包括以下步骤:
(1)称量0.3429g邻苯二甲酸和0.1571g硫脲(邻苯二甲酸:硫脲=5:5摩尔比)倒入20ml的烧杯内,向其加入10ml的n,n-二甲基甲酰胺(dmf)溶剂,超声5min,使其充分溶解,将溶解后的溶液转移到50ml的圆底烧瓶内;
(2)将圆底烧瓶放置微波化学反应仪内,在800w的功率下反应2.5min,待冷却至室温后,将反应好的溶液移至20ml的烧杯内;
(3)向步骤(2)得到的溶液中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;
(4)在步骤(3)的沉积物中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;反复提纯3次;
(5)将提纯后的沉积物放入80℃真空烘箱内进行烘干3h后,进行研磨得到固态碳点粉末样品。
实施例4
一种结晶诱导橙红色发光固态碳点的制备方法,包括以下步骤:
(1)称量0.3429g邻苯二甲酸和0.1571g硫脲(邻苯二甲酸:硫脲=5:5摩尔比)倒入20ml的烧杯内,向其加入10ml的n,n-二甲基甲酰胺(dmf)溶剂,超声5min,使其充分溶解,将溶液转移到50ml的圆底烧瓶内;
(2)将圆底烧瓶放置微波化学反应仪内,在800w的功率下反应3min,待冷却至室温后,将反应好的溶液移至20ml的烧杯内;
(3)向步骤(2)得到的溶液中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以12000rpm离心4min,移除上清液后,得到沉积物;
(4)在步骤(3)的沉积物中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以12000rpm离心4min,移除上清液后,得到沉积物;反复提纯3次;
(5)将提纯后的沉积物放入80℃真空烘箱内进行烘干3h后,进行研磨得到固态碳点粉末样品。
实施例5
一种结晶诱导橙红色发光固态碳点的制备方法,包括以下步骤:
(1)称量0.4068g邻苯二甲酸和0.0932g硫脲(邻苯二甲酸:硫脲=10:5摩尔比)倒入20ml的烧杯内,向其加入10ml的n,n-二甲基甲酰胺(dmf)溶剂,超声5min,使其充分溶解,将溶液转移到50ml的圆底烧瓶内;
(2)将圆底烧瓶放置微波化学反应仪内,在800w的功率下反应3min,待冷却至室温后,将反应好的溶液移至20ml的烧杯内;
(3)向步骤(2)得到的溶液中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;
(4)在步骤(3)的沉积物中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;反复提纯3次;
(5)将提纯后的沉积物放入85℃真空烘箱内进行烘干3h后,进行研磨得到固态碳点粉末样品。
实施例6
一种结晶诱导橙红色发光固态碳点的制备方法,包括以下步骤:
(1)称量0.2057g邻苯二甲酸和0.0943g硫脲(邻苯二甲酸:硫脲=5:5摩尔比)倒入20ml的烧杯内,向其加入10ml的n,n-二甲基甲酰胺(dmf)溶剂,超声5min,使其充分溶解,将溶解后的溶液转移到50ml的圆底烧瓶内;
(2)将圆底烧瓶放置微波化学反应仪内,在640w的功率下反应2.5min,待冷却至室温后,将反应好的溶液移至20ml的烧杯内;
(3)向步骤(2)得到的溶液中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;
(4)在步骤(3)的沉积物中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;反复提纯3次;
(5)将提纯后的沉积物放入80℃真空烘箱内进行烘干3h后,进行研磨得到固态碳点粉末样品。
对比例1(邻苯二甲酸和硫脲摩尔比低于本发明最低配比)
一种结晶诱导橙红色发光固态碳点的制备方法,包括以下步骤:
(1)称量0.0912g邻苯二甲酸和0.2088g硫脲(邻苯二甲酸:硫脲=1:5摩尔比)倒入20ml的烧杯内,向其加入10ml的n,n-二甲基甲酰胺(dmf)溶剂,超声5min,使其充分溶解,将溶解后的溶液转移到50ml的圆底烧瓶内;
(2)将圆底烧瓶放置微波化学反应仪内,在800w的功率下反应2.5min,待冷却至室温后,将反应好的溶液移至20ml的烧杯内;
(3)向步骤(2)得到的溶液中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;
(4)在步骤(3)的沉积物中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;反复提纯3次;
(5)将提纯后的沉积物放入80℃真空烘箱内进行烘干3h后,进行研磨得到固态碳点粉末样品。
对比例2(邻苯二甲酸和硫脲摩尔比高于本发明最高配比)
一种结晶诱导橙红色发光固态碳点的制备方法,包括以下步骤:
(1)称量0.2692g邻苯二甲酸和0.0308g硫脲(邻苯二甲酸:硫脲=20:5摩尔比)倒入20ml的烧杯内,向其加入10ml的n,n-二甲基甲酰胺(dmf)溶剂,超声5min,使其充分溶解,将溶解后的溶液转移到50ml的圆底烧瓶内;
(2)将圆底烧瓶放置微波化学反应仪内,在800w的功率下反应3min,待冷却至室温后,将反应好的溶液移至20ml的烧杯内;
(3)向步骤(2)得到的溶液中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;
(4)在步骤(3)的沉积物中加入10ml的去离子水超声搅拌均匀,将所得悬浊液以10000rpm离心5min,移除上清液后,得到沉积物;反复提纯3次;
(5)将提纯后的沉积物放入80℃真空烘箱内进行烘干3h后,进行研磨得到固态碳点粉末样品。
对比例3
一种结晶诱导橙红色发光固态碳点的制备方法,同实施例6的制备方法,所不同的是,步骤(2)微波反应在800w反应4min。
对比例4
一种结晶诱导橙红色发光固态碳点的制备方法,同实施例6的制备方法,所不同的是,步骤(2)微波反应在400w反应3min。
对比例5
一种结晶诱导橙红色发光固态碳点的制备方法,同实施例6的制备方法,所不同的是,步骤(2)微波反应在800w反应1.5min。
一、上述不同原料摩尔比及制备条件的各实施例及对比例汇总如下表1;日光下照射及uv紫外激光下照射照片如图1-2所示。
表1各实施例
由图1可以看出,本发明制备方法制得的产物为固态粉末状,在uv照射下呈橙红色,固态发光,稳定性优异;经测定,固态发光量子产率达17%。由图2可以看出,原料配比发生改变的对比例、及制备条件调整的对比例所制得的产物均为液态或半固态,且uv下发蓝光居多,对比例1和对比例3显示发黄色和橙色光,但发光效果变差(亮度,颜色均有改变)。本发明制得的橙红色发光固态碳点相较现有蓝绿发光碳点更稀有,且性能稳定,产率明显优于现有已发现的红光碳点。
二、选择本发明的最优实施例3,对制备的产物粉末进行化学表征分析,如图3-7所示。其中,图3为产物对应的粉末衍射图及结构解析;由图3可以看出,各衍射峰均具有非常窄的半高宽值,几乎不存在杂峰,说明本发明方法制备的复合物结晶程度高,经过jade晶体软件指标化进一步证实且给出了具体的晶体结构。
图4为实施例3产物固体碳点复合物的透射电镜tem(hrtem)和扫描电镜sem形貌结构。由图4可以看出,本发明制备的碳点复合物在乙醇溶剂中被溶解(邻苯二甲酸溶于乙醇),但是多数碳点仍旧处于被包覆状态;并且碳点内部有良好的晶格条纹,说明碳点具有高石墨化结构,而该固态状复合物呈细棒状。
图5为实施例3产物固体碳点复合物的xps和ft-ir光谱。由图5可以看出,本发明方法制备的复合物拥有丰富的官能团,尤其是conh类基团的出现,证实了硫脲和邻苯二甲酸脱水缩合反应的发生。通过各元素的xps光谱分析得到,硫和氮元素(硫脲中)成功掺杂在碳点中,这也是本发明制得的碳点橙红色发光的有力证据。
图6为实施例3产物固体碳点粉末的日光下和紫外光365nm激发下的实物图。由图6(b1)、图6(b2),可看到本发明方法制备的固态发光粉末能够发出橙红色的光,并且环境条件下放置9个月,发光与现制备产品的发光相比,几乎没有发生任何变化;存储稳定性优异。
图7为图6实物a2对应的光致发光谱。由图7可以看出,本发明方法制备的复合物的发射光谱在580,630和690nm处存在峰值,说明了该发光的多重性。
由以上各实施例及对比例分析可知,本发明制备方法制得的固态碳点为结晶橙红色发光粉末,突破了现有发光碳点液态发光、固态不易发光,或者现有固态发光碳点基本为蓝绿光、存储稳定性差的问题;该结晶橙红色发光粉末存储稳定,固态发光量子产率达17%,制备过程具有溶剂用量少、工艺简单、可重复性高的有益效果。
上述具体实施方式不能作为对本发明保护范围的限制,对于本技术领域的技术人员来说,对本发明实施方式所做出的任何替代改进或变换均落在本发明的保护范围内。
本发明未详述之处,均为本技术领域技术人员的公知技术。
- 还没有人留言评论。精彩留言会获得点赞!