高效采收油藻的功能化石墨烯基磁性纳米材料及制备方法与流程
本发明属于纳米材料及其应用技术领域,特别涉及一种用于油藻高效采收的功能化石墨烯基磁性纳米材料及其制备方法。
背景技术:
生物质能源具有可再生性、替代性好、生物相容性高、碳收支平衡,被认为是解决能源危机、促进可持续发展并减少温室气体排放的最有潜力的新能源(纪占武, 郑文范. 关于发展生物能源化解能源危机的思考[J].东北大学学报(社会科学版), 2009, 11(6): 490-495)。作为生物质能源的一种,微藻具有光合效率高,细胞增殖快,生产周期短,油脂含量高,不占用耕地,副产品价值高等优点,同时可以通过去除水中氮磷应用于污水处理过程,是制备生物柴油的理想原材料(蒋晓菲, 周红茹, 金青哲,等. 微藻油脂制取技术的研究进展[J].中国油脂, 2012, 37(10): 62-66)。微藻细胞体积小、含水量高、自然培养密度低,对于工业规模的微藻养殖,从藻液中采收微藻一直是个瓶颈。絮凝法、过滤法、浮选法、离心法等传统采收方法存在成本高、能源使用量大、操作方法复杂、技术不完善、收获效率较低等缺陷,在应用上有一定的局限性。利用磁性纳米材料磁性分离收获微藻技术具备操作简单、分离迅速、运行成本低以及节约能源等各种优势。但裸露的磁性纳米材料通常不具备良好的水溶性,在应用中由于其本身的磁性会发生团聚,使其性能急剧下降。石墨烯作为一种新型的二维碳材料,具有巨大的比表面积,易于合成,材料丰富,合成成本低,热学和化学稳定性好。石墨烯片层和纳米粒子的有效结合,可以使纳米粒子作为石墨稀片层间的间隔物质,减小石墨稀片层的凝聚和重堆积;同时,可以有效减少存在磁相互作用的磁性颗粒的团聚。磁性纳米颗粒具有较大的比表面积、优异的磁学性质,修饰在石墨烯片层上形成石墨烯基磁性纳米材料,不但能够增强吸附材料的吸附性能,而且通过外加磁场从水体中分离,在磁性石墨烯纳米材料表面功能化修饰相关基团,可通过静电吸附作用及表面官能团的化学键合作用对产油藻进行高效捕集,同时可通过外加磁场实现从水体中的分离回收,是一种高效、安全、经济的微藻采收方法,具有巨大的应用前景与研究价值,但目前并没有相关报道涉及产油藻高效采收的磁性石墨烯材料。
技术实现要素:
本发明的目的是提供一种功能化石墨烯基磁性纳米材料。
本发明的第二个目的是提供一种用于油藻高效采收的功能化石墨烯基磁性纳米材的制备方法。
本发明的第三个目的是提供任一所述的功能化石墨烯基磁性纳米材料作为油藻高效采收材料的应用。
一种功能化石墨烯基磁性纳米材料,该材料由磁性石墨烯纳米材料和修饰所述磁性石墨烯纳米材料的阳离子聚合物聚二甲基二烯丙基氯化铵(PDDA)组成。
在本发明中,所述的功能化石墨烯基磁性纳米材料(以下称该材料)具备海绵状结构、比表面积大、吸附性能好。该材料具备较好的磁学性能,可通过磁性分离提高对油藻的收获效率,产物易于提取和回收利用、降低成本。该材料表面活性位点多,带有大量的正电基团,有效抵抗外界酸碱变化对微藻采收的影响。
本发明中所述功能化石墨烯基磁性纳米材料中磁性纳米粒子粒径为3-16nm,在中性条件下具有20-25mV的正电位。
本发明提供的功能化石墨烯基磁性纳米材料的制备方法包括如下步骤:
1)采用改进Hummers法制备氧化石墨,液相超声分散制得氧化石墨烯纳米材料;
2)采用化学共沉淀法制备四氧化三铁纳米颗粒;
3)对所述四氧化三铁纳米粒子进行酸化预处理后,与所述氧化石墨烯复合得到磁性石墨烯纳米材料;
4)对所述磁性石墨烯纳米材料进行改性修饰,得到功能化石墨烯基磁性纳米材料。
在本发明的方案中,步骤1)具体为:称取1g石墨粉加入23mL浓硫酸,搅拌12-24h,搅拌状态下加入0.5gNaNO3固体粉末,继续反应3h,在低于10°C的条件下缓慢搅拌加入3gKMnO4固体。升温至35°C反应0.5h,继续升温至38°C反应2h。缓慢逐滴加入去离子水3mL,5min后再加入3mL,继续反应5min后加入40mL,15min后加入140mL。最后加入30%的过氧化氢溶液10mL终止反应。反应结束后静置4h以上得到固体产物,用5%的稀盐酸溶液洗涤2-5次后继续用去离子水洗涤,得到纯净的氧化石墨产物,冷冻干燥18-24h。称取一定质量的干燥后的氧化石墨材料,加入去离子水超声1-3h,得氧化石墨烯分散液。其中,所述的氧化石墨的质量与去离子水的体积比为1mg : 2mL。
在本发明的方案中,步骤2)具体为:称取一定质量的FeCl2∙4H2O和FeCl3∙6H2O粉末溶于去离子水中,在氮气保护下加热到80 °C,逐滴加入25w%的氨水使溶液pH值为10-11,保持此温度继续搅拌反应30min,溶液颜色由棕色变为黑色。冷却至室温,用去离子水洗涤2-5次,冷冻干燥12-20小时,得产物四氧化三铁纳米粒子。其中,所述的FeCl2∙4H2O和FeCl3∙6H2O的摩尔比为1:2。
在本发明的方案中,步骤3)具体为:取一定量的步骤2)得到的四氧化三铁纳米粒子,加入1mol/L的硝酸溶液,超声15-30min,磁性分离后用去离子水洗涤2-3次,得酸化的四氧化三铁纳米粒子。其中,所述的四氧化三铁的质量与硝酸的体积比为1mg : 1mL。
进一步地,称取一定质量酸化后的四氧化三铁纳米材料,加入一定体积的步骤1)所得的氧化石墨烯分散液,超声0.5-1h后搅拌反应3h。用去离子水洗涤2-5次,冷冻干燥12-20小时,得磁性石墨烯纳米材料。其中,所述的酸化后的四氧化三铁的质量与氧化石墨烯分散液的体积比为1mg:1mL。
在本发明的方案中,步骤4)具体为:取20 wt%的分子量为100000-200000的聚二甲基二烯丙基氯化铵(PDDA)溶液2-3mL加入80-100mL去离子水搅拌8-10h。称取一定质量的步骤3)所得的磁性石墨烯纳米材料,加入去离子水超声5-10min使之充分分散。将磁性石墨烯分散液逐滴滴入PDDA溶液中,超声0.5h后搅拌3-8小时,用去离子水洗涤2-5次,冷冻干燥10-15小时。其中,所述的磁性石墨烯纳米材料的质量与去离子水的体积比为1mg:1mL。所述的磁性石墨烯分散液与20 wt% PDDA溶液的体积比为 (30-50) : 1。
本发明生产工艺简单,操作方便,成本低廉,产品无毒害,安全环保,具备良好的应用前景。
附图说明
图1为实施例1中功能化石墨烯基磁性纳米材料及其与藻细胞形成絮凝体的SEM图,(a)为功能化石墨烯基纳米材料,(b)为材料与藻细胞的絮凝体。
图2为实施例1中四氧化三铁纳米粒子的粒径分布图,(a)为TEM统计的石墨烯片层上的Fe3O4纳米粒子的粒径分布,(b)为分散在水中的Fe3O4纳米粒子的水合动力学直径分布。
图3为实施例1中材料的XRD图及UV-vis图,(a)为氧化石墨烯(GO)-A,Fe3O4-B及功能化石墨烯基纳米材料(GFP)-C的XRD图,(b)为氧化石墨烯(GO)-A,磁性石墨烯纳米材料(GF)-B及功能化石墨烯基磁性纳米材料(GFP)-C的UV-vis图。
图4为实施例1中材料的FT-IR图,A-Fe3O4, B-氧化石墨烯(GO), C-磁性石墨烯纳米材料(GF)及D-功能化石墨烯基磁性纳米材料(GFP)。
图5为实施例1中材料的XPS图,(a)为Fe3O4, 氧化石墨烯(GO), 磁性石墨烯纳米材料(GF), 功能化石墨烯基纳米材料(GFP),(b)为功能化石墨烯基纳米材料(GFP)中的C 1s ,(c)为功能化石墨烯基纳米材料(GFP)中的Fe 2p ,(d)为功能化石墨烯基纳米材料(GFP)中的 N 1s。
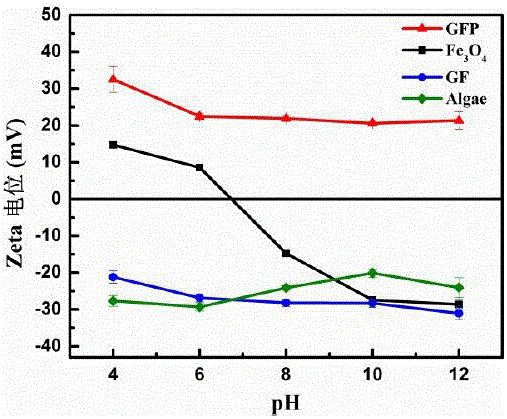
图6为实施例1中的Fe3O4, 磁性石墨烯纳米材料(GF), 功能化石墨烯基纳米材料(GFP)及藻细胞在不同pH条件下的Zeta电位图。
图7为实施例5中在pH 8.0,藻液浓度为0.2g/L的条件下Fe3O4, 磁性石墨烯纳米材料(GF), 功能化石墨烯基磁性纳米材料(GFP)三种材料投加剂量的影响,(a)为对收获效率的影响,(b)为对收获容量的影响图。
图8为实施例6中在藻液浓度为0.2g/L,材料投加量为70mg/L的条件下pH值对Fe3O4,磁性石墨烯纳米材料(GF),功能化石墨烯基磁性纳米材料(GFP)三种材料收获效率的影响图。
图9为实施例7中功能化石墨烯基磁性纳米材料对微藻的吸附热力学模型拟合图。
图10为实施例8中功能化石墨烯基磁性纳米材料对微藻的吸附动力学模型拟合图。
图11为实施例9中循环再生的功能化石墨烯基磁性纳米材料对微藻的收获效率及收获生物量的变化图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明的实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例中使用的藻种为前期分离筛选到的高产油小球藻HQ(保藏单位中国微生物菌种保藏管理委员会普通微生物中心,保藏地址北京市朝阳区北辰西路1号院3号,保藏日期2013年5月7日,保藏编号No. CGMCC7601,分类命名小球藻Chlorella sp.),进行自养培养20天,初始藻液浓度为0.2g/L(以藻细胞干重计)。建立藻细胞干重与波长690nm处吸光度的标准曲线,通过测量收获前后上清液的吸光值来计算藻液浓度,从而测定收获生物量、收获效率和收获容量。
实施例1。
称取1g石墨粉加入23mL浓硫酸,搅拌24h,搅拌状态下加入0.5gNaNO3固体粉末,继续反应3h,在低于10 °C的条件下缓慢搅拌加入3gKMnO4固体。升温至35 °C反应0.5h,继续升温至38 °C反应2h。缓慢逐滴加入去离子水3mL,5min后再加入3mL,继续反应5min后加入40mL,15min后加入140mL。最后加入30%的过氧化氢溶液10mL终止反应。反应结束后静置4h以上得到固体产物,用5%的稀盐酸溶液洗涤5次后继续用去离子水洗涤,得到纯净的氧化石墨产物,冷冻干燥20h。取干燥后的氧化石墨材料,加入去离子水超声2h,得氧化石墨烯分散液。
称取0.99g的FeCl2∙4H2O和2.7g的FeCl3∙6H2O粉末溶于去离子水中,在氮气保护下加热到80 °C,逐滴加入25w%的氨水使溶液pH值为10-11,保持此温度继续搅拌反应30min,溶液颜色由棕色变为黑色。冷却至室温,用去离子水洗涤3次,冷冻干燥20小时,得产物四氧化三铁纳米粒子。
取1g上述四氧化三铁纳米粒子,加入1mol/L的硝酸溶液1000mL,超声20min,磁性分离后用去离子水洗涤3次,得酸化的四氧化三铁纳米粒子。
将上述酸化后的四氧化三铁纳米粒子分散于所述氧化石墨烯分散液,超声1h后搅拌反应3h。用去离子水洗涤4次,冷冻干燥20小时,得磁性石墨烯纳米材料。
取20 wt%的分子量为100000-200000的聚二甲基二烯丙基氯化铵(PDDA)溶液2mL加入100mL去离子水搅拌10h。取100mg上述磁性石墨烯纳米材料,加入100mL去离子水超声10min使之充分分散。将磁性石墨烯分散液逐滴滴入PDDA溶液中,超声0.5h后搅拌5小时,用去离子水洗涤3次,冷冻干燥15小时,得到功能化石墨烯基磁性纳米材料。
如图1所示,上述制备的功能化石墨烯基磁性纳米材料呈现出蓬松的海绵状结构,当材料与微藻结合后可形成明显的絮凝体。
如图2所示,上述制备的GO-Fe3O4/PDDA磁性纳米材料中Fe3O4纳米粒子的平均直径在8.54±2.41 nm(TEM测试统计),而其水合动力学直径达到70-240 nm,功能化的磁性石墨烯纳米材料可以有效提高磁性纳米粒子的分散性。
如图3所示,曲线中各组元的特征峰与标准峰位相吻合,氧化石墨烯与四氧化三铁纳米粒子通过共价键成功结合,同时PDDA的修饰没有改变Fe3O4纳米粒子的晶格结构,保证了材料的磁性能。
如图4所示,氧化石墨烯中主要存在环氧基、烷氧基和羧基,Fe-O键的存在进一步证明四氧化三铁通过化学键合作用与氧化石墨烯片层结合,C-N键和N-H键的出现表明PDDA成功修饰到磁性石墨烯纳米材料表面。
如图5所示,GO-Fe3O4/PDDA磁性纳米材料主要包含C,N,O,Fe四种特征元素,同时对C元素的分峰拟合结果进一步证明了石墨烯片层与四氧化三铁和PDDA通过化学键合作用成功结合。
如图6所示,石墨烯片层的存在增加了材料的稳定性,进一步修饰PDDA后,材料在较宽的pH范围内均保持较高的正电性,可以通过架桥网捕和静电吸附作用高效采收微藻,并抵抗pH变化的影响。
实施例2。
称取1g石墨粉加入23mL浓硫酸,搅拌24h,搅拌状态下加入0.5gNaNO3固体粉末,继续反应3h,在低于10 °C的条件下缓慢搅拌加入3gKMnO4固体。升温至35 °C反应0.5h,继续升温至38 °C反应2h。缓慢逐滴加入去离子水3mL,5min后再加入3mL,继续反应5min后加入40mL,15min后加入140mL。最后加入30%的过氧化氢溶液10mL终止反应。反应结束后静置4h以上得到固体产物,用5%的稀盐酸溶液洗涤5次后继续用去离子水洗涤,得到纯净的氧化石墨产物,冷冻干燥20h。取干燥后的氧化石墨材料,加入去离子水超声1h,得氧化石墨烯分散液。
称取0.99g的FeCl2∙4H2O和2.7g的FeCl3∙6H2O粉末溶于去离子水中,在氮气保护下加热到80 °C,逐滴加入25w%的氨水使溶液pH值为10-11,保持此温度继续搅拌反应30min,溶液颜色由棕色变为黑色。冷却至室温,用去离子水洗涤3次,冷冻干燥20小时,得产物四氧化三铁纳米粒子。
取1g上述四氧化三铁纳米粒子,加入1mol/L的硝酸溶液1000mL,超声20min,磁性分离后用去离子水洗涤3次,得酸化的四氧化三铁纳米粒子。
将上述酸化后的四氧化三铁纳米粒子分散于所述氧化石墨烯分散液,超声1h后搅拌反应3h。用去离子水洗涤4次,冷冻干燥20小时,得磁性石墨烯纳米材料。
取20 wt%的分子量为100000-200000的聚二甲基二烯丙基氯化铵(PDDA)溶液2mL加入100mL去离子水搅拌10h。取100mg上述磁性石墨烯纳米材料,加入100mL去离子水超声10min使之充分分散。将磁性石墨烯分散液逐滴滴入PDDA溶液中,超声0.5h后搅拌5小时,用去离子水洗涤3次,冷冻干燥15小时,得到功能化石墨烯基磁性纳米材料。
实施例3。
称取1g石墨粉加入23mL浓硫酸,搅拌24h,搅拌状态下加入0.5gNaNO3固体粉末,继续反应3h,在低于10 °C的条件下缓慢搅拌加入3gKMnO4固体。升温至35 °C反应0.5h,继续升温至38 °C反应2h。缓慢逐滴加入去离子水3mL,5min后再加入3mL,继续反应5min后加入40mL,15min后加入140mL。最后加入30%的过氧化氢溶液10mL终止反应。反应结束后静置4h以上得到固体产物,用5%的稀盐酸溶液洗涤5次后继续用去离子水洗涤,得到纯净的氧化石墨产物,冷冻干燥20h。取干燥后的氧化石墨材料,加入去离子水超声2h,得氧化石墨烯分散液。
称取0.99g的FeCl2∙4H2O和2.7g的FeCl3∙6H2O粉末溶于去离子水中,在氮气保护下加热到80°C,逐滴加入25w%的氨水使溶液pH值为10-11,保持此温度继续搅拌反应30min,溶液颜色由棕色变为黑色。冷却至室温,用去离子水洗涤3次,冷冻干燥20小时,得产物四氧化三铁纳米粒子。
取1g上述四氧化三铁纳米粒子,加入1mol/L的硝酸溶液1000mL,超声10min,磁性分离后用去离子水洗涤3次,得酸化的四氧化三铁纳米粒子。
将上述酸化后的四氧化三铁纳米粒子分散于所述氧化石墨烯分散液,超声1h后搅拌反应3h。用去离子水洗涤4次,冷冻干燥20小时,得磁性石墨烯纳米材料。
取20 wt%的分子量为100000-200000的聚二甲基二烯丙基氯化铵(PDDA)溶液2mL加入100mL去离子水搅拌10h。取100mg上述磁性石墨烯纳米材料,加入100mL去离子水超声10min使之充分分散。将磁性石墨烯分散液逐滴滴入PDDA溶液中,超声0.5h后搅拌5小时,用去离子水洗涤3次,冷冻干燥15小时,得到功能化石墨烯基磁性纳米材料。
实施例4。
称取1g石墨粉加入23mL浓硫酸,搅拌24h,搅拌状态下加入0.5gNaNO3固体粉末,继续反应3h,在低于10 °C的条件下缓慢搅拌加入3gKMnO4固体。升温至35 °C反应0.5h,继续升温至38 °C反应2h。缓慢逐滴加入去离子水3mL,5min后再加入3mL,继续反应5min后加入40mL,15min后加入140mL。最后加入30%的过氧化氢溶液10mL终止反应。反应结束后静置4h以上得到固体产物,用5%的稀盐酸溶液洗涤5次后继续用去离子水洗涤,得到纯净的氧化石墨产物,冷冻干燥20h。取干燥后的氧化石墨材料,加入去离子水超声2h,得氧化石墨烯分散液。
称取0.99g的FeCl2∙4H2O和2.7g的FeCl3∙6H2O粉末溶于去离子水中,在氮气保护下加热到80 °C,逐滴加入25w%的氨水使溶液pH值为10-11,保持此温度继续搅拌反应30min,溶液颜色由棕色变为黑色。冷却至室温,用去离子水洗涤3次,冷冻干燥20小时,得产物四氧化三铁纳米粒子。
取1g上述四氧化三铁纳米粒子,加入1mol/L的硝酸溶液1000mL,超声20min,磁性分离后用去离子水洗涤3次,得酸化的四氧化三铁纳米粒子。
将上述酸化后的四氧化三铁纳米粒子分散于所述氧化石墨烯分散液,超声1h后搅拌反应3h。用去离子水洗涤4次,冷冻干燥20小时,得磁性石墨烯纳米材料。
取20 wt%的分子量为100000-200000的聚二甲基二烯丙基氯化铵(PDDA)溶液2mL加入100mL去离子水搅拌10h。取100mg上述磁性石墨烯纳米材料,加入100mL去离子水超声10min使之充分分散。将磁性石墨烯分散液逐滴滴入PDDA溶液中,超声0.5h后搅拌3小时,用去离子水洗涤3次,冷冻干燥15小时,得到功能化石墨烯基磁性纳米材料。
实施例5。
各取10 mL藻液在锥形瓶中,初始藻液浓度为0.2g/L,用0.1 M HCl 和 0.1 M NaOH 将藻液pH 均调为8.0,分别加入10-450 mg/L实施例1中的Fe3O4纳米材料、磁性石墨烯纳米材料、功能化石墨烯基磁性纳米材料,于250 rpm摇床中震荡 20min,形成纳米材料和藻细胞的复合物。然后用永磁铁于瓶外进行磁性收获,复合物从悬浮基质中分离出来,此时测定上清液在最大吸收波长处的吸光度,最后根据生物量-吸光度标准曲线计算收获效率和收获容量。
如图7所示,随着纳米材料用量的增加,几种材料的收获效率提高,但收获容量逐渐降低。在达到95 %收获效率的条件下,材料的最佳投加量由Fe3O4的300 mg/L,降低到GO-Fe3O4的200 mg/L,并进一步降低到GO-Fe3O4/PDDA的70 mg/L,显著降低了采收成本。
实施例6。
各取10 mL藻液在锥形瓶中,初始藻液浓度为0.2g/L,用0.1 M HCl 和 0.1 M NaOH 将藻液pH 调为4.0/8.0/12.0,分别加入70 mg/L实施例1中的Fe3O4纳米材料、磁性石墨烯纳米材料、功能化石墨烯基磁性纳米材料,于250 rpm摇床中震荡 20min,形成纳米材料和藻细胞的复合物。然后用永磁铁于瓶外进行磁性收获,复合物从悬浮基质中分离出来,此时测定上清液在最大吸收波长处的吸光度,最后根据生物量-吸光度标准曲线计算收获效率。
如图8所示,在不同的pH条件下,对于裸露的四氧化三铁纳米粒子,收获效率随pH值的升高而降低,而石墨烯片层的存在显著提高了材料对微藻的采收效率,PDDA修饰后的功能化石墨烯基磁性纳米材料在较宽的酸碱范围内均可保持较高的吸附效率。
实施例7。
各取10 mL藻液在锥形瓶中,初始藻液浓度设置为0.25-1.8g/L,用0.1 M HCl 和 0.1 M NaOH 将藻液pH 调为8.0,加入70 mg/L实施例1中的功能化石墨烯基磁性纳米材料,于250 rpm摇床中震荡 20min,形成纳米材料和藻细胞的复合物。然后用永磁铁于瓶外进行磁性收获,复合物从悬浮基质中分离出来,此时测定上清液在最大吸收波长处的吸光度,根据生物量-吸光度标准曲线计算平衡时藻液残留生物量,进行吸附热力学拟合。
如图9所示,功能化石墨烯基磁性纳米材料对微藻的收获过程符合Langmuir吸附模型,吸附过程为单分子层吸附。
实施例8。
各取10 mL藻液在锥形瓶中,初始藻液浓度为0.2g/L,用0.1 M HCl和 0.1 M NaOH 将藻液pH 调为8.0,加入70 mg/L实施例1中的功能化石墨烯基磁性纳米材料,于250 rpm摇床中震荡 20min,形成纳米材料和藻细胞的复合物。然后用永磁铁于瓶外进行磁性收获,复合物从悬浮基质中分离出来,每10min设置一个时间梯度,测定上清液在最大吸收波长处的吸光度,根据生物量-吸光度标准曲线计算收获容量,进行吸附动力学拟合。
如图10所示,功能化石墨烯基磁性纳米材料对微藻的收获过程符合二级动力学吸附模型,电子配对和交换的化学过程为吸附中的主要控制因素。
实施例9。
各取10 mL藻液在锥形瓶中,初始藻液浓度为0.2g/L,用0.1 M HCl 和 0.1 M NaOH 将藻液pH 调为8.0,加入70 mg/L实施例1中的功能化石墨烯基磁性纳米材料,于250 rpm摇床中震荡 20min,形成纳米材料和藻细胞的复合物。然后用永磁铁于瓶外进行磁性收获,复合物从悬浮基质中分离出来,测定上清液在最大吸收波长处的吸光度,根据生物量-吸光度标准曲线计算收获效率。收获后向分离的复合物中加入2mL氯仿和甲醇的混合溶液(体积比2:1),60w超声15min后磁性分离,加入500 µL去离子水使回收后的材料再分散,并加入30mg/L未使用的原材料,进行下一次的收获实验,循环进行5次。
如图11所示,经过5次循环再生后,功能化石墨烯基磁性纳米材料对微藻仍可保持较高的收获效率(88.7 %),具备较好的循环稳定性,可以实现再生利用。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!