一种含金属原子纳米碳材料及其制备方法和应用以及一种烃脱氢反应方法与流程
本发明涉及一种含金属原子纳米碳材料,本发明还涉及一种含金属原子纳米碳材料的制备方法以及由该方法制备的含金属原子纳米碳材料,本发明又涉及一种通过将上述含金属原子纳米碳材料进行焙烧而制备的含金属原子纳米碳材料,本发明进一步涉及根据本发明的含金属原子纳米碳材料作为烃脱氢反应的催化剂的应用、以及一种烃脱氢反应方法。
背景技术:
烃类物质的脱氢反应是一类重要的反应类型,例如大部分低碳链烯烃是通过低碳链烷烃的脱氢反应而获得的。脱氢反应根据氧气是否参与可以划分为直接脱氢反应(即,氧气不参与)和氧化脱氢反应(即,氧气参与)两类。
多种类型的纳米碳材料已被证明对烃类物质的直接脱氢反应和氧化脱氢反应均具有催化效果,在纳米碳材料中引入氧原子和/或氮原子则可以改善其催化活性。
在纳米碳材料中引入氧原子,可以在纳米碳材料表面形成羟基、羰基、羧基、酯基和酸酐等含氧官能团。
可以通过对纳米碳材料进行氧化处理实现在纳米碳材料中引入氧原子,从而增加纳米碳材料中含氧官能团的含量。例如,可以将纳米碳材料在强酸(如hno3、h2so4)和/或强氧化性溶液(如h2o2、kmno4)中进行回流反应,在回流反应的同时还可以辅助进行微波加热或超声振荡,以增强氧化反应的效果。但是,在强酸和/或强氧化性溶液中进行回流反应可能会对纳米碳材料的骨架结构产生不利影响,甚至破坏纳米碳材料的骨架结构。例如:将纳米碳材料在硝酸中进行回流反应,虽然可以在纳米碳材料表面引入大量含氧官能团,但是极易造成纳米碳材料被切断和/或明显增加石墨网络结构中的缺陷位,从而降低纳米碳材料的性能,如热稳定性。另外,通过在强酸和/或强氧化性溶液中进行回流反应,以引入氧原子时,氧原子的引入量对反应操作条件的依赖性高,波动范围较宽。
在纳米碳材料中引入氮原子时,根据氮原子在纳米碳材料中所处化学环境的不同,通常将氮原子划分为化学氮和结构氮。化学氮主要是以表面官能团的形式出现在材料的表面,如氨基或亚硝酰基等表面含氮官能团。结构氮是指进入纳米碳材料的骨架结构与碳原子键合的氮原子。结构氮主要包括石墨型氮(即,
可以通过在纳米碳材料合成过程中引入含氮的功能性气氛(如氨气、氮气、尿素、三聚氰胺),利用高温和/或高压在纳米碳材料的合成过程中将氮元素同时引入到纳米碳材料的骨架结构和/或表面 中;也可以通过将纳米碳材料置于含氮的功能性气氛(如氨气、氮气、尿素、三聚氰胺)中,利用高温和/或高压将氮元素引入到纳米碳材料的表面。高温和/或高压尽管可以在纳米碳材料中形成结构氮,但是含氮物种的类型依赖于反应条件,不易控制;并且,如此产生的不同类型的含氮物种在纳米碳材料的表面分布不均匀,导致含氮纳米碳材料的性能不稳定。还可以通过将纳米碳材料进行氧化处理,然后与胺反应,从而在纳米碳材料表面引入氮原子,如此引入的氮原子基本为化学氮。
尽管有关纳米碳材料的掺杂改性及其催化性能的研究取得了诸多进展,但是对于其中的一些基本问题仍未形成共识,依然需要对掺杂改性纳米碳材料及其制备方法和催化性能进行深入研究。
技术实现要素:
本发明的一个目的在于提供一种含金属原子纳米碳材料的制备方法,采用该方法不仅能在纳米碳材料表面引入金属原子,而且能稳定地提高纳米碳材料中杂原子的含量,同时对纳米碳材料本身的结构影响不大。
本发明的另一个目的在于提供一种含金属原子纳米碳材料,该含金属原子纳米碳材料用于烃类物质的脱氢反应时,能获得更高的原料转化率和产物选择性。
本发明的又一目的在于提供一种烃脱氢反应方法,该方法能获得更高的原料转化率和产物选择性。
根据本发明的第一个方面,本发明提供了一种含金属原子纳米碳材料,该含金属原子纳米碳材料含有c元素、o元素、n元素和至少一种金属元素,以该含金属原子纳米碳材料的总量为基准并以元素计,o元素的含量为1-8重量%,n元素的含量为1-10重量%,所述金属元素的总量为1-10重量%,c元素的含量为72-97重量%;该含金属原子纳米碳材料中,由x射线光电子能谱确定的氧元素的总含量为iot,由x射线光电子能谱中529.5-530.8ev范围内的峰确定的o元素的含量为iom,iom/iot在0.02-0.3的范围内;该含金属原子纳米碳材料中,由x射线光电子能谱中531.0-532.5ev范围内的峰确定的o元素的量为ioc,由x射线光电子能谱中532.6-533.5ev范围内的峰确定的o元素的量为ioe,ioc/ioe在0.2-0.8的范围内;该含金属原子纳米碳材料中,由x射线光电子能谱确定该含金属原子纳米碳材料中的n元素的总量为int,由x射线光电子能谱中398.5-400.1ev范围内的峰确定的n元素的量为inc,inc/int在0.6-0.95的范围内,由x射线光电子能谱中403.5-406.5ev范围内的峰确定的n元素的含量为inn,inn/int在0.05-0.35的范围内。
根据本发明的第二个方面,本发明提供了一种含金属原子纳米碳材料的制备方法,该方法包括将一种分散有原料纳米碳材料、至少一种含氮化合物以及至少一种硝酸金属盐的水分散液于密闭容器中进行反应,所述含氮化合物选自nh3、肼和尿素,反应过程中,所述水分散液的温度保持在80-310℃的范围内。
根据本发明的第三个方面,本发明提供了一种由根据本发明第二个方面的方法制备的含金属原子纳米碳材料。
根据本发明的第四个方面,本发明提供了一种含金属原子纳米碳材料,该含金属原子纳米碳材 料是将根据本发明第一个方面或者第三个方面的含金属原子纳米碳材料进行焙烧而制得的。
根据本发明的第五个方面,本发明提供了根据本发明第一个方面的含金属原子纳米碳材料、根据本发明第三个方面的含金属原子纳米碳材料、或者根据本发明第四个方面的含金属原子纳米碳材料作为烃脱氢反应的催化剂的应用。
根据本发明的第六个方面,本发明提供了一种烃脱氢反应方法,该方法包括在存在或不存在氧气的条件下,在烃脱氢反应条件下,将烃与根据本发明第一个方面的含金属原子纳米碳材料、根据本发明第三个方面的含金属原子纳米碳材料、或者根据本发明第四个方面的含金属原子纳米碳材料接触。
根据本发明的含金属原子纳米碳材料的制备方法,不仅能稳定地调控和/或提高纳米碳材料中金属原子和杂原子的含量,同时对纳米碳材料本身的结构影响小。并且,根据本发明的含金属原子纳米碳材料的制备方法,制备的含金属原子纳米碳材料具有稳定的性能。
根据本发明的含金属原子纳米碳材料在烃类物质的脱氢反应中显示出良好的催化性能,能明显提高原料转化率和产物选择性。
附图说明
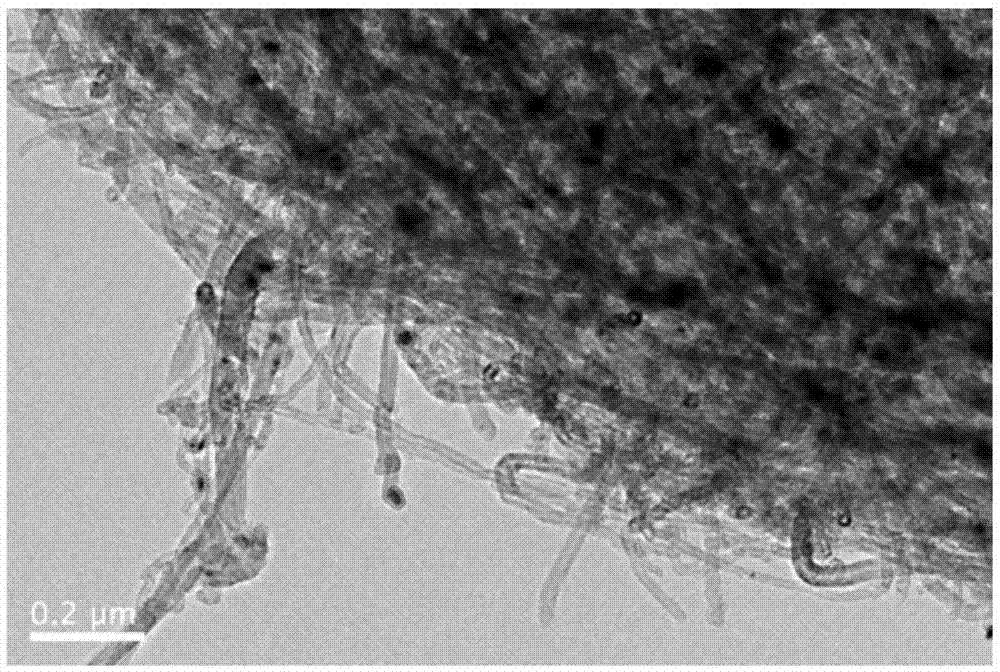
图1为实施例1制备的含金属原子纳米碳材料的透射电子显微镜照片。
图2为实施例1使用的原料纳米碳材料的透射电子显微镜照片。
具体实施方式
本发明中,纳米碳材料是指分散相尺度至少有一维小于100nm的碳材料。
根据本发明的第一个方面,本发明提供了一种含金属原子纳米碳材料,该含金属原子纳米碳材料含有c元素、o元素、n元素以及至少一种金属元素。本发明中,“至少一种”表示一种或两种以上。
根据本发明的含金属纳米碳材料,所述金属元素选自可以形成硝酸金属盐的金属元素,如选自元素周期表中第ia族金属元素、第iia族金属元素、第iiib族金属元素、第ivb族金属元素、第vb族金属元素、第vib族金属元素、第viib族金属元素、第viii族金属元素、第ib族金属元素、第iib族金属元素、第iiia族金属元素、第iva族金属元素和第va族金属元素。所述金属元素的具体实例可以包括但不限于锂、钠、钾、镁、钙、锶、钪、钇、稀土金属元素(如镧、铈、镨)、钛、锆、钒、铌、铬、钼、钨、锰、铁、钌、钴、铑、镍、钯、铂、铜、银、金、锌、铝、锗、锡、铅和锑。优选地,所述金属元素选自第ia族金属元素、第iia族金属元素、第viii族金属元素、第ib族金属元素、第iib族金属元素和第iva族金属元素,此时将该含金属纳米碳材料用作烃脱氢反应的催化剂时,能获得更高的催化活性。更优选地,所述金属元素选自第viii族金属元素。进一步优选地,所述金属元素选自铁、钌、钴、铑、镍、钯和铂,此时将该含金属纳米碳材料用作烃脱氢反应的催化剂时,能获得进一步提高的催化活性。
根据本发明的含金属原子纳米碳材料,以该含金属原子纳米碳材料的总量为基准并以元素计,o元素的含量为1-8重量%,优选为2-8重量%,更优选为3.5-6重量%;n元素的含量为1-10重量%,优选为1.5-5重量%,更优选为2-4重量%;金属元素的总量为1-10重量%,优选为2-5重量%,更优选为2-4.5重量%;c元素的含量为72-97重量%,优选为82-94.5重量%,更优选为85.5-92.5重量%。其中,各元素的含量采用x射线光电子能谱法测定。样品在测试前在150℃的温度下于氦气气氛中干燥3小时。
本发明中,x射线光电子能谱分析在thermoscientific公司的配备有thermoavantagev5.926软件的escalab250型x射线光电子能谱仪上进行测试,激发源为单色化alkαx射线,能量为1486.6ev,功率为150w,窄扫描所用通透能为30ev,分析测试时的基础真空为6.5×10-10mbar,电子结合能用单质碳的c1s峰(284.0ev)校正,在thermoavantage软件上进行数据处理,在分析模块中采用灵敏度因子法进行定量分析。
根据本发明的含金属原子纳米碳材料,该含金属原子纳米碳材料中,由x射线光电子能谱确定的氧元素的总含量为iot,由x射线光电子能谱中529.5-530.8ev范围内的峰确定的o元素(即,与金属原子键合的氧原子)的含量为iom,iom/iot在0.02-0.3的范围内,优选在0.05-0.3的范围内,更优选在0.06-0.2的范围内。根据本发明的含金属原子纳米碳材料,由x射线光电子能谱中531.0-533.5ev范围内的峰确定的o元素的含量为ionm,ionmiot在0.7-0.98的范围内,优选在0.7-0.95的范围内,更优选在0.8-0.94的范围内。本发明中,在表示数值范围时,“在×-×的范围内”包括两个边界数值。
本发明中,将x射线光电子能谱中的o1s谱峰的面积记为ao1,将o1s谱峰分成两组峰,将在529.5-530.8ev范围内的谱峰(对应于与金属原子相连的氧物种)的面积记为ao2,将在531.0-533.5ev范围内的谱峰(对应于不与金属原子相连的氧物种)的面积记为ao3,其中,iom/iot=ao2/ao1,ionm/iot=ao3/ao1。
根据本发明的含金属原子纳米碳材料,由x射线光电子能谱中531.0-532.5ev范围内的峰确定的o元素(即,c=o)的量为ioc,由x射线光电子能谱中532.6-533.5ev范围内的峰确定的o元素(即,c-o)的量为ioe,ioc/ioe在0.2-0.8的范围内,优选在0.4-0.6的范围内。本发明中,将x射线光电子能谱中在531.0-533.5ev范围内的谱峰(对应于不与金属原子相连的氧物种)进一步分成两组峰,即在531.0-532.5ev范围内的谱峰(对应于c=o物种)以及在532.6-533.5ev范围内的谱峰(对应于co物种),将在531.0-532.5ev范围内的谱峰的面积记为ao4,将在532.6-533.5ev范围内的谱峰的面积记为ao5,ioc/ioe=ao4/ao5。
根据本发明的含金属原子纳米碳材料,以该含金属原子纳米碳材料中由x射线光电子能谱确定的c元素的总量为基准,由x射线光电子能谱中284.7-284.9ev范围内的峰确定的c元素(即,石墨型碳)的含量可以为60-95重量%,优选为70-92重量%,更优选为75-90重量%;由x射线光电子能谱中286.0-288.8ev范围内的峰确定的c元素的总含量可以为5-40重量%,优选为8-30重量%,更优选为10-25重量%。本发明中,由x射线光电子能谱中的c1s谱峰的面积ac1确定c元素的总量,将 x射线光电子能谱中的c1s谱峰分成两组峰,即在284.7-284.9ev范围内的谱峰(对应于石墨型碳物种)以及在286.0-288.8ev范围内的谱峰(对应于非石墨型碳物种),将在284.7-284.9ev范围内的谱峰的面积记为ac2,将在286.0-288.8ev范围内的谱峰的面积记为ac3,由x射线光电子能谱中284.7-284.9ev范围内的峰确定的c元素的含量=ac2/ac1,由x射线光电子能谱中286.0-288.8ev范围内的峰确定的c元素的总含量=ac3/ac1。
根据本发明的含金属原子纳米碳材料,该含金属原子纳米碳材料中,由x射线光电子能谱中288.6-288.8ev范围内的峰确定的c元素的量为icc,由x射线光电子能谱中286.0-286.2ev范围内的峰确定的c元素的量为ice,icc/ice在0.2-1的范围内,优选在0.3-0.8的范围内。本发明中,将x射线光电子能谱中在286.0-288.8ev范围内的谱峰(对应于非石墨碳物种)进一步划分为两组峰,即在286.0-286.2ev范围内的谱峰(对应于羟基和醚型碳物种)以及在288.6-288.8ev范围内的谱峰(对应于羧基、酐和酯型碳物种),将在286.0-286.2ev范围内的谱峰的面积记为ac4,将在288.6-288.8ev范围内的谱峰的面积记为ac5,icc/ice=ac5/ac4。
根据本发明的含金属原子纳米碳材料,由x射线光电子能谱确定该含金属原子纳米碳材料中的n元素的总量为int,由x射线光电子能谱中398.5-400.1ev范围内的峰确定的n元素(即,除石墨型氮以及-no2型氮外的氮物种)的量为inc,inc/int在0.6-0.95的范围内,优选在0.65-0.8的范围内。
根据本发明的含金属原子纳米碳材料,由x射线光电子能谱确定该含金属原子纳米碳材料中的n元素的总量为int,由x射线光电子能谱中403.5-406.5ev范围内的峰确定的n元素(即,-no2物种)的含量为inn,inn/int在0.05-0.35的范围内,优选在0.06-0.25的范围内。
根据本发明的含金属原子纳米碳材料,由x射线光电子能谱中400.6-401.5ev范围内的峰确定的n元素(即,石墨型氮)含量较低甚至不含。一般地,根据本发明的含金属原子纳米碳材料中,由x射线光电子能谱中400.6-401.5ev范围内的峰确定的n元素的量为ing,ing/int为不高于0.3,一般在0.02-0.2的范围内,优选在0.05-0.15的范围内。
本发明中,由x射线光电子能谱中的n1s谱峰的面积确定n元素的总量an1,将x射线光电子能谱中的n1s谱峰分成三组峰,即在403.5-406.5ev范围内的谱峰(对应于-no2物种)、在400.6-401.5ev范围内的谱峰(对应于石墨型氮物种)以及398.5-400.1ev范围内的谱峰(除石墨型氮以及-no2型氮外的氮物种),将在400.6-401.5ev范围内的谱峰的面积记为an2,将在398.5-400.1ev范围内的谱峰的面积记为an3,将在403.5-406.5ev范围内的谱峰的面积记为an4,inc/int=an3/an1,ing/int=an2/an1,inn/int=an4/an1,在得到的比值为低于0.01时,认为不含该类物种,并将该类物种的含量记为0。
本发明中,各峰的位置由该峰的峰顶所对应的结合能确定,由上文所述范围确定的峰是指峰顶所对应的结合能处于该范围内的峰,在该范围内可以包括一个峰,也可以包括两个以上的峰。例如:398.5-400.1ev范围内的峰是指峰顶所对应的结合能处于398.5-400.1ev的范围内的全部峰。
根据本发明的含金属原子纳米碳材料可以以常见的各种形态存在,具体可以为但不限于含金属 原子碳纳米管、含金属原子石墨烯、含金属原子薄层石墨、含金属原子纳米碳颗粒、含金属原子纳米碳纤维、含金属原子纳米金刚石和含金属原子富勒烯中的一种或两种以上的组合。所述含金属原子的碳纳米管可以为含金属原子单壁碳纳米管、含金属原子双壁碳纳米管和含金属原子多壁碳纳米管中的一种或两种以上的组合。根据本发明的含金属原子纳米碳材料,优选为含金属原子多壁碳纳米管。
根据本发明的含金属原子纳米碳材料,优选地,所述含金属原子多壁碳纳米管的比表面积为50-500m2/g,这样能进一步提高该含金属原子纳米碳材料的催化性能,特别是作为烃类物质脱氢反应的催化剂的催化性能。更优选地,该含金属原子多壁碳纳米管的比表面积为80-300m2/g。进一步优选地,该含金属原子多壁碳纳米管的比表面积为90-250m2/g。更进一步优选地,该含金属原子多壁碳纳米管的比表面积为120-180m2/g。本发明中,所述比表面积由氮气吸附bet法测定。
根据本发明的含金属原子纳米碳材料,所述含金属原子多壁碳纳米管在400-800℃的温度区间内的失重率为w800,在400-500℃的温度区间内的失重率为w500,w500/w800优选在0.01-0.5的范围内,这样能够获得更好的催化效果,特别是用作烃类物质脱氢反应的催化剂时,能获得更好的催化效果。更优选地,w500/w800在0.05-0.4的范围内。进一步优选地,w500/w800在0.05-0.15的范围内。本发明中,w800=w800-w400,w500=w500-w400,w400为在400℃的温度下测定的质量损失率,w800为在800℃的温度下测定的质量损失率,w500为在500℃的温度下测定的质量损失率;所述失重率采用热重分析仪在空气气氛中测定,测试起始温度为25℃,升温速率为10℃/min;样品在测试前在150℃的温度和1标准大气压下于氦气气氛中干燥3小时。
在本发明的一种优选的实施方式中,所述含金属原子纳米碳材料优选为含金属原子多壁碳纳米管,该含金属原子多壁碳纳米管的比表面积为50-500m2/g,优选为80-300m2/g,更优选为90-250m2/g,进一步优选为120-180m2/g;并且,w500/w800在0.01-0.5的范围内,优选在0.05-0.4的范围内,更优选在0.05-0.15的范围内。
根据本发明的含金属原子纳米碳材料,对于除氧原子和氮原子外的其它非金属杂原子,如硫原子和磷原子,其含量可以为常规含量。一般地,根据本发明的含金属原子纳米碳材料中,除氧原子和氮原子外的其它非金属杂原子(如硫原子和磷原子)的总量可以为0.5重量%以下,优选为0.2重量%以下,更优选为0.1重量%以下,进一步优选为0.05重量%以下。根据本发明的含金属原子纳米碳材料,除选自前述金属元素外,还可以含有其它金属原子,所述其它金属原子例如可以为来源于制备纳米碳材料时使用的催化剂。所述其它金属原子的含量一般为2.5重量%以下,优选为2重量%以下,进一步优选为1重量%以下,更进一步优选为0.5重量%以下,特别优选为0.2重量%以下。
根据本发明的第二个方面,本发明提供了一种含金属原子纳米碳材料的制备方法,该方法包括将一种分散有原料纳米碳材料、至少一种含氮化合物以及至少一种硝酸金属盐的水分散液于密闭容器中进行反应。
所述含氮化合物选自nh3、肼和尿素。
根据本发明的方法,所述硝酸金属盐中的金属元素选自可以形成硝酸金属盐的金属元素,如选 自元素周期表中第ia族金属元素、第iia族金属元素、第iiib族金属元素、第ivb族金属元素、第vb族金属元素、第vib族金属元素、第viib族金属元素、第viii族金属元素、第ib族金属元素、第iib族金属元素、第iiia族金属元素、第iva族金属元素和第va族金属元素。所述硝酸金属盐中的金属元素的具体实例可以包括但不限于锂、钠、钾、镁、钙、钡、锶、钪、钇、稀土金属元素(如镧、铈、镨)、钛、锆、钒、铌、铬、钼、钨、锰、铁、钌、钴、铑、镍、钯、铂、铜、银、金、锌、铝、锗、锡和锑。优选地,所述硝酸金属盐中的金属元素选自第ia族金属元素、第iia族金属元素、第viii族金属元素、第ib族金属元素、第iib族金属元素和第iva族金属元素,由此制备的含金属纳米碳材料在用作烃脱氢反应的催化剂时,能获得更高的催化活性。更优选地,所述硝酸金属盐中的金属元素选自第viii族金属元素,进一步优选选自铁、钌、钴、铑、镍、钯和铂,由此制备的含金属纳米碳材料用作烃脱氢反应的催化剂时,能获得进一步提高的催化活性。
根据本发明的方法,所述含氮化合物和所述硝酸金属盐的用量可以根据预期在原料纳米碳材料中引入的氮元素、氧元素以及金属元素的含量以及种类进行选择。在最终制备的含金属原子纳米碳材料在用作烃脱氢反应的催化剂时,优选地,原料纳米碳材料:含氮化合物:硝酸金属盐的重量比在1:0.01-10:0.01-10的范围内,由此制备的含金属原子纳米碳材料在用作烃脱氢反应的催化剂时能获得进一步提高的原料转化率和产物选择性。更优选地,原料纳米碳材料:含氮化合物:硝酸金属盐的重量比在1:0.05-8:0.05-8的范围内。进一步优选地,原料纳米碳材料:含氮化合物:硝酸金属盐的重量比在1:1-5:0.5-3的范围内。
根据本发明的方法,所述含氮化合物与所述硝酸金属盐的摩尔比优选在1:0.001-10的范围内,由此制备的含金属原子纳米碳材料在用作烃脱氢反应的催化剂时也具有更为优异的催化活性。更优选地,所述含氮化合物与所述硝酸金属盐的摩尔比在1:0.01-8的范围内。进一步优选地,所述含氮化合物与所述硝酸金属盐的摩尔比在1:0.02-4的范围内。更进一步优选地,所述含氮化合物与所述硝酸金属盐的摩尔比在1:0.05-1的范围内。特别优选地,所述含氮化合物与所述硝酸金属盐的摩尔比在1:0.05-0.8的范围内。
根据本发明的方法,可以根据原料纳米碳材料的量对水的用量进行选择。优选地,原料纳米碳材料:h2o的重量比在1:2-500的范围内,在水的用量处于该范围之内时,纳米碳材料在处理过程中的结构形态保持性更好,例如:对于碳纳米管而言,在处理过程中基本不会被切断。更优选地,原料纳米碳材料:h2o的重量比在1:5-300的范围内。进一步优选地,原料纳米碳材料:h2o的重量比在1:10-50的范围内。
根据本发明的方法,在一种优选的实施方式中,所述含氮化合物为氨,所述硝酸金属盐中的金属原子选自铁、钴和镍,由此制备的含金属原子纳米碳材料在用作烃脱氢反应的催化剂时能获得更好的催化反应效果。原料纳米碳材料:含氮化合物:硝酸金属盐的重量比优选在1:0.02-8:0.05-3的范围内,更优选在1:0.2-6:0.1-2的范围内,进一步优选在1:1.5-5:0.5-1的范围内。在该优选的实施方式中,含氮化合物:硝酸金属盐的摩尔比优选在1:0.001-1的范围内,更优选在1:0.01-0.5的 范围内,进一步优选在1:0.015-0.05的范围内。在该优选的实施方式中,原料纳米碳材料:h2o的重量比优选在1:5-100的范围内,更优选在1:10-80的范围内,进一步优选在1:15-50的范围内。
根据本发明的方法,在另一种优选的实施方式中,所述含氮化合物为肼,所述硝酸金属盐中的金属原子选自钯和铂,由此制备的含金属原子纳米碳材料在用作烃脱氢反应的催化剂时能获得更好的催化反应效果。原料纳米碳材料:含氮化合物:硝酸金属盐的重量比优选在1:0.02-5:0.05-6的范围内,更优选在1:0.05-2:0.5-5的范围内,进一步优选在1:0.8-1.2:0.4-0.8的范围内。在该优选的实施方式中,含氮化合物:硝酸金属盐的摩尔比优选在1:0.01-1的范围内,更优选在1:0.05-0.1的范围内。在该优选的实施方式中,原料纳米碳材料:h2o的重量比优选在1:5-100的范围内,更优选在1:10-80的范围内,进一步优选在1:15-50的范围内。
根据本发明的方法,在又一种优选的实施方式中,所述含氮化合物为尿素,所述硝酸金属盐中的金属原子选自钌和铑,由此制备的含金属原子纳米碳材料在用作烃脱氢反应的催化剂时能获得更好的催化反应效果。原料纳米碳材料:含氮化合物:硝酸金属盐的重量比优选在1:0.02-5:0.01-5的范围内,更优选在1:0.1-4:0.05-4的范围内,进一步优选在1:1-3:1-3的范围内。在该优选的实施方式中,含氮化合物:硝酸金属盐的摩尔比优选在1:0.01-10的范围内,更优选在1:0.02-5的范围内,进一步优选在1:0.05-1的范围内。在该优选的实施方式中,原料纳米碳材料:h2o的重量比优选在1:5-200的范围内,更优选在1:10-100的范围内,进一步优选在1:15-50的范围内。
根据本发明的方法,所述反应的条件以足以提高原料纳米碳材料中的氧原子、氮原子和金属原子的含量为准。优选地,反应过程中,所述水分散液的温度在80-310℃的范围内。在所述水分散液的温度处于上述范围之内时,不仅能有效地提高原料纳米碳材料中的氧原子、氮原子和金属原子含量,而且不会对原料纳米碳材料的结构形态产生明显影响。更优选地,反应过程中,所述水分散液的温度在90-300℃的范围内。进一步优选地,反应过程中,所述水分散液的温度在100-230℃的范围内。
根据本发明的方法,所述反应的持续时间可以根据反应的温度进行选择,以能够在原料纳米碳材料中引入足量的氧原子、氮原子和金属原子为准。一般地,所述反应的持续时间可以在0.5-96小时的范围内,优选在2-72小时的范围内,更优选在12-48小时的范围内。
根据本发明的方法,可以采用常用的各种方法来形成所述水分散液,例如可以将原料纳米碳材料分散在水(优选为去离子水)中,然后加入所述含氮化合物和所述硝酸金属盐,从而得到所述水分散液。为了进一步提高原料纳米碳材料的分散效果,同时缩短分散的时间,可以采用超声振荡的方法将原料纳米碳材料分散在水中。所述超声振荡的条件可以为常规选择,一般地,所述超声振荡的频率可以为10-200khz,优选为90-140khz;所述超声振荡的持续时间可以为0.1-6小时,优选为1-4小时。根据本发明的方法,所述含氮化合物和所述硝酸金属盐根据种类各自可以以溶液(优选为水溶液)的形式提供,也可以各自以纯物质的形式提供,没有特别限定。
根据本发明的方法,所述原料纳米碳材料中氧元素和氮元素的含量没有特别限定,可以为常规选择。一般地,所述原料纳米碳材料中氧元素的含量为不高于1.5重量%,优选为不高于1.2重量%, 更优选为不高于0.5重量%,进一步优选为不高于0.3重量%;氮元素的含量为不高于0.5重量%,优选为不高于0.2重量%,更优选为不高于0.1重量%,进一步优选为不高于0.05重量%,更进一步优选为不高于0.02重量%。根据本发明的方法,所述原料纳米碳材料中除氧原子和氮原子外的其余非金属杂原子(如磷原子和硫原子)的总量(以元素计)可以为常规含量。一般地,所述原料纳米碳材料中除氧元素和氮元素外的其余非金属杂原子的总量(以元素计)为不高于0.5重量%,优选为不高于0.2重量%,更优选为不高于0.1重量%,进一步优选为不高于0.05重量%。根据本发明的方法,所述原料纳米碳材料根据来源的不同,可能含有一些金属元素,例如来源于制备原料纳米碳材料时使用的催化剂中的金属原子。所述原料纳米碳材料中金属原子的含量(以元素计)一般为2.5重量%以下,优选为1.8重量%以下,进一步优选为0.5重量%以下,更进一步优选为0.1重量%以下。
根据本发明的方法,原料纳米碳材料在使用前可以采用本领域常用的方法进行预处理(如洗涤),以除去原料纳米碳材料表面的一些杂质;也可以不进行预处理,直接使用。本发明公开的实施例中,原料纳米碳材料在使用前均未进行预处理。
根据本发明的方法,可以对各种存在形态的纳米碳材料进行处理,从而提高该纳米碳材料中的氧原子、氮原子和金属原子含量。所述原料纳米碳材料可以为但不限于碳纳米管、石墨烯、纳米金刚石、薄层石墨、纳米碳颗粒、纳米碳纤维和富勒烯中的一种或两种以上的组合。所述碳纳米管可以为单壁碳纳米管、双壁碳纳米管和多壁碳纳米管中的一种或两种以上的组合。优选地,所述原料纳米碳材料为碳纳米管,更优选为多壁碳纳米管。
根据本发明的方法,在一种优选的实施方式中,所述原料纳米碳材料为多壁碳纳米管,所述多壁碳纳米管的比表面积可以为50-500m2/g,优选为80-300m2/g,更优选为100-260m2/g,进一步优选为120-190m2/g。
在所述原料纳米碳材料为多壁碳纳米管时,所述多壁碳纳米管在400-800℃的温度区间内的失重率为w800,在400-500℃的温度区间内的失重率为w500,w500/w800可以在0.01-0.5的范围内,优选在0.02-0.4的范围内,更优选在0.05-0.35的范围内,进一步优选在0.05-0.15的范围内。
在本发明的一种更为优选的实施方式中,所述原料纳米碳材料为多壁碳纳米管,所述多壁碳纳米管的比表面积为50-500m2/g,优选为80-300m2/g,更优选为100-260m2/g,进一步优选为120-190m2/g;所述多壁碳纳米管在400-800℃的温度区间内的失重率为w800,在400-500℃的温度区间内的失重率为w500,w500/w800在0.01-0.5的范围内,优选在0.02-0.4的范围内,更优选在0.05-0.35的范围内,进一步优选在0.05-0.15的范围内。
根据本发明的方法,所述反应在密闭容器中进行。所述反应可以在自生压力(即,不额外施加压力)下进行,也可以在加压的条件下进行。优选地,所述反应在自生压力下进行。所述密闭容器可以为常见的能实现密封和加热的反应器,如高压反应釜。
根据本发明的方法,还可以包括从反应得到的混合物中分离出固体物质,并将分离出的固体物质进行干燥,从而得到所述含金属原子纳米碳材料。
可以采用常用的固液分离方法从反应得到的混合物中分离出固体物质,如离心、过滤和倾析中的一种或两种以上的组合。
所述干燥的条件可以为常规选择,以能脱除分离出的固体物质中的挥发性物质为准。一般地,所述干燥可以在50-200℃的温度下进行,优选在80-180℃的温度下进行,更优选在120-160℃的温度下进行。所述干燥的持续时间可以根据干燥的温度和方式进行选择。一般地,所述干燥的持续时间可以为0.5-48小时,优选为6-24小时,更优选为10-12小时。所述干燥可以在常压(即,1标准大气压)下进行,也可以在减压的条件下进行。从进一步提高干燥的效率的角度出发,所述干燥优选在减压的条件下进行。
根据本发明的方法,能有效地提高原料纳米碳材料中的氧原子、氮原子和金属原子含量,同时不会对原料纳米碳材料的结构形态产生明显影响。
根据本发明的第三个方面,本发明提供了一种由根据本发明的方法制备的含金属原子纳米碳材料。
根据本发明的第四个方面,本发明提供了一种含金属原子纳米碳材料,该含金属原子纳米碳材料是将根据本发明第一个方面的含金属原子纳米碳材料或者根据本发明第三个方面的含金属原子纳米碳材料进行焙烧而制得的。
所述焙烧可以在常规条件下进行。优选地,所述焙烧在250-500℃的温度下进行。更优选地,所述焙烧在300-450℃的温度下进行。所述焙烧的持续时间可以根据焙烧的温度进行选择。一般地,所述焙烧的持续时间可以为1-24小时,优选为2-12小时。所述焙烧可以在含氧气氛中进行,也可以在由惰性气体形成的气氛中进行。所述含氧气氛可以为空气气氛;还可以为氧气与惰性气体混合形成的混合气氛,所述混合气氛中,氧气的含量可以为0.1-22体积%。所述惰性气体可以包括但不限于氮气和/或稀有气体,所述稀有气体可以为氩气和/或氦气。从便利性和成本等角度考虑,优选地,所述焙烧在含氧气氛(如空气气氛)中进行。
根据本发明的含金属原子纳米碳材料或者由本发明的方法制备的含金属原子纳米碳材料具有良好的催化性能,特别是在烃类物质脱氢反应中显示出较高的催化活性。
根据本发明的含金属原子纳米碳材料或者由本发明的方法制备的含金属原子纳米碳材料可以直接用作催化剂,也可以以成型催化剂的形式使用。所述成型催化剂可以含有根据本发明的含金属原子纳米碳材料或者由本发明的方法制备的含金属原子纳米碳材料以及粘结剂。所述粘结剂可以根据该成型催化剂的具体使用场合进行选择,以能够满足使用要求为准,例如可以为有机粘结剂和/或无机粘结剂。所述有机粘结剂可以为常见的各种聚合物型粘结剂,所述无机粘结剂可以为常见的各种耐热无机氧化物,如氧化铝和/或氧化硅。在所述成型催化剂为对烃脱氢反应(如直接脱氢反应和氧化脱氢反应)、特别是对氧化脱氢反应具有催化作用的成型催化剂时,所述粘结剂优选为无机粘结剂。所述成型催化剂中,含金属原子纳米碳材料的含量可以根据具体使用要求进行选择,没有特别限定,一般地,以所述成型催化剂的总量为基准,所述含金属原子纳米碳材料的含量可以为5-95重量%。
根据本发明的第五个方面,本发明提供了根据本发明第一个方面的含金属原子纳米碳材料、根据本发明第三个方面的含金属原子纳米碳材料、或者根据本发明第四个方面的含金属原子纳米碳材料作为烃脱氢反应的催化剂的应用。
根据本发明的应用,所述含金属原子纳米碳材料可以直接用于烃脱氢反应,也可以成型后用于烃脱氢反应。所述脱氢反应可以在氧气存在下进行,也可以不在氧气存在下进行。优选地,所述脱氢反应在氧气存在下进行,这样能获得更好的催化效果。
根据本发明的第六个方面,本发明提供了一种烃脱氢反应方法,该方法包括在存在或不存在氧气的条件下,在烃脱氢反应条件下,将烃与根据本发明第一个方面的含金属原子纳米碳材料、根据本发明第三个方面的含金属原子纳米碳材料、或者根据本发明第四个方面的含金属原子纳米碳材料接触。
根据本发明的烃脱氢反应方法,所述含金属原子纳米碳材料可以直接用于与烃接触,也可以将所述含金属原子纳米碳材料成型后用于与烃接触。
根据本发明的烃脱氢反应方法可以对多种类型的烃进行脱氢,从而得到不饱和烃,如烯烃。根据本发明的方法特别适于对烷烃进行脱氢,从而得到不饱和烃,例如烯烃。
根据本发明的方法,所述烃优选为烷烃,如c2-c12的烷烃。具体地,所述烃可以为但不限于乙烷、丙烷、正丁烷、异丁烷、正戊烷、异戊烷、新戊烷、环戊烷、正己烷、2-甲基戊烷、3-甲基戊烷、2,3-二甲基丁烷、环己烷、甲基环戊烷、正庚烷、2-甲基己烷、3-甲基己烷、2-乙基戊烷、3-乙基戊烷、2,3-二甲基戊烷、2,4-二甲基戊烷、正辛烷、2-甲基庚烷、3-甲基庚烷、4-甲基庚烷、2,3-二甲基己烷、2,4-二甲基己烷、2,5-二甲基己烷、3-乙基己烷、2,2,3-三甲基戊烷、2,3,3-三甲基戊烷、2,4,4-三甲基戊烷、2-甲基-3-乙基戊烷、正壬烷、2-甲基辛烷、3-甲基辛烷、4-甲基辛烷、2,3-二甲基庚烷、2,4-二甲基庚烷、3-乙基庚烷、4-乙基庚烷、2,3,4-三甲基己烷、2,3,5-三甲基己烷、2,4,5-三甲基己烷、2,2,3-三甲基己烷、2,2,4-三甲基己烷、2,2,5-三甲基己烷、2,3,3-三甲基己烷、2,4,4-三甲基己烷、2-甲基-3-乙基己烷、2-甲基-4-乙基己烷、3-甲基-3-乙基己烷、3-甲基-4-乙基己烷、3,3-二乙基戊烷、1-甲基-2-乙基环己烷、1-甲基-3-乙基环己烷、1-甲基-4-乙基环己烷、正丙基环己烷、异丙基环己烷、三甲基环己烷(包括三甲基环己烷的各种异构体,如1,2,3-三甲基环己烷、1,2,4-三甲基环己烷、1,2,5-三甲基环己烷、1,3,5-三甲基环己烷)、正癸烷、2-甲基壬烷、3-甲基壬烷、4-甲基壬烷、5-甲基壬烷、2,3-二甲基辛烷、2,4-二甲基辛烷、3-乙基辛烷、4-乙基辛烷、2,3,4-三甲基庚烷、2,3,5-三甲基庚烷、2,3,6-三甲基庚烷、2,4,5-三甲基庚烷、2,4,6-三甲基庚烷、2,2,3-三甲基庚烷、2,2,4-三甲基庚烷、2,2,5-三甲基庚烷、2,2,6-三甲基庚烷、2,3,3-三甲基庚烷、2,4,4-三甲基庚烷、2-甲基-3-乙基庚烷、2-甲基-4-乙基庚烷、2-甲基-5-乙基庚烷、3-甲基-3-乙基庚烷、4-甲基-3-乙基庚烷、5-甲基-3-乙基庚烷、4-甲基-4-乙基庚烷、4-丙基庚烷、3,3-二乙基己烷、3,4-二乙基己烷、2-甲基-3,3-二乙基戊烷、苯乙烷、1-苯基丙烷、2-苯基丙烷、1-苯基丁烷、2-苯基丁烷、1-苯基戊烷、2-苯基戊烷和3-苯基戊烷中的一种或两种以上的组合。更优选地,所述烃为丙烷、正丁烷、异丁烷和苯乙烷中的一种或两种以上。进一步优 选地,所述烃为正丁烷。
根据本发明的烃脱氢反应方法,所述反应可以在存在氧气的条件下进行,也可以在不存在氧气的条件下。优选地,根据本发明的烃脱氢反应方法,在存在氧气的条件下进行。在本发明的方法在存在氧气的条件下进行时,氧气的用量可以为常规选择。一般地,烃与氧气的摩尔比可以为0.01-100:1,优选为0.1-10:1,更优选为0.2-5:1,最优选为0.5-2:1。
根据本发明的烃脱氢反应方法,可以通过载气将烃和氧气送入反应器中与含金属原子纳米碳材料接触反应。所述载气可以为常用的在反应条件下不会与反应物和反应生成物发生化学相互作用并且不会发生分解的气体,如氮气、二氧化碳、稀有气体和水蒸气中的一种或两种以上的组合。所述载气的用量可以为常规选择。一般地,载气的含量可以30-99.5体积%,优选为50-99体积%,更优选为70-98体积%。
根据本发明的烃脱氢反应方法,所述接触的温度可以为常规选择,以足以使烃发生脱氢反应为准。一般地,所述接触可以在200-650℃的温度下进行,优选在300-600℃的温度下进行,更优选在350-550℃的温度下进行,进一步优选在400-450℃的温度下进行。
根据本发明的烃脱氢反应方法,所述接触可以在固定床反应器中进行,也可以在流化床反应器中进行,没有特别限定。优选地,所述接触在固定床反应器中进行。
根据本发明的烃脱氢反应方法,所述接触的持续时间可以根据接触的温度进行选择,如所述接触在固定床反应器中进行时,可以用进料的气体的体积空速来表示接触的持续时间。一般地,进料的气体的体积空速可以为0.1-10000h-1,优选为1-6000h-1,更优选为5-5000h-1,进一步优选为10-4000h-1,例如300-600h-1。
以下结合实施例详细说明本发明,但并不因此限制本发明的范围。
以下实施例和对比例中,x射线光电子能谱分析在thermoscientific公司的配备有thermoavantagev5.926软件的escalab250型x射线光电子能谱仪上进行测试,激发源为单色化alkαx射线,能量为1486.6ev,功率为150w,窄扫描所用通透能为30ev,分析测试时的基础真空为6.5×10-10mbar,电子结合能用单质碳的c1s峰(284.0ev)校正,在thermoavantage软件上进行数据处理,在分析模块中采用灵敏度因子法进行定量分析。样品在测试前在150℃的温度和1标准大气压下于氦气气氛中干燥3小时。
以下实施例和对比例中,热重分析在ta5000热分析仪上进行,测试条件为空气气氛,升温速度为10℃/min,温度范围为室温(25℃)至1000℃。样品在测试前在150℃的温度和1标准大气压下于氦气气氛中干燥3小时。采用美国micromertrics公司的asap2000型n2物理吸附仪测定比表面积。采用美国fei公司生产的高分辨透射电镜分析原料纳米碳材料以及含金属原子纳米碳材料的微观形貌。
以下实施例和对比例中,含氮化合物以25重量%水溶液的形式提供,硝酸金属盐以固体物质的形式提供。
实施例1-44用于说明本发明的含金属原子纳米碳材料及其制备方法。
实施例1
(1)将20g作为原料纳米碳材料的多壁碳纳米管(比表面积为136m2/g,氧原子含量为0.3重量%,氮原子含量为0.02重量%,除氮和氧外的其余非金属杂原子(磷和硫)的总含量为0.01重量%,金属原子总含量为0.1重量%,在400-800℃温度区间内的失重率为w800,在400-500℃温度区间内的失重率为w500,w500/w800为0.12,购自中国科学院成都有机化学有限公司)分散在去离子水,分散在超声振荡条件下进行,超声振荡条件包括:频率为140khz,时间为1小时。然后,加入nh3和硝酸铁,从而得到水分散液,其中,按原料纳米碳材料:nh3:硝酸铁:h2o的重量比为1:1.6:1:17.4的比例投料。
(2)将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于100℃的温度下,在自生压力下反应24小时。反应结束后,待高压反应釜内的温度降至室温后,打开反应釜,将反应混合物进行过滤和洗涤,并收集固体物质。将收集到的固体物质在常压(1标准大气压,下同)、120℃的温度下干燥12小时后,得到含金属原子纳米碳材料,该含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
图1为制备的含金属原子纳米碳材料的透射电子显微镜照片,图2为作为原料的多壁碳纳米管的透射电子显微镜照片。从图1和图2可以看出,含金属原子纳米碳材料的微观形态良好,表明反应过程对纳米碳材料的结构影响不大。
对比例1
将与实施例1相同的水分散液置于配备冷凝管的三口烧瓶中,将该三口烧瓶置于温度为100℃的油浴中,于常压下回流反应24小时。反应结束后,待三口烧瓶内的温度降至室温后,将反应混合物进行过滤和洗涤,并收集固体物质。将收集到的固体物质在常压、120℃的温度下干燥12小时后,得到含金属原子纳米碳材料。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
对比例2
采用与实施例1相同的方法制备纳米碳材料,不同的是,步骤(1)配制的水分散液不含硝酸铁,即,将作为原料纳米碳材料分散在去离子水中,然后加入nh3,从而得到水分散液,其中,nh3、原料纳米碳材料和水的用量均与实施例1相同。制备的纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
对比例3
采用与实施例1相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)配制的水分散液不含nh3,也就是,将作为原料纳米碳材料分散在去离子水中,然后加入硝酸铁,从而得到水分散液, 其中,硝酸铁的摩尔量与实施例1中nh3和硝酸铁的总摩尔量相同,原料纳米碳材料和水的用量均与实施例1相同。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
对比例4
采用与实施例1相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)配制的水分散液不含nh3,也就是,将作为原料纳米碳材料分散在去离子水中,然后加入硝酸铁,从而得到水分散液,其中,硝酸铁、原料纳米碳材料和水的用量均与实施例1相同。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
对比例5
将采用与对比例2相同的方法制备的含金属原子纳米碳材料分散在去离子水中,分散在超声振荡条件下进行,超声振荡条件包括:频率为110khz,时间为2小时。然后,加入硝酸铁,从而得到水分散液,其中,按原料纳米碳材料(对比例2中使用的原料纳米碳材料):硝酸铁:h2o的重量比为1:1:17.4的比例投料。
将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于100℃的温度下,在自生压力下反应24小时。反应结束后,待高压反应釜内的温度降至室温后,打开反应釜,将反应混合物进行过滤和洗涤,并收集固体物质。将收集到的固体物质在常压、120℃的温度下干燥12小时后,得到含金属原子纳米碳材料,该含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例2
采用与实施例1相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,作为原料纳米碳材料的多壁碳纳米管(购自山东大展纳米材料有限公司)的比表面积为251m2/g,在400-800℃的温度区间内的失重率为w800,在400-500℃的温度区间内的失重率为w500,w500/w800为0.33,氧原子含量为0.62重量%,氮原子含量为0.01重量%,除氮原子和氧原子外的其余非金属杂原子(磷原子和硫原子)的总含量为0.01重量%,金属原子总含量为0.08重量%。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例3
采用与实施例1相同的方法制备含金属原子纳米碳材料,不同的是,步骤(2)中,将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于80℃的温度下,在自生压力下反应24小时。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例4
采用与实施例1相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,nh3和硝酸 铁的总重量保持不变的条件下,使nh3:硝酸铁的摩尔量为1:0.02。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例5
将20g作为原料纳米碳材料的多壁碳纳米管(比表面积为183m2/g,氧原子含量为0.2重量%,氮原子含量为0.01重量%,除氮原子和氧原子外的其余非金属杂原子(磷原子和硫原子)的总含量为0.04重量%,金属原子总含量为0.03重量%,在400-800℃的温度区间内的失重率为w800,在400-500℃的温度区间内的失重率为w500,w500/w800为0.07,购自中国科学院成都有机化学有限公司)分散在去离子水中,分散在超声振荡条件下进行,超声振荡条件包括:频率为90khz,时间为4小时。然后,加入nh3和硝酸钯,从而得到水分散液,其中,按原料纳米碳材料:nh3:硝酸钯:h2o的重量比为1:5:1:44的比例投料。
(2)将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于200℃的温度下,在自生压力下反应48小时。反应结束后,待高压反应釜内的温度降至室温后,打开反应釜,将反应混合物进行过滤和洗涤,并收集固体物质。将收集到的固体物质在常压、160℃的温度下干燥10小时后,得到含金属原子纳米碳材料,该含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
对比例6
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)配制的水分散液不含nh3,也就是,将原料纳米碳材料分散在去离子水中,然后加入硝酸钯,从而得到水分散液,其中,硝酸钯的摩尔量与实施例5中nh3和硝酸钯的总摩尔量相同,原料纳米碳材料和水的用量与实施例5相同。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
对比例7
采用与实施例5相同的方法制备纳米碳材料,不同的是,步骤(1)配制的水分散液不含硝酸钯,也就是,将原料纳米碳材料分散在去离子水中,然后加入nh3,从而得到水分散液,其中,nh3的摩尔量与实施例5中nh3和硝酸钯的总摩尔量相同。制备的纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例6
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,作为原料纳米碳材料的多壁碳纳米管(购自山东大展纳米材料有限公司)的比表面积为103m2/g,w500/w800为0.23,氧原子含量为1.1重量%,氮原子含量为0.03重量%,除氮和氧外的其余非金属杂原子(磷和硫)的总含量为0.01重量%,金属原子总含量为1.6重量%。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例7
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(2)中,将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于310℃的温度下,在自生压力下反应48小时。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例8
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,原料纳米碳材料:nh3:硝酸钯:h2o的重量比为1:0.2:1:44。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例9
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,按原料纳米碳材料:nh3:硝酸钯:h2o的重量比为1:5:0.1:44的比例投料。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例10
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,硝酸钯用等摩尔量的硝酸镍代替。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例11
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,硝酸钯用等摩尔量的硝酸钡代替。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例12
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,硝酸钯用硝酸钾、硝酸钙和硝酸钠代替,硝酸钾、硝酸钙和硝酸钠的总重量与实施例5中硝酸钯的重量相同,硝酸钾、硝酸钙与硝酸钠的摩尔比为1:1:1。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例13
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,硝酸钯用等摩尔量的硝酸铅代替。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例14
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,硝酸钯用等摩尔量的硝酸钴代替。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例15
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,硝酸钯用硝酸铜和硝酸锌代替,硝酸铜和硝酸锌的总重量与实施例5中硝酸钯的重量相同,硝酸铜与硝酸锌的摩尔比为2:1。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例16
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,硝酸钯用等摩尔量的硝酸铂代替。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表1中列出。
实施例17
采用与实施例1相同的方法制备含金属原子纳米碳材料,不同之处如下:步骤(1)中,将原料纳米碳材料分散在去离子水中,然后加入肼和硝酸钯,从而得到水分散液,其中,按原料纳米碳材料:肼:硝酸钯:h2o的重量比为1:1:0.6:18.4的比例投料;步骤(2)中,将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于110℃的温度下,在自生压力下反应36小时。
制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
对比例8
将与实施例17相同的水分散液置于配备冷凝管的三口烧瓶中,将该三口烧瓶置于温度为110℃的油浴中,于常压下回流反应36小时。反应结束后,待三口烧瓶内的温度降至室温后,将反应混合物进行过滤和洗涤,并收集固体物质。将收集到的固体物质在常压、120℃的温度下干燥12小时后,得到含金属原子纳米碳材料。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
对比例9
采用与实施例17相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)配制的水分散液不含肼,也就是,将原料纳米碳材料分散在去离子水中,然后加入硝酸钯,其中,硝酸钯的摩尔量与实施例17中肼和硝酸钯的总摩尔量相同,原料纳米碳材料以及水的用量均与实施例17相同。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
对比例10
采用与实施例17相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)配制的水分散 液不含肼,也就是,将原料纳米碳材料分散在去离子水中,然后加入硝酸钯,其中,硝酸钯、原料纳米碳材料以及水的用量均与实施例17相同。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
对比例11
采用与实施例17相同的方法制备纳米碳材料,不同的是,步骤(1)配制的水分散液不含硝酸钯,也就是,将原料纳米碳材料分散在去离子水中,然后加入肼,从而得到水分散液,其中,原料纳米碳材料、肼以及水的用量均与实施例17相同。制备的纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例18
采用与实施例17相同的方法制备含金属原子纳米碳材料,不同的是,步骤(2)中,将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于90℃的温度下,在自生压力下反应36小时。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例19
采用与实施例17相同的方法制备含金属原子纳米碳材料,不同的是,作为原料纳米碳材料的多壁碳纳米管与实施例2相同。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例20
采用与实施例17相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,肼和硝酸钯的总重量相同的条件下,使肼:硝酸钯的摩尔比为1:0.05。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例21
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同之处如下:步骤(1)中,将原料纳米碳材料分散在去离子水中,然后加入肼和硝酸铂,从而得到水分散液,其中,按原料纳米碳材料:肼:硝酸铂:h2o的重量比为1:1:0.5:48.5的比例投料;步骤(2)中,将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于180℃的温度下,在自生压力下反应12小时。
制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例22
采用与实施例21相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,作为原料 纳米碳材料的多壁碳纳米管与实施例6相同。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例23
采用与实施例21相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,按原料纳米碳材料:肼:硝酸铂:h2o的重量比为1:0.05:0.5:48.5的比例投料。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例24
采用与实施例21相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,按原料纳米碳材料:肼:硝酸铂:h2o的重量比为1:1:5:48.5的比例投料。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例25
采用与实施例21相同的方法制备含金属原子纳米碳材料,不同的是,步骤(2)中,将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于350℃的温度下,在自生压力下反应12小时。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例26
采用与实施例21相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,硝酸铂用等摩尔量的硝酸铁代替。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例27
采用与实施例21相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,硝酸铂用等摩尔量的硝酸钌代替。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例28
采用与实施例1相同的方法制备含金属原子纳米碳材料,不同之处如下:步骤(1)中,将原料纳米碳材料分散在去离子水中,然后加入尿素和硝酸钌,从而得到水分散液,其中,按原料纳米碳材料:尿素:硝酸钌:h2o的重量比为1:3:1:16的比例投料;步骤(2)中,将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于120℃的温度下,在自生压力下反应12小时。
制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
对比例12
将与实施例28相同的水分散液置于配备冷凝管的三口烧瓶中,将该三口烧瓶置于温度为120℃的油浴中,于常压下回流反应12小时。反应结束后,待三口烧瓶内的温度降至室温后,将反应混合物进行过滤和洗涤,并收集固体物质。将收集到的固体物质在常压、120℃的温度下干燥12小时后,得到含金属原子纳米碳材料。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
对比例13
采用与实施例28相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)配制的水分散液不含尿素,也就是,将原料纳米碳材料分散在去离子水中,然后加入硝酸钌,从而得到水分散液,其中,原料纳米碳材料、水以及硝酸钌的用量均与实施例28相同。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
对比例14
采用与实施例28相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)配制的水分散液不含尿素,也就是,将原料纳米碳材料分散在去离子水中,然后加入硝酸钌,从而得到水分散液,其中,硝酸钌的用量与实施例28中尿素和硝酸钌的总摩尔量相同,原料纳米碳材料以及水的用量均与实施例28相同。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
对比例15
采用与实施例28相同的方法制备纳米碳材料,不同的是,步骤(1)配制的水分散液不含硝酸钌,也就是,将原料纳米碳材料分散在去离子水中,然后加入尿素,从而得到水分散液,其中,尿素的摩尔量与实施例28中尿素和硝酸钌的总摩尔量相同,原料纳米碳材料和水的用量均与实施例28相同。制备的纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例29
采用与实施例28相同的方法制备含金属原子纳米碳材料,不同的是,步骤(2)中,将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于80℃的温度下,在自生压力下反应12小时。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例30
采用与实施例28相同的方法制备含金属原子纳米碳材料,不同的是,作为原料纳米碳材料的多壁碳纳米管同实施例2。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例31
采用与实施例28相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,尿素和硝酸钌的总重量保持不变的条件下,使尿素:硝酸钌的摩尔比为1:0.05。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例32
采用与实施例5相同的方法制备含金属原子纳米碳材料,不同之处如下:步骤(1)中,将原料纳米碳材料分散在去离子水中,然后加入尿素和硝酸铑,从而得到水分散液,其中,按原料纳米碳材料:尿素:硝酸铑:h2o的重量比为1:1:3:46的比例投料;步骤(2)中,将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于230℃的温度下,在自生压力下反应36小时。
制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例33
采用与实施例32相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,作为原料纳米碳材料的多壁碳纳米管与实施例6相同。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例34
采用与实施例32相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,按原料纳米碳材料:尿素:硝酸铑:h2o的重量比为1:1:0.05:46的比例投料。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例35
采用与实施例32相同的方法制备含金属原子纳米碳材料,不同的是,步骤(1)中,按原料纳米碳材料:尿素:硝酸铑:h2o的重量比为1:0.1:3:46的比例投料。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例36
采用与实施例32相同的方法制备含金属原子纳米碳材料,不同的是,步骤(2)中,将得到的水分散液置于带有聚四氟乙烯内衬的高压反应釜中,于310℃的温度下,在自生压力下反应36小时。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例37
采用与实施例32相同的方法制备含金属原子纳米碳材料,不同的是,硝酸铑用等摩尔量的硝酸镍代替。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例38
采用与实施例32相同的方法制备含金属原子纳米碳材料,不同的是,硝酸铑用等摩尔量的硝酸铂代替。制备的含金属原子纳米碳材料的组成、比表面积以及w500/w800在表2中列出。
实施例39
将实施例1制备的含金属原子纳米碳材料在350℃的温度下空气气氛中焙烧4小时。
对比例16
将对比例1制备的含金属原子纳米碳材料在350℃的温度下空气气氛中焙烧4小时。
对比例17
将对比例2制备的含金属原子纳米碳材料在350℃的温度下空气气氛中焙烧4小时。
对比例18
将对比例3制备的含金属原子纳米碳材料在350℃的温度下空气气氛中焙烧4小时。
对比例19
将对比例4制备的含金属原子纳米碳材料在350℃的温度下空气气氛中焙烧4小时。
对比例20
将对比例5制备的含金属原子纳米碳材料在350℃的温度下空气气氛中焙烧4小时。
实施例40
将实施例2制备的含金属原子纳米碳材料在350℃的温度下空气气氛中焙烧4小时。
实施例41
将实施例3制备的含金属原子纳米碳材料在350℃的温度下空气气氛中焙烧4小时。
实施例42
将实施例4制备的含金属原子纳米碳材料在350℃的温度下空气气氛中焙烧4小时。
实施例43
将实施例17制备的含金属原子纳米碳材料在450℃的温度下空气气氛中焙烧2小时。
实施例44
将实施例32制备的含金属原子纳米碳材料在300℃的温度下空气气氛中焙烧12小时。
实施例45-88用于说明本发明根据本发明的含金属原子纳米碳材料的应用以及烃脱氢反应方法。
实施例45-82
分别将0.2g(装填体积为1.9ml)实施例1-38制备的含金属原子纳米碳材料作为催化剂装填在通用型固定床微型石英管反应器中,微型石英管反应器两端封有石英砂,在0.15mpa和430℃条件下,将含有烃和氧气的气体(正丁烷的浓度为1.98体积%,正丁烷和氧气摩尔比0.5:1,余量为作为载气的氮气)以总体积空速为400h-1通入反应器中进行反应,连续监测从反应器中输出的反应混合物的组成,并计算正丁烷转化率、总烯烃选择性和2-丁烯选择性,反应3小时和24小时的结果列于表3中。
对比例21-35
采用与实施例45-82相同的方法进行反应,不同的是,分别使用对比例1-15制备的含金属原子纳米碳材料作为催化剂。反应结果在表3中列出。
对比例36
采用与实施例45-82相同的方法进行反应,不同的是,使用与实施例1相同的原料纳米碳材料作为催化剂。反应结果在表3中列出。
对比例37
采用与实施例45-82相同的方法进行反应,不同的是,使用与实施例5相同的原料纳米碳材料作为催化剂。反应结果在表3中列出。
实施例83-88
采用与实施例45-82相同的方法进行反应,不同的是,使用实施例39-44制备的含金属原子碳纳米管作为催化剂。反应结果在表4中列出。
对比例38-42
采用与实施例45-82相同的方法进行反应,不同的是,使用对比例16-20制备的含金属原子碳纳米管作为催化剂。反应结果在表4中列出。
对比例43
采用与实施例45-82相同的方法进行反应,不同的是,催化剂为将与实施例1相同的原料纳米碳材料在350℃的温度下空气气氛中焙烧4小时而得到的。反应结果在表4中列出。
对比例44
采用与实施例45-82相同的方法进行反应,不同的是,催化剂为将与实施例5相同的原料纳米碳材料在300℃的温度下空气气氛中焙烧12小时而得到的。反应结果在表4中列出。
表3
表4
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种含金属原子纳米碳材料及其制备方法和应用以及一种烃脱氢反应方法与流程
- 一种含金属原子纳米碳材料及其制备方法和应用以及一种烃脱氢反应方法与流程
- 一种含金属原子纳米碳材料及其制备方法和应用以及一种烃脱氢反应方法与流程
- 一种含金属原子纳米碳材料及其制备方法和应用以及一种烃脱氢反应方法与流程
- 一种通过快速贴附双相层碳纳米管进行碳纤维表面改性的装置的制作方法
- 一种金属掺杂的光热碳纳米材料及其制备方法和应用与流程
- 一种伊利石负载纳米碳复合吸附材料及其制备方法与流程
- 一种填充金属硫化物的碳纳米管的制备方法及在锂离子电池中的应用与流程
- 一种碳纳米管表面均匀包覆金属银的方法及装置与流程
- 氨基化碳纳米管掺杂的碳纳米纤维吸附材料及其制备方法与流程