用于稀燃压缩天然气发动机的氧化催化剂的制作方法
本发明总体涉及用于处理含烃的废气流的氧化催化剂领域以及制备和使用所述催化剂以氧化甲烷和其它低级烃的方法。
背景技术:
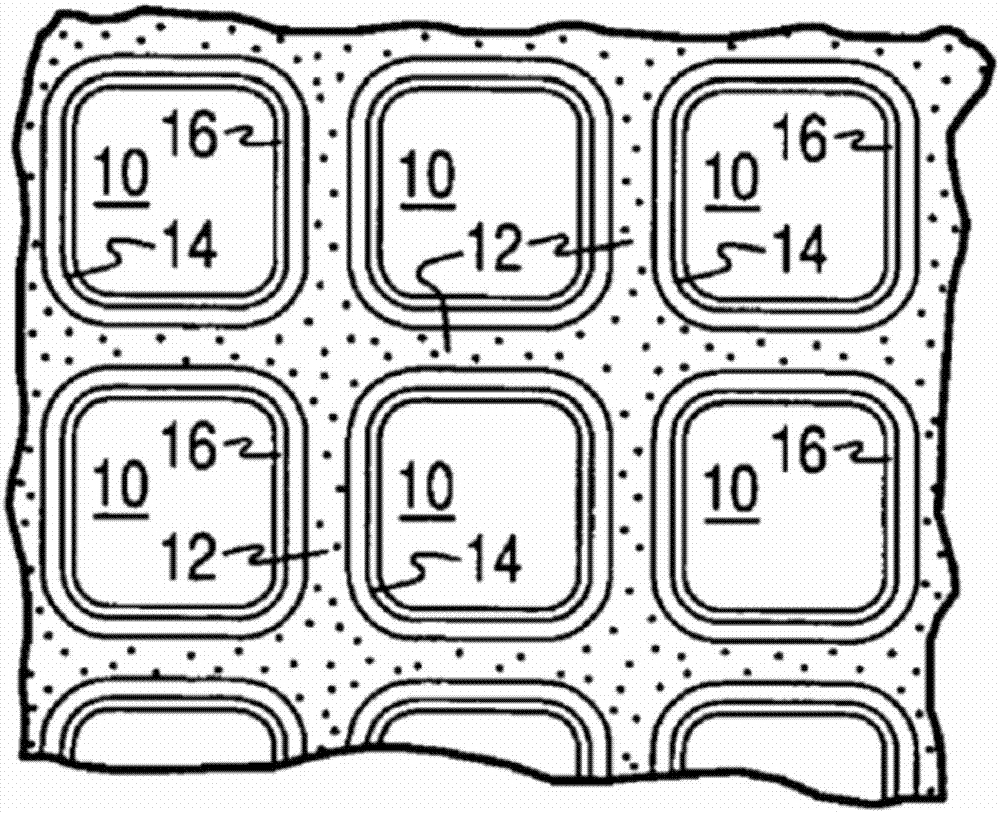
:大多数客车和卡车发动机(机动车辆发动机)通过汽油或柴油燃料操作,但是,因为天然气被视为“干净的”燃料,所以将其用作机动车辆的燃料受到越来越多的关注。天然气主要包含甲烷(ch4)和其它低分子量气态烃类。已发现,与通过汽油操作的发动机相比,用甲烷或天然气作为燃料操作的发动机每英里产生较少量的导致烟雾的那种类型的未燃烧烃类。因为地面层臭氧的形成相应地减少,所以较低量的此烃类排放被认为是特别有利的。天然气可用于火花点火式、化学计量操作的、汽油类型的发动机;或者用于压缩点火式、稀燃(lean)操作的、柴油类型的发动机。因为汽油、柴油和天然气都是烃燃料,因此,似乎是针对汽油和柴油发动机开发的操作实践和废气处理技术可直接地用于以甲烷作燃料的发动机。但是,情况不是这样的。已发现,以化学计量的天然气(甲烷,ch4)作燃料的发动机当根据以汽油作燃料的发动机实践用三效催化剂操作时,表现出通过排气系统以未氧化形式通向大气中的未燃烧甲烷。类似地,以稀燃的天然气作燃料的发动机当根据以柴油做燃料的发动机实践用柴油氧化催化剂操作时也表现出以未氧化形式通向大气的未燃烧甲烷。虽然甲烷无毒并从在低高度下促进臭氧形成的意义上来讲,其不是反应性烃,但是它是一种温室效应气体。甲烷保留在大气中并具有二氧化碳的大气热反射效应的约30倍。与较高分子量的烃气体不同,甲烷在传统贵金属催化剂上不易氧化,甚至在富氧的废气中。这些催化剂直到加热到非常高的温度(例如,600℃或更高)才会变得“活泼”以氧化甲烷,因为来自很多稀燃天然气发动机的废气温度很少超过500℃,所以仍存在防止未燃烧甲烷从车辆的排气系统逸出进入大气的问题。就此而言,对设计和开发用于稀燃压缩天然气发动机的催化剂系统存在大量未满足的需求。技术实现要素:本发明提供一种适于甲烷和其它气态烃排放物的至少部分转化的氧化催化剂组合物。这些氧化催化剂组合物可用于稀燃压缩天然气发动机,其中来自这些发动机的废气流包括多种烃和硫物质(sox)。废气中的硫物质主要呈二氧化硫so2形式,并来源于燃料和油中存在的含有机硫的烃的燃烧。这些硫物质可随时间使催化剂中毒并因此降低其对烃氧化的催化活性。但是,与该领域中已知的氧化催化剂相比,在脱硫处理后,本发明的催化剂组合物显示出改善的耐硫性和更好的催化活性的恢复性。本发明的一个方面描述了一种被构造成在含烃流中用于烃转化的催化剂组合物,该催化剂组合物包含负载在含氧化锆的材料上的铂族金属(pgm)组分,其中氧化锆为至少90重量%呈单斜晶相。在一个实施方案中,铂族金属(pgm)组分选自由钯、铂及其组合组成的组。在一个实施方案中,铂族金属(pgm)组分包括以约1:1至0:1的重量比存在的铂和钯。在一个实施方案中,钯和铂中的至少一种呈胶体沉积的纳米颗粒形式。在一个实施方案中,含氧化锆的材料包含不超过10%的四方晶相。在一个实施方案中,含氧化锆的材料具有约0.15至约0.4ml/g的孔体积。在一个实施方案中,含氧化锆的材料具有约25至约100m2/g的bet表面积。在一个实施方案中,含氧化锆的材料具有约2nm至约15nm的孔半径。在一个实施方案中,组合物包含以金属计算并基于最终的金属浸渍的含氧化锆的材料的总重量约0.01重量%至约6.0重量%的铂族金属(pgm)组分。在一个实施方案中,含氧化锆的材料在600℃下水热老化12小时后具有数量为约50至约150微摩尔/克范围内的强路易斯酸位点。在一个实施方案中,在硫暴露后,组合物包含通过co-drift试验测量的桥接-co信号相对于线性-co吸收信号的峰强度比率,其中峰强度比率在约0.5至约2的范围内。本发明的另一个方面描述了一种催化剂制品,其包括:催化剂基材,其具有适于气体流动的多个通道,每个通道具有壁表面;和催化涂层,其包含根据本发明的实施方案的催化剂组合物。在一个实施方案中,催化剂基材为一种流通型蜂窝基材。在一个实施方案中,蜂窝包括壁流式过滤基材。在一个实施方案中,催化涂层以至少约1.0g/in3的负载量存在于基材上。在一个实施方案中,基材上催化剂组合物的铂族金属(pgm)组分的总负载量为至少约30g/ft3。本发明的另一方面描述了一种转化废气流中的烃的方法,其包括使所述气流与催化剂组合物在一定温度下接触一段时间,所述温度和时间足以降低气流中的烃的水平。在一个实施方案中,催化剂组合物具有改善的耐硫性并更容易地从硫暴露恢复以用于废气流中甲烷的转化。附图说明图1是可包含根据本发明的催化剂制品(即,氧化催化剂)载体涂料(washcoat)组合物的蜂窝型基材载体的透视图;图2是相对于图1放大并沿着平行于图1的基材载体端面的平面的部分横截面视图,其示出了图1中所示的多个气流通道的放大视图,在其中基材是整体式流通型基材的实施方案中;图3为相对于图1放大的截面的剖视图,其中图1中的蜂窝型基材载体表示壁流式过滤基材整体料;图4为线图,其显示了在轻微老化(degreen)处理后及在硫酸化前,实施例1、实施例2和实施例3中所制备的催化剂组合物的co-drifts数据;图5为线图,其显示了在硫酸化后,实施例1、实施例2和实施例3中所制备的催化剂组合物的co-drifts数据;及图6为线图,其显示了在650℃下柴油发动机老化50小时后,实施例1和实施例2中所制备的催化剂组合物的co-drifts数据。具体实施方式现在将在下面更详细地描述本发明。但是,本发明可以多种不同形式呈现并且不应视为限制在文中所述的实施方案;而是,提供这些实施方案以使本公开彻底并完整,并向本领域技术人员充分地传达本发明范围。如本说明书及权利要求中所用,单数形式“一个/一种(a)”、“一个/一种(an)”和“所述(the)”包括复数指示物,除非上下文中明确地另外指出。本发明提供一种适于气态烃排放物(例如甲烷)的至少部分转化的氧化催化剂组合物。该氧化催化剂组合物包括负载在多孔高熔点氧化物材料上的至少一种铂族金属(pgm)组分。在一些实施方案中,多孔高熔点氧化物载体根据其结晶相定义。在一些实施方案中,pgm组分同样根据以pgm沉积在载体上的方式得到的其形式定义。本发明的氧化催化剂组合物可用于稀燃压缩天然气发动机,其中来自所述发动机的废气流包括烃和少量硫物质(sox)。硫物质可随时间使催化剂中毒并从而降低其对烃转化的催化活性。但是,与该领域中已知的氧化催化剂相比,在脱硫处理后,本发明的催化剂组合物显示了改善的耐硫性及更好的催化活性。如文中所用,术语“催化剂”或“催化剂组合物”指促进反应的材料。可使催化剂“轻微老化”,意指使催化剂暴露于高温或废气流一段短时间以达到稳定的初始活性。催化剂可为“新鲜的”,意指催化剂是新的并且没有暴露在废气流下。如文中所用,术语“脱硫”广泛地指与硫化过程相反的过程,意指从催化剂中至少部分地去除含硫物质。通常,此去除需要高能量并在高温条件下完成。此处,表面硫物质在较低温度下主要包括硫酸根(so42-)和亚硫酸根(so32-);并且从催化剂去除的硫主要呈二氧化硫的形式(so2)。如文中所用,术语“流”广泛地指也可包含固体或液体颗粒物质的流动气体的任何组合。术语“气流”或“废气流”意指气态组分流,例如稀燃发动机的废气,其也可包含夹带的非气态组分例如液滴、固体颗粒等。稀燃发动机的废气流通常还包含燃烧产物(co2和h2o)、不完全燃烧产物(co和hc)、氮氧化物(nox)、也称为(烟尘)的可燃和/或含碳颗粒物质(pm)和未反应的氧和氮。如本文所用的,术语“基材”是指通常以含有多个含有在其上的催化剂组合物的颗粒的载体涂料的形式将催化剂组合物放置于其上的整体式材料(monolithicmaterial)。通过在液体载剂中制备含有一定固体含量(例如,按重量计10%至60%)的颗粒的浆料来形成载体涂料,然后将其涂覆在基材上并干燥和煅烧以提供载体涂料层。该方法称为载体涂料法,其中用浆料涂覆基材以在基材上形成催化涂层。如本文所用的,术语“载体涂料”具有其在施加于基材材料的催化剂材料或其他材料的薄的、粘附涂层领域中的通常含义,如蜂窝型载体膜,其是足够多孔的以允许待处理的气流通过。如本文所用的,术语“催化剂制品”是指用于促进期望的反应的元件。例如,催化剂制品可包含载体涂料,所述载体涂料含有在基材上的催化剂组合物。催化剂制品可以“进行轻微老化”意指将催化剂暴露于高温下短的一定量时间。催化剂制品还可以进行“老化”意指将催化剂暴露于高温下较长的一定量的时间,其代表“完全使用寿命”。如本文所用的,“浸渍的”或“浸渍”是指可溶性催化剂材料到载体材料的多孔结构中的渗透。如文中所用,“胶体”指由以阻止它们容易地过滤或快速地沉降的方式分散在另一种物质中的颗粒组成的物质,例如,铂族金属的胶体悬浮液/溶液。例如,“胶体”铂族金属(pgm)为由在均质溶液中的分散颗粒(例如纳米颗粒)组成的pgm。在另一个实例中,“胶体”铂族金属(pgm)为由沉积在另一种材料(例如呈颗粒形式的高熔点氧化物材料)上的分散颗粒(例如,纳米颗粒)组成的pgm。因此,术语“非胶体”指一种铂族金属,其在浸渍过程期间完全地溶解在溶液中并且不呈颗粒的形式。因此,相同的铂族金属可以纳米颗粒(即胶体形式)存在或可以溶液形式存在,例如,不以颗粒存在但以溶解在溶剂中的可溶性物质存在(非胶体形式)。通过胶体溶液沉积的pgm因此在材料上表现出与由非胶体溶液(例如溶解盐)沉积的pgm不同的性质。例如,当使用胶体pgm溶液时,初始pgm粒度与载体涂料负载量无关;并且,pgm颗粒呈还原性更强状态(金属性更强)。胶体铂族金属纳米颗粒可选自由pt、pd、au、ag、ru、rh、ir、os、其合金、及其混合物组成的组。在一个或多个实施方案中,胶体铂族金属纳米颗粒可通过以下中的一种或多种定义:铂族金属中约90%或更多呈完全还原形式;纳米颗粒具有约1至约10nm的平均尺寸;纳米颗粒中至少90%具有在约2nm(以上或以下)的平均粒度内的粒度。当通过浸渍方法转移到高熔点金属氧化物的表面时,铂金属纳米颗粒保持大致相同的平均尺寸及粒度分布。由非胶体,可溶性铂金属溶液前体制得的比较催化剂通常表现出显著数量的颗粒在尺寸上<1nm,及一些颗粒>10nm的粒度的双峰分布。催化剂组合物氧化催化剂组合物可包含至少一种pgm组分和至少一种高熔点氧化物材料。如文中所用,“铂族金属”或“pgm”指铂族金属或其氧化物,包括铂(pt)、钯(pd)、钌(ru)、铑(rh)、锇(os)、铱(ir)、金(au)、银(ag)及其混合物。在优选的实施方案中,pgm组分可包含铂或钯中一者或两者。在一个非限制性实施例中,铂和钯可以约0.1:10至约10:0.1、约1:10至约10:1、约3:1至约1:8、约2:1至约1:6或约1:1至约1:4的重量比组合。pgm组分(例如,pt、pd或其组合)的浓度可变化,但是通常为相对于全部浸渍的高熔点氧化物材料的重量约0.1重量%至约10重量%(例如,相对于浸渍的载体材料约0.5重量%至约6重量%),其中全部浸渍的高熔点氧化物载体包括整个pgm组分及高熔点氧化物载体。如文中所用,“高熔点氧化物材料”指显示可识别的晶体结构或“相”的多孔的含金属的氧化物材料,其在与稀燃压缩天然气发动机废气相关的温度下化学及物理稳定。示例性高熔点氧化物材料包括氧化铝、二氧化硅、氧化锆、氧化钛、二氧化铈及其物理混合物或化学组合,包括原子掺杂的组合。在优选的实施方案中,高熔点氧化物材料为氧化锆。该含氧化锆的材料可定义为至少一部分氧化锆呈单斜晶形。更优选地,大部分的高熔点氧化物材料呈单斜晶形的氧化锆。以下讨论单斜晶形的氧化锆的其它特定比例。单斜晶形的氧化锆可不是高熔点氧化物材料的仅有可得形式;但是,根据本公开的实施方案使用单斜晶形是有利的。例如,单斜氧化锆天然以矿物斜锆石存在。单斜晶体结构提供具有与四方晶体结构的传统的稳定氧化锆不同的暴露的晶体面的氧化锆并因此表现出不同的催化性质。根据本领域已知的方法,利用温度和压力的变化,氧化锆高熔点氧化物材料可由四方晶相转换成单斜晶相。例如,在一些实施方案中,使金属浸渍的高熔点氧化物载体在600℃下煅烧以促进转换成单斜晶相。在其它实施方案中,高熔点氧化物材料(例如,含氧化锆的材料)为至少50重量%、或至少60重量%、或至少70重量%、或至少80重量%、或至少90重量%、或至少95重量%、或至少98重量%、或至少99重量%、或至少99.5重量%单斜的。在其它实施方案中,高熔点氧化物材料为不超过10重量%、或不超过9重量%、或不超过8重量%、或不超过7重量%、或不超过6重量%、或不超过5重量%、或不超过4重量%、或不超过3重量%、或不超过2重量%、或不超过1重量%呈四方晶相。高熔点氧化物材料通常以高结晶材料的形式存在,所述材料为至少约75%结晶的,至少约80%结晶的,至少约85%结晶的,至少约90%结晶的,至少约95%结晶的,至少约98%结晶的,至少约99%结晶的,或至少约99.5%结晶的。高熔点氧化物材料的粒度可变化。通常,粒度可由约10微米至约40微米,优选地约10微米至约20微米,更优选地约5微米至约10微米,最优选地约6微米至约8微米的d90粒度表征。d90定义为90%的颗粒具有更细粒度的粒度。高熔点氧化物材料通常显示出超过1m2/g,至约200m2/g,优选地约25m2/g至约100m2/g的bet表面积。“bet表面积”具有其常见含义,参见通过n2吸附测定表面积的brunauer,emmett,teller方法。在一个或多个实施方案中,bet表面积为至少约25m2/g,至少约50m2/g,或至少约95m2/g。高熔点氧化物材料通常显示出约0.05ml/g至约1.5ml/g,优选地约0.1ml/g至约0.6ml/g,更优选地约0.15ml/g至约0.5ml/g,更优选地0.15ml/g至约0.4ml/g的孔体积。高熔点氧化物材料通常表现出约1至约20nm,优选地约4nm至约15nm,更优选地约7.5nm至约12nm的孔半径。在一些实施方案中,孔半径可以埃表示,其中高熔点氧化物材料通常表现出约10埃至约200埃,优选地约40埃至约150埃,更优选地约75埃至约120埃的孔半径。在本发明的一些方面中,高熔点氧化物材料含有路易斯酸位点,其为可吸附sox物质的配位不饱和位点。这些路易斯酸位点因为用于组成高熔点氧化物材料的金属原子(例如锆)而携载正电荷。存在的路易斯酸位点的强度和浓度部分地取决于所用的金属原子类型及高熔点氧化物材料的结晶状态。例如,取决于温度,氧化锆以单斜晶系、四方晶系或立方晶系存在。氧化物的表面酸度和碱度取决于存在什么相及显示什么晶面。吸附研究显示单斜氧化锆的碱性比四方氧化锆较强,因为其与co2形成较强的键。co吸附显示四方晶相具有酸性比单斜晶相酸性较强的路易酸位点,但是其具有较低浓度的这些路易斯酸位点。通常,表面pgm物质具有催化活性,但也发生硫中毒,即吸附sox物质。如果表面硫位点密度可通过使路易斯酸位点的数量最小化而减小,则可保持pgm的催化活性较长时间。在一些实施方案中,路易斯酸位点的数量通常在约50至约1000微摩尔/g,优选地约50微摩尔/g至约550微摩尔/g,更优选地约50微摩尔/g至约300微摩尔/g,最优选地约50至约150微摩尔/g范围内。在一些实施方案中,包含高熔点氧化物材料的催化剂组合物是新鲜的。在其它实施方案中,使包含高熔点氧化物材料的催化剂组合物轻微老化(例如,600℃/12h,用蒸汽空气)。在另一个实施方案中,将包含高熔点氧化物材料的催化剂组合物暴露于硫物质,然后用脱硫条件进行处理。本发明的另一个方面描述了包含其中至少90重量%呈单斜晶相的高熔点氧化物材料的氧化催化剂组合物的耐硫中毒性。与四方晶相相比,呈单斜晶相的氧化锆高熔点氧化物载体较不强烈地吸附so3(t.kanougi等,j.mol.catal.a,2002,177,289-298)。在载体上强的硫吸附可导致pgm位点被硫物质更多覆盖,以及通过载体相互作用改变pgm颗粒表面的电子性质。在本发明的另一个方面中,胶体pgm组分的表面积可用co/drift光谱测量。通常使用一氧化碳(co)吸附以探测催化剂表面结构及所负载的金属、金属合金、金属氧化物和金属硫化物中可利用的吸附位点。ftir研究确定能够吸附到金属表面上的co分子的量并可确定co分子与金属表面的配位。例如,2090cm-1附近的吸附信号对应于金属表面上的线性吸附的co分子(即,一个co分子与一个金属离子缔合)。1970cm-1附近的吸附信号对应于吸附到金属表面的桥接的co分子(即,两个金属离子与一个co分子缔合)。线性co吸附信号更可能发生在具有低配位的位点的小微晶上(例如,具有与更大数量的吸附物分子配位的能力,如具有较小几何限制的co),而桥接的co分子更可能发生在存在高配位的平面位点的大微晶上。在一些实施方案中,在暴露于硫物质(例如,硫酸化)后,包含pgm组分的催化组合物(其中至少一种pgm来源于胶体pgm前体)与具有相同pgm组分的催化组合物(其中该pgm来源于非胶体可溶性pgm前体)相比,显示出在约1970cm-1处更强的co吸附信号。在一些实施方案中,对于包含pgm组分的催化剂组合物(其中至少一种pgm来源于胶体pgm前体)而言,与包含相同pgm组分的催化剂组合物(其中该pgm来源于非胶体可溶性前体)相比,在约1970cm-1处的co吸附信号为至少25%更多,至少50%更多,至少75%更多,或至少100%更多。在一些实施方案中,包含pgm组分的催化组合物(其中至少一种pgm是胶体pgm(例如,胶体pd))表现出约0.5至约2,优选地约0.8至约1.2,最优选地约1的桥接的co吸附信号相对于co线性吸附信号的峰强度比。在一些实施方案中,桥接及线性co的吸附信号的总co强度因较少量的可利用的pd颗粒表面积而较低。在一些实施方案中,改善的耐s性可归因于以下的组合:使用高熔点氧化物材料(例如,包含锆)及还采用具有胶体pgm前体的该材料以增加组合物的整体催化活性(例如,在发动机老化或硫老化后),此可因为在胶体前体中呈现的较大初始pgm颗粒更耐老化和/或更耐来自硫的负面相互作用。基材根据一个或多个实施方案,用于负载用于稀燃压缩天然气发动机的氧化催化剂组分的基材可由通常用于制备汽车催化剂的任何材料构成并且通常包含金属或陶瓷蜂窝状结构。基材通常提供其中将载体涂料组合物施加并粘附于其上的多个壁表面,从而用作催化剂组合物的载体基材。示例性金属基材包括耐热金属和金属合金,如钛和不锈钢以及其中铁是基本或主要组分的其他合金。这样的合金可包含镍、铬和/或铝的一种或多种,并且这些金属的总量可有利地占合金的至少约15重量%,例如,约10重量%至25重量%的铬,约3重量%至8重量%的铝和多至约20重量%的镍。合金还可包含少量或痕量的一种或多种其他金属,如锰、铜、钒、钛等。表面或金属载体可在高温下(例如,1000℃或更高)被氧化以在基材的表面上形成氧化物层,改善合金的耐腐蚀性并促进载体涂料层到金属表面的粘附。用于构建基材的陶瓷材料可包括任何合适的耐火材料,例如,堇青石、莫来石、堇青石-α氧化铝、氮化硅、碳化硅、锆莫来石、锂辉石、氧化铝-二氧化硅氧化镁、钛酸铝、硅酸锆、硅线石、硅酸镁、锆石、叶长石、α氧化铝、铝硅酸盐等。可使用任何合适的基材,如整体式流通型基材,其具有从基材的入口面向出口面延伸的多个细的、平行气流通道以使得通道对流体流是开放的。从入口到出口基本上为直路径的通道由壁限定,其中将催化剂材料作为载体涂料涂覆在所述壁上以使得流经通道的气体接触催化剂材料。整体式基材的流动通道可以是薄壁通道,其可以为任何合适的横截面形状,如梯形、矩形、正方形、正弦形、六边形、椭圆形、圆形等。这样的结构可包含约60至约1200或更多的气体入口开口(即,“小室”)/平方英寸横截面积(cpsi),更通常地为约300cpsi至约600cpsi。流通型基材的壁厚度可变化,其通常范围为0.002英寸至0.1英寸。代表性的市售流通型基材是具有400cpsi且壁厚度为6mil,或者具有600cpsi且壁厚度为4mil的堇青石基材。然而,应当理解,本发明并不限于特定的基材类型、材料或几何形状。在可替代的实施方案中,基材可以为壁流式基材,其中在基材的一端用无孔塞封闭各通道,在相反的端面封闭交替的通道。这需要气体流经壁流式基材的多孔壁以到达出口。这样的整体式基材可包括多至约700或更多cpsi,如约100cpsi至400cpsi并且更通常为约200cpsi至约300cpsi。如上所述,小室的横截面形状可变化。壁流式基材的壁厚度通常为0.002英寸至0.1英寸。代表性市售壁流式基材由多孔堇青石构成,其中的实例具有200cpsi和10mil壁厚度或者具有300cpsi和8mil壁厚度,并且壁孔隙率为45%至65%。其他陶瓷材料如钛酸铝、碳化硅和氮化硅也可用于壁流式过滤基材上。然而,应当理解,本发明并不限于特定的基材类型、材料或几何形状。应注意,当基材是壁流式基材时,催化剂组合物除了可沉积在壁的表面上之外还可渗透到多孔壁的孔结构中(即,部分或完全阻塞孔开口)。图1和2示出了如本文所述的以涂覆有载体涂料组合物的流通型基材的形式的示例性基材2。参照图1,示例性基材2具有圆柱形形状和圆柱形外表面4、上游端面6和相应的下游端面8,其中端面8与端面6相同。基材2具有在其中形成的多个细的、平行气流通道10。如图2中所见,流动通道10由壁12形成并且经过载体2从上游端面6延伸到下游端面8,通道10是通畅的以便允许流体(例如,气流)通过其气流通道10纵向地经过载体2流动。如在图2中更加容易地见到的,壁12的尺寸和构造成使得气流通道10具有基本上规则的多边形形状。如所示出的,若有需要,可将载体涂料施加在多个、不同层中。在所示实施方案中,载体涂料由粘附于载体部件的壁12上的独立底部涂料载体层14和涂覆在底部载体涂料层14上的第二独立顶部载体涂料层16两者组成。本发明可用一个或多个(例如,2个、3个或4个)载体涂料层实施并且不限于所示的两层实施方案。或者,图1和3示出了如本文所述的以涂覆有载体涂料组合物的壁流式过滤基材的形式的示例性基材2。如图3中所见,示例性基材2具有多个通道52。通道由过滤基材的内壁53呈管状封闭。基材具有入口端54和出口端56。交替的通道在入口端用入口塞58塞住并且在出口端用出口塞60塞住以形成在入口54和出口56处的相对的棋盘图案。气流62经过未堵塞的通道入口64进入,由出口塞60阻止并经过通道壁53(其是多孔的)扩散到出口侧66。由于入口塞58,气体不能返回到壁的入口侧。用于本发明的多孔壁流式过滤器之所以起催化作用是在于所述元件的壁在其上具有或在其中包含一种或多种催化材料。催化材料可仅存在于元件壁的入口侧上、仅存在于出口侧上、存在于入口侧和出口侧两者上或者壁本身可由所有或部分催化材料组成。在本发明的一些实施方案中,壁本身可部分填充有催化材料。本发明包括使用催化材料的一个或多个层,所述催化材料是在壁中或在元件的入口壁和/或出口壁上,或者壁内和壁上位置的任何组合。在描述组合物的载体涂料或催化剂金属组分或其他组分的量时,方便的是使用组分的重量单位/单位体积的催化剂基材。因此,本文使用单位,克/立方英寸(“g/in3”)和克/立方英尺(“g/ft3”)来意指组分的重量/基材的体积,包括基材的空隙空间的体积。有时也使用重量/体积的其他单位如g/l。在载体基材如整体式流通型基材上的催化剂组合物的总负载量通常为约0.5g/in3至约6g/in3,约1g/in3至约5g/in3,约1g/in3至约4g/in3,或约1至约3g/in3。没有载体材料(即,pt或pd或其组合)的pgm组分的总负载量通常为约30至约200g/ft3,优选地50至约150g/ft3,最优选地50至约100g/ft3(例如,至少约30g/ft3或至少约50g/ft3)。对于包含存在于单载体涂料层或多个载体涂料层中的铂和钯的组合的pgm组分,存在于基材上的每种金属的总量以铂和钯的重量比表示,其可以约1:0至约0:1,约1:1至约0:1,或约1:2至约1:8的重量比组合。应注意,这些重量/单位体积通常通过在用催化剂载体涂料组合物处理之前和之后对催化剂基材进行称重来计算,并且由于处理过程包括在高温下干燥和煅烧催化剂基材,所以这些重量表示基本上不含溶剂的催化剂涂层,因为已经基本上移除了载体涂料浆料中的所有水。制造催化剂组合物的方法非胶体的pgm-浸渍的高熔点氧化物材料的制备通常包括用pgm溶液(例如铂溶液或钯溶液,或其组合)浸渍以颗粒形式的高熔点氧化物载体材料。多种pgm组分(例如,铂和钯)可同时或分开浸渍,并可用例如初始润湿技术,浸渍到相同的载体颗粒或分开的载体颗粒中。也称为毛细管浸渍或干法浸渍的初始润湿浸渍技术通常用于非均相材料(即催化剂)的合成。通常,将金属前体溶于水溶液或有机溶液中并随后将含金属的溶液加入催化剂载体中,该催化剂载体含有与所加入的溶液体积相同的孔体积。毛细管作用将溶液吸入载体的孔中。超出载体孔体积的添加溶液使溶液运输从毛细管作用过程变化到扩散过程。然后对催化剂进行干燥及煅烧以去除溶液内的挥发性组分,使金属沉积在催化剂表面上。最大负载量受限于前体在溶液中的溶解度。浸渍材料在高熔点金属氧化物的孔中的浓度分布取决于在浸渍及干燥期间在孔内的传质条件。高熔点氧化物载体颗粒通常足够干燥以吸附基本上所有的pgm浸渍溶液以形成潮湿固体。可用水溶性化合物的水溶液或pgm组分的复合物,例如硝酸钯或硝酸铂、四氨合硝酸钯或铂、或pt胺络合物、或醋酸铂。用pgm溶液处理载体颗粒后,使颗粒任选地干燥,例如通过在升高的温度下(例如,约100-150℃)热处理颗粒一段时间(例如,约1-3小时),然后任选地煅烧以将pgm组分转化成更加催化活性的形式。示例性煅烧过程涉及在约400-600℃的温度下在空气中热处理约10分钟至约3小时。以上过程可根据需要重复以达到pgm浸渍的所期望水平。可将所得材料以干粉或浆料形式储存。在优选的实施方案中,与载体颗粒组合的pgm的至少一部分呈胶体前体形式。包含胶体pgm-组分(即,呈胶体沉积的纳米颗粒形式的pgm组分)的高熔点氧化物材料的制备通常包括将含水或有机pgm-胶体悬浮液(例如,铂胶体悬浮液或钯胶体悬浮液或其组合)施加于高熔点氧化物材料。呈胶体沉积的纳米颗粒形式的多种pgm组分(例如,铂和钯)可同时地或分开地施加,并可用初始润湿技术,沉积在相同的载体颗粒或单独的载体颗粒上,或也可直接加入浆料中。在一些实施方案中,高熔点氧化物材料在暴露于胶体pgm(即,pgm组分,其中至少一种pgm呈胶体沉积的纳米颗粒形式)前已用非胶体pgm预先浸渍以产生pgm改性的高熔点氧化物材料。当高熔点氧化物材料已经用胶体pgm-组分处理时,所得的经pgm改性的高熔点氧化物材料以干粉或浆料形式储存。基材涂覆过程呈包含pgm-浸渍的高熔点氧化物材料的载体颗粒形式(其可具有胶体或非胶体pgm-组分)的上述催化剂组合物与水混合以形成用于涂覆催化剂载体基材(例如,蜂窝型基材)的浆料。除了催化剂颗粒外,浆料可任选地包含呈乙酸锆、胶体氧化锆或氢氧化锆形式的粘合剂、缔合的增稠剂和/或表面活性剂(包括阴离子、阳离子、非离子型或两性表面活性剂)。浆料的ph通常为约2.5至约5。将酸性或碱性物质加入浆料中可相应地调整ph。例如,在一些实施方案中,浆料的ph通过添加氢氧化铵或硝酸水溶液加以调整。当存在时,粘合剂通常使用量为总载体涂料负载量的约1-5重量%。粘合剂可例如为勃姆石、γ-氧化铝或δ/θ氧化铝。或者,粘合剂可基于氧化锆或基于二氧化硅,例如乙酸锆、氧化锆溶胶或二氧化硅溶胶。可研磨浆料以增强颗粒的混合及均质材料的形成。研磨可在球磨机、连续磨机或其它类似装置中进行,并且浆料的固体含量可例如为约20-60重量%,更具体地约30-45重量%。在一个实施方案中,研磨后的浆料的特征在于约5至约40微米,优选地5至约25微米,更优选地约5至约10微米的d90粒度。将d90定义为90%的颗粒具有更细粒度的粒度。然后使用本领域已知的载体涂料技术将浆料涂覆在催化剂基材上。在一个实施方案中,将催化剂基材在浆料中浸渍一次或多次用浆料涂覆。其后,将经涂覆的基材在升高的温度(例如,100℃至150℃)下干燥一定时间段(例如,约1小时至3小时),然后通过例如在400℃至600℃下加热通常约10分钟至约3小时进行煅烧。在干燥和煅烧后,最终的载体涂料涂层可视为基本上不含溶剂。在煅烧之后,通过上述载体涂料技术得到的催化剂负载量可通过计算经涂覆的基材重量与未经涂覆的基材重量的差来测定。如对于本领域技术人员而言显而易见的是,催化剂负载量可通过改变浆料流变学来修改。此外,如有需要,可重复进行用于产生载体涂料的涂覆/干燥/煅烧过程从而建立期望的负载水平或厚度的涂层,这意味着可施加大于一个的载体涂料层。催化剂组合物可作为单层或多层进行施加。在一个实施方案中,催化剂以单层(例如,图2中仅层16)施加。在一个实施方案中,催化剂组合物以多层施加,每个层具有相同或不同的组成。例如,底层(例如,图2中的层14)可包含本发明的氧化催化剂组合物,该氧化催化剂组合物包含浸渍到高熔点氧化物材料中的pgm组分并且顶层(例如,图2中的层16)可包含本发明的催化剂组合物,该本发明的催化剂组合物包含浸渍到高熔点氧化物组分中的相同或不同的pgm组分,其中任一pgm组分可为胶体或非胶体的。在每个层中氧化催化剂组合物的相对量可变化,一个示例性双层涂层在底层(与基材表面相邻)中包含含有pgm组分的氧化催化剂组合物的总重量约10-60重量%和在顶层中包含氧化催化剂组合物的总重量约10-90重量%。烃转化的方法通常,可根据以下所示等式,将任意发动机的废气流中所存在的烃转化为二氧化碳和水:o2+[烃]→co2+h2o通常,发动机废气流中所存在的烃包括c1-c6烃(即,低级烃),例如甲烷,但是也可检测到高级烃(大于c6)。如前所述,烃通常通过未修饰的废气流移动且当排放到大气中时,存在环境危害。因此本发明的这些方面涉及一种用于转化废气流中的烃(例如甲烷)的方法,其包括使气流与所公开的实施方案所描述的催化剂组合物在一定温度下接触一段时间,所述温度和时间足以降低废气流中的烃(例如甲烷)的水平。在一些实施方案中,与接触催化剂组合物之前的废气流中存在的烃的水平相比,废气流中存在的烃(例如甲烷)的水平降低至少约60%,或至少约70%,或至少约75%,或至少约80%,或至少约90%,或至少约95%。在一些实施方案中,用本发明实施方案所描述的催化剂组合物,转化烃(例如甲烷)所需的温度为约300℃至约650℃,约400℃至约600℃,或约450℃至约550℃。在一些实施方案中,催化剂组合物是新鲜的。在其它实施方案中,已将催化剂组合物轻微老化。在其它实施方案中,催化剂组合物已暴露于硫物质,所述硫物质可吸附到催化剂组合物上,从而降低了其对烃转化的催化活性。在其它实施方案中,催化剂组合物已暴露于硫物质,然后用脱硫条件,将硫物质部分地从催化剂中去除。在一些实施方案中,催化组合物包含含有氧化锆的高熔点氧化物材料。在一些实施方案中,高熔点氧化物材料为至少90重量%的高熔点氧化物载体为单斜的。本发明的另一个方面描述了一种用于用本发明的催化剂组合物转化烃(例如甲烷)的方法,其中将转化效率作为起燃温度(light-offtemperature)(即,t50)的函数测定。起燃温度为这样一种温度,在此温度下,催化剂组合物能够将50%的烃转化为二氧化碳和水。通常,针对任意给定的催化剂组合物,所测量的起燃温度越低,催化剂组合物进行催化反应(例如,烃转化)越有效。在一些实施方案中,烃(例如甲烷)的转化包括包含pgm-组分的催化组合物(其中至少一种pgm来源于呈胶体形式的pgm)比包含相同pgm组分的参考催化剂组合物(其中无pgm来源于呈胶体形式的pgm)更低的起燃温度。在一些实施方案中,包含pgm-组分的催化组合物(其中至少一种pgm来源于呈胶体形式的pgm)的起燃温度比包含相同pgm组分的参考催化剂组合物(其中无pgm来源于呈胶体形式的pgm)低至少约1%、至少约2%、至少约3%、至少约4%、至少约6%、至少约7%、至少约8%、至少约9%或至少约10%。在一些实施方案中,包含pgm-组分的催化组合物(其中至少一种pgm来源于呈胶体形式的pgm)的起燃温度比包含相同pgm组分的参考催化剂组合物(其中无pgm来源于呈胶体形式的pgm)低至少约20-30℃。在一些实施方案中,包含pgm-组分的催化组合物(其中至少一种pgm来源于呈胶体形式的pgm)的起燃温度比包含相同pgm组分的参考催化剂组合物(其中无pgm来源于呈胶体形式的pgm)低至少20-30℃。在一些实施方案中,催化剂组合物包含单斜的氧化锆高熔点氧化物材料。在一些实施方案中,高熔点氧化物材料为至少90重量%单斜的。在一些实施方案中,该单斜的催化组合物显示了比包含不是至少90重量%单斜的高熔点氧化物载体的催化剂组合物低至少5%,更优选地至少10%,甚至更优选地至少15%的起燃温度。在一些实施方案中,该单斜的催化组合物显示出与包含不是至少90重量%单斜的高熔点氧化物载体的催化剂组合物相比低至少约10℃至约30℃的起燃温度。在一些实施方案中,使包含参考组合物的催化组合物轻微老化。在其它实施方案中,催化组合物和参考组合物在与废气流接触前已暴露于硫物质。在其它实施方案中,催化组合物和参考组合物已暴露于硫物质,然后在与废气流接触前,用脱硫条件将硫物质从催化剂中部分地去除。本发明的另一个方面涉及包含pgm-组分的催化组合物(其中至少一种pgm呈胶体沉积的纳米颗粒形式)的水热稳定性与包含相同pgm-组分的参考催化剂组合物(其中无pgm呈胶体沉积的纳米颗粒形式)相比增加。例如,将催化组合物和参考组合物在650℃下暴露于柴油发动机老化50小时然后检测它们对烃的催化转化。在一些实施方案中,包含至少一种呈胶体沉积的纳米颗粒形式的pgm的催化组合物与包含相同pgm-组分的参考催化剂组合物(其中无pgm呈胶体沉积的纳米颗粒形式)相比显示出对于烃(例如,甲烷)转化的更低的起燃温度。例如,在一些实施方案中,催化组合物的起燃温度与参考催化剂组合物相比低至少约5%,或至少约10%,或至少约15%。在另一个实例中,在一些实施方案中,催化组合物的起燃温度与参考催化剂组合物相比低至少约20至约30℃。实施例对比实施例:催化剂制品的制备使γ-al2o3材料用稀释的pt胺络合物溶液初始润湿浸渍,然后在空气中以110℃/4h进行干燥。使干燥的pt/al2o3粉末用稀释的pd硝酸盐溶液初始润湿浸渍。在充分地混合约15分钟后,将浸渍的粉末加入二次离子化水(di)水中以形成浆料悬浮液。用1:1wt/wthno3:水将浆料ph调整至ph4.5,进行研磨直到最终粒度d90达到12-15μm。然后,使浆料在40-45%固体含量下涂覆到400/4蜂窝基材上。干燥后,使催化剂在590℃下在空气中煅烧1小时。所得的载体涂料负载量为1.57g/in3,pgm负载量为50g/ft3及pt/pd比率为1:4。实施例1:催化剂制品的制备用初始润湿技术,将zro2-a用稀释的pt胺络合物溶液浸渍以得到pt-浸渍的zro2-a材料。然后,将该材料加入二次离子化(di)水中以形成约50%固体含量的浆料悬浮液中。将10-15%pd硝酸盐溶液逐滴加入浆料中,然后以全部载体涂料负载量的2.5%添加乙酸锆溶液。如果浆料的ph低于2.5-3,则用碱调整。温和地研磨浆料以破坏一些较大的聚集物,并且最终的粒度为d90=6-8μm。然后,将浆料在40-45%固体含量下涂覆到400/4蜂窝基材上。干燥后,使催化剂在590℃下在空气中煅烧1小时。所得的载体涂料负载量为1.57g/in3,并且铂族金属(pgm)负载量为50g/ft3及pt/pd比率为1/4。实施例2:催化剂制品的制备使zro2-a用稀释的pt胺络合物溶液初始润湿浸渍,然后加入到稀释的胶体pd溶液(例如,纳米pd粒度为1-4nm)中以形成浆料悬浮液。用于胶体pd制备的程序可见于国际专利申请号pct/us2015/054525,该申请的全文以引用的方式并入本文中。将乙酸锆溶液以全部载体涂料负载量的2.5%加入浆料中,并且如果浆料的ph低于2.5-3,则用碱调整。温和地研磨浆料以破坏一些较大的聚集物,直到最终的粒度d90达到6-8μm。然后,将浆料在37-42%固体含量下涂覆到400/4蜂窝基材上。干燥后,使催化剂在590℃下在空气中煅烧1小时。所得的载体涂料负载量为1.57g/in3,pgm负载量为50g/ft3及pt/pd比率为1/4。实施例3:催化剂制品的制备使zro2-a用稀释的pd硝酸盐溶液初始润湿浸渍,然后加入di水中以形成浆料悬浮液;将胶体pt溶液加入浆料中,然后以全部载体涂料负载量的2.5%添加乙酸锆溶液(作为粘合剂)。如果浆料的ph低于2.5-3,则用碱调整。温和地研磨浆料以破坏一些较大的聚集物,直到最终的粒度d90达到6-8μm。然后,将浆料在40-45%固体含量下涂覆到400/4蜂窝基材上。干燥后,使催化剂在590℃下在空气中煅烧1小时。所得的载体涂料负载量为1.57g/in3,并且pgm负载量为50g/ft3及pt/pd比率为1/4。用于胶体pd制备的程序可见于国际专利申请号pct/us2015/054525,纳米pt金属粒度为1-3nm。实施例4:催化剂制品的制备将zro2-a缓慢地加入稀释的胶体pt溶液中以形成浆料悬浮液,在充分地混合后,将胶体pd溶液逐滴地加入浆料中,然后以全部载体涂料负载量的2.5%添加乙酸锆溶液。如果浆料的ph低于3,则用碱调整。温和地研磨浆料以破坏一些较大的聚集物,直到最终的粒度d90达到6-8μm。然后,将浆料在35-40%固体含量下涂覆到400/4蜂窝基材上。干燥后,使催化剂在590℃下在空气中煅烧1小时。所得的载体涂料负载量为1.57g/in3,并且pgm负载量为50g/ft3及pt/pd比率为1/4。实施例5:催化剂制品的制备将zro2-a缓慢地加入稀释的胶体pd溶液中以形成浆料悬浮液;以全部载体涂料负载量的2.5%添加乙酸锆溶液。如果浆料的ph低于3,则用碱调节。温和地研磨浆料以破坏一些较大的聚集物,直到最终的粒度d90达到6-8μm。然后,将浆料在35-40%固体含量下涂覆到400/4蜂窝基材上。干燥后,使催化剂在590℃下在空气中煅烧1小时。所得的载体涂料负载量为1.57g/in3,pgm负载量为50g/ft3且只含有pd。实施例6:催化剂粉末的制备如实施例1所述,使每种单独的载体材料,zro2-a(6a)、10%y2o3-zro2(6b)、5%ceo2-zro2(6c)及10%pr6o11-zro2(6d)用pd硝酸盐初始润湿浸渍,添加di水以将浆料设定在30%固体含量下。将pt胺络合物溶液逐滴地加入浆料中并用稀释的hno3(水溶液)将所得浆料的ph降到4。研磨浆料直到达到一致,然后在搅拌下加热至干燥。使单独的干粉在空气中在590℃下煅烧2小时,产生3重量%pgm催化剂粉末。实施例7:催化剂粉末的制备用两种不同的zro2材料:zro2-a和zro2-b,制备1/4pt/pd比率的包含2%pgm的催化剂。使zro2材料用稀释的pd硝酸盐溶液初始润湿浸渍;在剪切混合10分钟后,将潮湿的pd/zro2加入稀释的胶体pt溶液中,向该溶液中添加乙酸锆(总质量的3%)。用碱将ph调节到3后,将浆料在500rpm下研磨10分钟,然后在搅拌下在热板上干燥。使干粉在590℃下在空气中煅烧1小时。样品以pt/pd为1/4的比率包含2%pt/pd。实施例8:催化剂粉末的制备将zro2-a载体与在600℃/2小时下煅烧的预先煅烧的zro2-a载体比较。类似于实施例7和实施例8所述,制备以1/4pt/pd比率包含4%pgm的粉末催化剂。实施例9:催化剂粉末的制备使用与实施例7相同的制备程序制备分别具有pt/pd比率为1/2、1/4及1/8的实施例9a、9b及9c粉末催化剂。实施例10:氧化锆载体与氧化铝载体的比较通过在550℃下在10%蒸汽空气中热处理4小时,使具有1.5x2”尺寸的对比实施例和实施例1中制备的涂覆的整体催化剂组合物轻微老化。硫老化在10ppmso2、10体积%的h2o、其余为空气的进料气体混合物中在450℃/25h下进行。总气体流速为5l/min及总so2暴露量为1.7gs/l。甲烷起燃试验在1000ppmch4、400ppmc2h6、600ppmco、150ppmc3h6、240ppmno、5%co2、10%o2及7%h2o,其余为n2的进料气体混合物中在sv50,000/h下进行。使催化剂以15℃/min升温速率从100加热到600℃,使用t50值来比较活性。结果显示在表1中。在第一次起燃试验期间,轻微老化及硫酸化的催化剂实施例1(zro2-a)表现出比轻微老化及硫酸化的催化剂对比实施例(al2o3)低25℃的t50值,并且在第二次起燃试验期间,t50优势长到38℃。表1.ch4起燃t50,℃催化剂轻微老化硫酸化,第一次试验硫酸化,第二次试验对比实施例391514478实施例1409489440实施例11:使用胶体pgm前体对氧化锆的耐硫性的影响类似于以上所述,使具有1.5x2”尺寸的实施例1-4中制备的涂覆的整体催化剂组合物轻微老化及硫酸化。用与如上描述相同的起燃程序评价轻微老化及硫酸化的催化剂;对于硫酸化的催化剂,在第二次起燃试验期间,只将温度升至475℃,然后在相同温度下,稳定30分钟。然后,通过1分钟的o2-关闭及1分钟的o2-开启五个循环,然后在起燃气体混合物中最后稳定30分钟进行来短暂脱硫。结果显示在表2中。催化剂实施例2-4显示出在硫酸化后,比催化剂实施例1的ch4起燃t50低接近30-40℃。此数据表明胶体pgm前体导致甲烷氧化催化剂改善的耐s性。胶体pgm前体的益处也表现在脱硫处理后的活性,其中催化剂实施例2-4的ch4t50值几乎恢复。并且,脱硫后,催化剂实施例2和实施例3(分别79%和80%)相对于催化剂实施例1(69%)而言,在475℃下的稳态甲烷转化率高出约10%。使用胶体pt和胶体pd二者的催化剂实施例4显示出进一步改善,其中硫化后,在475℃下,观察到87%的甲烷转化率。表2.胶体pgm前体对耐s性&脱硫效率的影响为了理解胶体pgm前体的有利机理,通过co-drifts技术,用从涂覆的整体样品刮下的载体涂料层,分析轻微老化或硫酸化的催化剂实施例1-3。在配备有mct检测器和具有znse窗的piketechnology环境室的bio-radexcaliburfts3000ir光谱仪上进行co-drifts试验。用研钵和研杵,将样品研磨成细粉,然后填入样品杯中。先使样品粉末在400℃下在40ml/min的流速下的流动ar中脱水1小时,然后冷却到30℃。收集样品的红外光谱,没有将探针分子吸附用作参照。将含1%co的ar引入室中。总co暴露时间为30分钟。然后,通过在30℃下,将样品通过流动ar吹扫2分钟并且测量co吸附光谱。在图4中,因为使用基于溶液的pd硝酸盐前体导致较高的pgm分散,所以催化剂实施例1和3显示了比催化剂实施例2整体较高的co吸附强度。通常,具有预形成的纳米金属颗粒的胶体pgm前体通常产生较低的初始分散及较少量的co吸附。使用胶体pd前体所产生的独特特征为桥接吸附的co相对于线性吸附的co的比率明显较高,这被认为是因为直接归因于胶体pd前体的起始较大的pd颗粒。在图5中,在硫酸化试验(1.7g/l总硫暴露量)后,催化剂实施例2和实施例3的总co吸附强度此时明显高于催化剂实施例1。值得注意的是,催化剂实施例2也表现出在1900cm-1附近的显著峰,这是因为桥接-吸附的co。co-drifts结果与表2中所示的活性数据完全符合。实施例12:使用胶体pgm前体对氧化锆的老化稳定性的影响用超低硫柴油燃料,使涂覆的整体催化剂实施例1、2和5在650℃下进行柴油发动机老化50小时。在表3中对比甲烷起燃活性。表3.在柴油发动机老化650℃/50h后,ch4起燃t50对比实施例1实施例2实施例5ch4转化t50(℃)462420423使用胶体pd前体的催化剂实施例2显示出低42℃的t50值。只使用胶体pd且不包含任何pt的催化剂实施例5也表现出与催化剂实施例2类似的ch4t50。用co-drifts表征催化剂实施例1和实施例2的载体涂料,比较示于图6。两个实施例都显示了线性吸附的co的类似强度,但是催化剂实施例2显示了明显较高强度的桥连吸附的co,此特征在图4中所示的轻微老化样品中发现。此数据表明,与硝酸钯相比,胶体pd前体在水热老化后得到较多可利用的pd表面。当使用胶体pd时,在催化剂处理的所有阶段(例如,轻微老化、硫酸化或发动机老化),观察到桥连吸附的co相对于线性吸附的co的较高比率,看来此特征与包含胶体pd的催化剂所观察到的较高活性具有很强的相关性。下表显示了对桥连吸附的co相对于线性吸附的co的比率的总结。表4.对比:桥连吸附的co相对于线性吸附的co的比率催化剂轻微老化硫酸化发动机老化实施例10.690.420.54实施例20.960.771.3实施例13:氧化锆材料的相结构的影响通过高通量测试单元测试实施例6中制备的粉末样品。将每个样品成形为250-500μm粒度,通常使用100-300mg成型样品并用刚砂稀释到1ml体积以供测试。对于催化剂测试,气体组合物由1500ppmch4、200ppmno、10%o2、10%h2o和5%co2、其余为n2组成。轻微老化在550℃下在反应气体中进行(每个位置1小时)。分别在400、450、500及550℃的稳态温度下,以80,000/h空间速度进行催化测试。使每个样品在每个温度下暴露于模拟的气体混合物10分钟,并将在最后30秒收集的数据用于对比。在轻微老化条件中试验后,使样品在10ppmso2、100ppmno、10体积%h2o/空气的气体混合物中在450℃下硫酸化15h,然后在不含so2的相同条件下再次测试。表5中显示了结果。硫酸化后,基于单斜的zro2-a的催化剂在硫酸化后表现出比基于掺杂的氧化锆(例如,10%y2o3-zro2、5%ceo2-zro2、10%pr6o11-zro2)的催化剂组合物低得多的起燃温度。如通过粉末x-射线衍射研究(xrd)所确定,这些掺杂的氧化锆载体看起来部分地(5%ceo2)或完全地(10%y2o3或10%pr6o11)呈四方晶相。表5.zro2-a与掺杂的氧化锆材料的比较(ch4t50,℃)zro2-a10%y2o3-zro25%ceo2-zro210%pr6o11-zro2轻微老化428452440500硫酸化504>550>550>550实施例14:氧化锆中单斜晶相的纯度的影响使实施例7的粉末催化剂在600℃下在10%蒸汽空气中水热老化12小时,并类似于以上实施例6中所描述进行硫酸化。另外,硫酸化后,在450℃和500℃下应用贫/富(l/r)再生,并且这是由1分钟的贫混合物(相同的试验进料)和5秒的富混合物(λ=0.95),循环10次组成。富混合物的组成为co/h2(3/1;针对λ=0.95调整的浓度)、1500ppmch4、200ppmno、10%h2o、1%o2、5%co2、及其余为n2。在按以下顺序的每个步骤后,在450℃下进行比较试验:-硫酸化前-第一次硫酸化后-在450℃下l/r再生后-第二次硫酸化后-在450℃下l/r再生后-在500℃下l/r再生后结果显示在表6中。zro2-a和zro2-b催化剂显示了在硫酸化前、在硫酸化后及在再生后的不同的甲烷转化情况。基于zro2-a的催化剂表现出改善的耐硫性和脱硫效率。在第一次硫酸化后,基于zro2-a的催化剂表现出与基于zro2-b的催化剂所观察到的82%(29%→5%)相对降低相比,甲烷转化率33%的相对降低(42%→28%)。在450℃或500℃下l/r再生后,基于zro2-a的催化剂观察到较高的(接近100%)活性恢复率。表6.在各种硫酸化&脱硫处理后在450℃下的ch4%为了理解催化活性的差异,用各种表面及整体分析技术表征两种载体。如下表7中所示,zro2-a载体具有比zro2-b略微较高的bet表面积;但是在600℃下水热老化12小时后,两种氧化锆载体均显示出明显降低的表面积且bet值变得几乎相同。表7.zro2-a与zro2-b的bet表面积也比较两种载体的路易斯位点的量,路易斯位点为可吸附sox物质的配位不饱和位点。下表8比较了在600℃/12小时下水热老化前后的强路易斯酸位点的数目,其显示了在水热老化后,zro2-a具有比zro2-b少50%的强路易斯酸位点。此结果表明具有较少强路易斯酸位点的氧化锆表面可改善催化剂的耐s性。路易斯酸测量基于用漫反射红外傅立叶变换(drifts)光谱的吡啶吸附-解吸方法。在配备有mct检测器和具有kbr窗允许气体例如n2流过的spectra-tech漫反射高温室的perkin-elmerparagon1000pc光谱仪进行测量。用玛瑙研钵将约40mg样品研磨成细粉并转移到铝样品杯中。在吡啶吸附前,先将样品在450℃下在流动的干燥n2中脱水1小时。然后,将样品在流动的n2下加热到180℃和400℃并保持50分钟,冷却并扫描。在180℃和400℃下解吸的吡啶的量对应于弱或强路易斯位点。数据以微摩尔/克报道且试验误差为±20%。表8.zro2-a与zro2-b的强路易斯酸位点而且,粉末xrd图比较显示了zro2-a(7%)在新鲜状态下具有比zro2-b(14%)较少的四方晶相;但是基于氧化锆的高熔点金属氧化物在水热老化后达到100%单斜晶相。为了评价单斜晶相的重要性,使zro2-a载体在600℃下预先煅烧2小时以导致100%纯度的单斜晶相(由xrd确定)。在表5中的实施例6所述的相同条件下,测试实施例8的比较粉末样品(硫酸化前及后测试)。在表9中,用预先煅烧的zro2-a(转化为100%单斜晶相)的催化剂的t50值在轻微老化后或在硫酸化后低14-20℃。此数据表明纯的单斜氧化锆相有利于改善耐硫性,可能是因为so3在单斜表面上的吸附最弱的事实。表9.纯的单斜氧化锆相的影响zro2-a,按现状zro2-a,煅烧600℃硫酸化前428412硫酸化460440实施例15:pt/pd比率对氧化锆的影响使具有不同pt/pd比率,1/2、1/4或1/8的实施例9的粉末催化剂在10体积%h2o空气中在650℃下老化12小时,然后如实施例7所述,进行硫酸化和贫/富脱硫试验(在表6中)。数据总结在表10中。较高的pt/pd似乎利于富再生(比率1/2和1/4)并具有较好的耐s性;但是增加pt量(比率1/2)可在硫不存在下得到较低的甲烷转化活性;最佳的pt/pd比率可接近1/4。表10.pt/pd比率对450℃下ch4%的影响pt/pd比率硫酸化前硫酸化l/r脱硫450℃1/23021481/44828551/8411328实施例16:在各种载体涂料负载量和pgm负载量下实施例3的比较通过按照对实施例3所述的相同制备程序,实施例3中涂覆的整体催化剂可在固定的1/4pt/pd比率下,随不同载体涂料负载量(1.5–6g/in3)和pgm负载量(50–150g/ft3),或两者的组合变化。下表11中显示了甲烷起燃活性t50的对比。在1000ppmch4、400ppmc2h6、600ppmco、150ppmc3h6、240ppmno、0.5ppmso2、5%co2、10%o2及7%h2o,其余为n2的进料气体混合物中在sv50,000/h下进行甲烷起燃试验。以15℃/min升温速率使催化剂从100℃加热到600℃。表11.在各种载体涂料负载量和pgm负载量下甲烷起燃t50的比较(pt/pd比率=1/4)虽然本文中本发明参照了具体实施方案加以描述,但是应理解,这些实施方案只是描述本发明的原理和应用。对于本领域技术人员显而易见的是,可在不脱离本发明的精神和范围下,对本发明的方法和设备进行多种修改和变化。因此,预期本发明包括属于随附权利要求范围及其等效项的范围内的修改和变化。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 高氯酸铵内嵌的碳/金属氧化物复合催化剂及其制备方法与制造工艺
- 一种复合氧化物负载杂多酸催化合成尿囊素的方法与制造工艺
- 一种无定形铁锌复合氧化物光芬顿催化剂的制备方法与制造工艺
- 具有杂原子的过渡金属化合物、包含该化合物的催化剂组合物和使用该催化剂组合物的聚合物的制备方法与制造工艺
- 配体化合物、过渡金属化合物和包含该化合物的催化剂组合物的制造方法与工艺
- 一种碳/金属氧化物纳米纤维复合催化剂的制备方法与制造工艺
- 改性无机耐热氧化物及其制备方法及加氢脱硫催化剂和降低加氢脱硫催化剂失活速度的方法与制造工艺
- 一种用于莫来石陶瓷晶须制备的氧化物催化剂的制造方法与工艺
- 一种中空金属蜂窝式氧化催化器的制造方法
- 一种处理汽车尾气的三元金属氧化物催化剂制备方法