一种聚吡咯功能化的钴配合物复合材料的合成方法及其应用与流程
本发明属于可见光催化材料合成领域,特别涉及一种聚吡咯功能化的钴配合物复合材料的合成方法及其应用。
背景技术:
印染工业过程中流失的染料占染料产量的15%,是工业废水的主要污染源之一。在各种染料中,亚甲基蓝可用于麻、蚕丝织物、纸张的染色和竹、木的着色;还可用于制造墨水和色淀及生物、细菌组织的染色等方面。由于该染料在的水溶性较好,因此在生产和使用过程中流失率高,易进入水体,对环境以及人体的皮肤、眼睛等均具有较大的危害作用。因此,开发新的功能性降解材料来降低或消除亚甲蓝染料污染物的危害具有重要的实际意义。.
基于芳香羧酸和有机胺混合配体的过渡金属配合物是一种无机-有机功能杂化材料。这类材料不仅结构多样而且应用广泛。比如该类材料可以作为紫外光催化降解有机染料的催化剂。但是,由于该类配合物材料具有较宽的带隙,因此在可见光范围内不够活跃,这使得在光催化过程中光生电子和空穴易发生复合,导致该类材料利用可见光的光催化效果不够高,因此,提高配合物在可见光范围内对染料污染物的催化降解具有十分重要的意义。
技术实现要素:
本发明要解决的技术问题是提供一种聚吡咯功能化的钴配合物复合材料的合成方法及其应用,该方法采用聚吡咯将钴配合物进行功能化,简易快速的,可扩大该类配合物的光响应范围,有效降低在光催化过程中光生电子和空穴之间的复合,使其在可见光下具有良好催化降解染料污染物效果,节约能源消耗,降低染料污染物对环境的污染。
本发明的技术解决方案是:
一种聚吡咯功能化的钴配合物复合材料的合成方法,其具体步骤如下:
(1)、基于有机配体功能钴配合物的合成
基于有机配体功能钴配合物分子式如下:
[Co(L)(1,3,5-HBTC)];
其中,L为N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺,1,3,5-HBTC为均苯三甲酸一氢根;
具体合成步骤如下:
将CoCl2·6H2O、N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺和均苯三甲酸,加入去离子水,搅拌形成悬浮混合物,用NaOH溶液调pH为4.3~6.4,倒入高压反应釜中,进行水热反应,得到块状晶体,洗涤,晾干,得到基于有机配体多功能钴配合物;
(2)聚吡咯功能化的钴配合物复合材料
聚吡咯功能化的钴配合物复合材料的表达式如下:
[Co(L)(1,3,5-HBTC)]/ppy;
其中,ppy为聚吡咯;
具体合成步骤如下:
a.将步骤(1)合成的基于有机配体多功能钴配合物]称取3克,研磨,然后分散到2毫升~4毫升的乙醇中,用球磨机研磨20分钟~40分钟,离心分离,保留滤饼,在60℃~80℃下干燥24小时,得到微晶态钴配合物[Co(L)(1,3,5-HBTC)]材料;
b.将0.03克~0.05克吡咯、15毫升~30毫升水、1.3克~1.7克微晶态钴配合物超声混合20分钟~40分钟,得到悬浊液;将含有0.11克~0.13克过硫酸胺和10毫升水的混合溶液加入到悬浊液中,搅拌20分钟后静止12小时,离心分离,洗涤,在65℃下真空干燥24h,得到聚吡咯功能化的钴配合物复合材料。
进一步的,所述N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺与均苯三甲酸的摩尔比为1:1~1:2。
进一步的,所述N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺与CoCl2·6H2O的摩尔比为1:1~1:3。
进一步的,所述N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺与去离子水的摩尔比为1:2800~1:8300。
进一步的,水热反应时,以5℃/h升温速率升温至110℃~130℃,水热条件下保温48h~96h,以5℃/h的降温速率降温到室温。
进一步的,将过硫酸胺水溶液加入到悬浊液中时,采用的加入方式为滴加,含有0.11克~0.13克过硫酸胺和10毫升水的混合溶液在5分钟内完成滴加。
进一步的,步骤(1)和(2)进行洗涤时,用去离子水和乙醇交替清洗3次~5次。
进一步的,步骤(2)研磨时,采用玛瑙研钵研磨2小时~4小时。
一种聚吡咯功能化的钴配合物复合材料作为可见光催化降解有机染料污染物材料的应用。
进一步的,聚吡咯功能化的钴配合物复合材料作为可见光催化降解有机染料污染物材料的应用,将50mg的[Co(L)(1,3,5-HBTC)]/ppy作为光催化剂,加入150mL浓度为0-10.0mg·L–1的亚甲蓝水溶液中,可见光照射下,进行光催化降解60分钟~75分钟。
本发明的有益效果是:
(1)钴配合物[Co(L)(1,3,5-HBTC)]具有二维片层结构;
(2)在钴配合物中引入N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺有机胺配体中间的噻吩基团,增加了配体的刚性和支撑能力,与金属钴离子配位后,有易于构筑高维开放骨架;配体中的酰胺基团的引入可以使配合物通过氢键作用形成高维超分子结构,此类超分子结构更有利于稳定配合物的结构和改善配合物的性能;配体中间噻吩上的S原子与酰胺基团类似,能增加配合物的亲水性,从而使合成的钴配合物对水溶性的有机污染物分子的亲和能力增强、催化降解效率高;通过调控反应体系的投料比例和pH值,成功的将均苯三甲酸中的1个羧基保护下来,只利用了两个羧基进行配位,而没有参与配位的羧基具有强氢键键合能力,有效的提高了配合物材料与聚吡咯的结合能力,从而提高了在可见光下对亚甲蓝的催化降解能力并极大程度上的实现了快速降解,在15分钟下对亚甲蓝有机污染物的可见光催化降解率可达54.4%,在75分钟下对亚甲蓝有机污染物的可见光催化降解率可达85.7%,可以作为亚甲蓝有机污染物的可见光催化降解材料使用。
附图说明
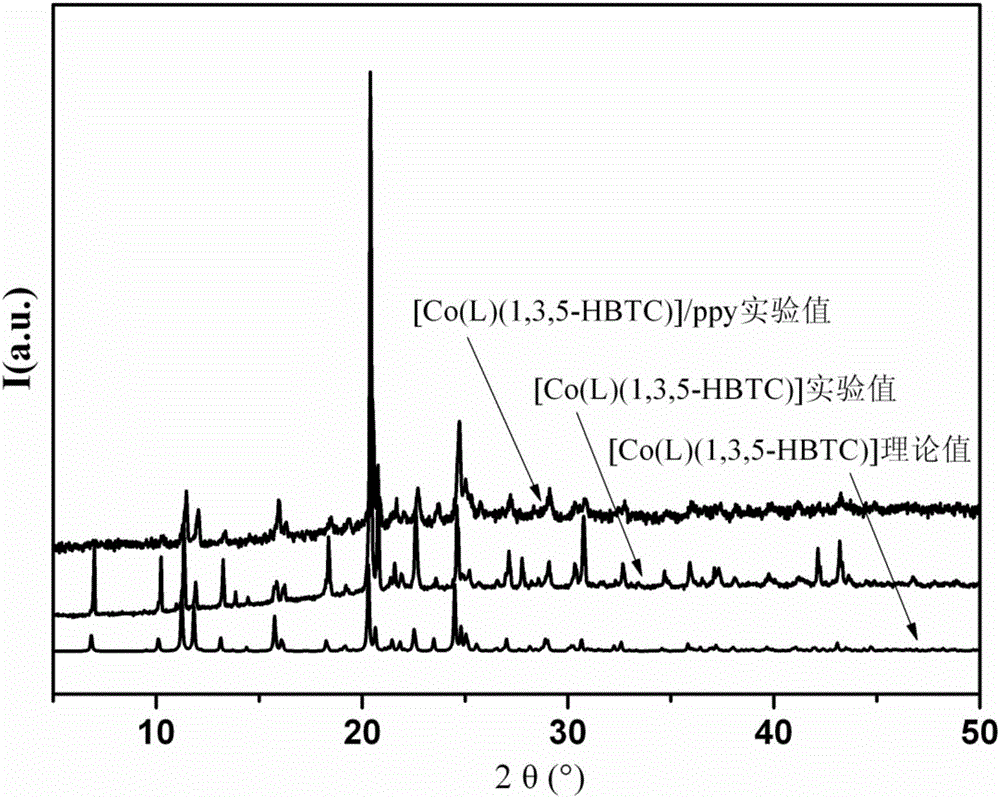
图1是本发明的[Co(L)(1,3,5-HBTC)]和[Co(L)(1,3,5-HBTC)]/ppy的XRD衍射图;
图2是本发明的[Co(L)(1,3,5-HBTC)]的热分析图;
图3是本发明的[Co(L)(1,3,5-HBTC)]和[Co(L)(1,3,5-HBTC)]/ppy的红外光谱图;
图4是本发明的[Co(L)(1,3,5-HBTC)]的SEM表面形貌图;
图5是本发明的[Co(L)(1,3,5-HBTC)]的固体漫反射图;
图6是本发明的[Co(L)(1,3,5-HBTC)]的tauc图;
图7是本发明的[Co(L)(1,3,5-HBTC)]/ppy的SEM表面形貌图;
图8是本发明的[Co(L)(1,3,5-HBTC)]/ppy的固体漫反射图;
图9是本发明的[Co(L)(1,3,5-HBTC)]/ppy的tauc图;
图10是本发明的[Co(L)(1,3,5-HBTC)]的配位环境图;
图11是本发明的[Co(L)(1,3,5-HBTC)]的二维结构正面图;
图12是本发明的[Co(L)(1,3,5-HBTC)]的二维结构侧面图;
图13是可见光照射下的亚甲蓝水溶液的180min内光催化降解紫外吸收图;
图14是可见光照射下加入本发明的[Co(L)(1,3,5-HBTC)]的亚甲蓝水溶液的150min内光催化降解紫外吸收图;
图15是可见光照射下加入本发明的[Co(L)(1,3,5-HBTC)]/ppy的亚甲蓝水溶液的75min内光催化降解紫外吸收图。
具体实施方式
实施例1~实施例3基于有机配体功能钴配合物的合成
实施例1合成[Co(L)(1,3,5-HBTC)],其中,L为N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺,结构式为:1,3,5-HBTC为均苯三甲酸一氢根将0.1mmol CoCl2·6H2O、0.10mmol N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺、0.1mmol均苯三甲酸和5mL H2O依次加入到25mL烧杯中,在室温下搅拌20min,得到悬浮混合物,用1mol/L的NaOH溶液调解悬浮混合物的pH至4.3后,转移到25mL的高压反应釜中,以5℃/h的加热速率升温至110℃,水热条件下保温48h,以5℃/h的降温速率将温度降至室温,得到粉色块状晶体,用去离子水和乙醇交替清洗3次,室温下自然晾干,得[Co(L)(1,3,5-HBTC)],产率为45%,其XRD衍射图谱如图1所示,其配位环境图如图10所示,其二维结构图如图11和图12所示。
实施例2合成[Co(L)(1,3,5-HBTC)],其中,L为N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺,1,3,5-HBTC为均苯三甲酸一氢根将0.2mmol CoCl2·6H2O、0.10mmol N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺、0.15mmol均苯三甲酸和10mL H2O依次加入到25mL烧杯中,在室温下搅拌30min,得到悬浮混合物,用1mol/L的NaOH溶液调解悬浮混合物的pH至5.3后,转移到25mL的高压反应釜中,以5℃/h的加热速率升温至120℃,水热条件下保温72h,以5℃/h的降温速率将温度降至室温,得到粉色块状晶体,用去离子水和乙醇交替清洗4次,室温下自然晾干,得[Co(L)(1,3,5-HBTC)],产率为65%,其XRD衍射图谱如图1所示,其配位环境图如图10所示,其二维结构图如图11和图12所示。
实施例3合成[Co(L)(1,3,5-HBTC)],其中,L为N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺,1,3,5-HBTC为均苯三甲酸一氢根将0.3mmol CoCl2·6H2O、0.10mmol N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺、0.2mmol均苯三甲酸和15mL H2O依次加入到25mL烧杯中,在室温下搅拌40min,得到悬浮混合物,用1mol/L的NaOH溶液调解悬浮混合物的pH至6.4后,转移到25mL的高压反应釜中,以5℃/h的加热速率升温至130℃,水热条件下保温96h,以5℃/h的降温速率将温度降至室温,得到粉色块状晶体,用去离子水和乙醇交替清洗5次,室温下自然晾干,得[Co(L)(1,3,5-HBTC)],产率为55%,其XRD衍射图谱如图1所示,其配位环境图如图10所示,其二维结构图如图11和图12所示。
本发明实施例1~实施例3的基于有机配体的多功能钴配合物的表征:
粉末衍射表征[Co(L)(1,3,5-HBTC)]相纯度:
在Rigaku Ultima IV粉末X射线衍射仪上收集完成粉末衍射数据,操作电流为40mA,电压为40kV。采用铜靶X射线。固定扫描,接收狭缝宽为0.1mm。密度数据收集使用2θ/θ扫描模式,扫描范围5°到50°,扫描速度为5°/s,跨度为0.02°/次。数据拟合使用Cerius2程序,单晶结构粉末衍射谱模拟转化使用Mercury 1.4.1。如图1所示,基于有机配体的多功能钴配合物的粉末X射线衍射谱图与拟合的XRD谱图基本吻合,表明配合物均为纯相。
热重表征[Co(L)(1,3,5-HBTC)]稳定性:
热稳定性采用PE-Pyris Diamond S-II热分析仪完成,加热速率10℃/min,温度范围20℃~800℃。图2表明本发明合成的配合物的分解温度范围为380℃~480℃。
[Co(L)(1,3,5-HBTC)]红外表征:
如图3所示[Co(L)(1,3,5-HBTC)]中存在来自N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺和均苯三甲酸一氢根的羧基、酰胺基、吡啶基等明显的特征吸收峰。
微米级[Co(L)(1,3,5-HBTC)]形貌:
如图4所示,由扫描电镜可观察到微米级钴配合物形貌为不规则形貌。
[Co(L)(1,3,5-HBTC)]的带隙宽度测试:
如图5和图6所示,经过测试后计算出带隙能量为2.94ev。说明[Co(L)(1,3,5-HBTC)]为半导体材料。
晶体结构测定:
用显微镜选取合适大小的单晶,室温下采用Bruker SMART APEX II衍射仪(石墨单色器,Mo-Ka)收集衍射数据。扫描方式衍射数据使用SADABS程序进行吸收校正。数据还原和结构解析分别使用SAINT和SHELXTL程序完成。最小二乘法确定全部非氢原子坐标,并用理论加氢法得到氢原子位置。采用最小二乘法对晶体结构进行精修。图10~图12展示出实施例1~实施例3中合成的基于有机配体的多功能钴配合物的基本配位情况和扩展结构。其晶体学衍射点数据收集与结构精修的部分参数如表1所示:
表1
实施例4~实施例6聚吡咯包裹的多功能钴配合物复合材料的制备
实施例4合成[Co(L)(1,3,5-HBTC)]/ppy,其中ppy为聚吡咯
a.对实施例1合成的[Co(L)(1,3,5-HBTC)]称取3克,采用玛瑙研钵研磨2小时,然后分散到2毫升的乙醇中,用球磨机研磨20分钟,离心分离,保留滤饼,在60℃下干燥24小时,得到微晶态配合物[Co(L)(1,3,5-HBTC)]材料;
b.将0.03克吡咯、15毫升水、1.3克微晶态配合物超声混合20分钟,得到悬浊液,将浓度为0.11克过硫酸钠和10毫升水的混合溶液以每秒钟一滴的速度加上到上述悬浊液中,5分钟内完成滴加,然后搅拌20分钟后静止12小时,离心分离,得到黑色聚吡咯包裹的配合物复合材料[Co(L)(1,3,5-HBTC)]/ppy,水和乙醇交替清洗3次,在65℃下真空干燥24h,产率95%。
实施例5合成[Co(L)(1,3,5-HBTC)]/ppy,其中ppy为聚吡咯
a.对实施例2合成的[Co(L)(1,3,5-HBTC)]称取3克,采用玛瑙研钵研磨3小时,然后分散到3毫升的乙醇中,用球磨机研磨30分钟,离心分离,保留滤饼,在70℃下干燥24小时,得到微晶态配合物[Co(L)(1,3,5-HBTC)]材料,。
b.将0.036克吡咯、20毫升水、1.7克微晶态配合物超声混合30分钟,得到悬浊液,将浓度为0.12克过硫酸钠和10毫升的混合溶液以每秒钟一滴的速度加上到上述悬浊液中,5分钟内完成滴加,然后搅拌20分钟后静止12小时,离心分离得到黑色聚吡咯包裹的配合物复合材料[Co(L)(1,3,5-HBTC)]/ppy,水和乙醇交替清洗3次,在65℃下真空干燥24h,产率98%。
实施例6合成[Co(L)(1,3,5-HBTC)]/ppy,其中ppy为聚吡咯
a.对实施例3合成的[Co(L)(1,3,5-HBTC)]称取3克,采用玛瑙研钵研磨4小时,然后分散到4毫升的乙醇中,用球磨机研磨40分钟,离心分离,保留滤饼,在80℃下干燥24小时,得到微晶态配合物[Co(L)(1,3,5-HBTC)]材料。
b.将0.05克吡咯、30毫升水、2.6克微晶态配合物超声混合40分钟,得到悬浊液,将浓度为0.13克过硫酸钠和10毫升水的混合溶液,以每秒钟1滴的速度加上到上述悬浊液中,5分钟内完成滴加,然后搅拌20分钟后静止12小时,得到黑色聚吡咯包裹的配合物复合材料[Co(L)(1,3,5-HBTC)]/ppy,水和乙醇交替清洗3次,在65℃下真空干燥24h,产率92%。
对实施例4~实施例6合成的[Co(L)(1,3,5-HBTC)]/ppy复合材料的表征:
如图1所示,经过聚吡咯包裹后,配合物的衍射峰基本没有变化,表明配合物在被包裹的过程中没有发生结构变化。
[Co(L)(1,3,5-HBTC)]/ppy红外表征:
如图3所示[Co(L)(1,3,5-HBTC)]/ppy中存在来自N,N'-双(3-吡啶)噻吩-2,5-二甲酰胺和均苯三甲酸一氢根的羧基、酰胺基、吡啶基、吡咯基等明显的特征吸收峰。表明聚吡咯与钴配合物的成功复合。
[Co(L)(1,3,5-HBTC)]/ppy的形貌表征:
如图7所示,经过扫描电镜测试,实施例4~实施例6合成的[Co(L)(1,3,5-HBTC)]/ppy为微米级的复合材料。
[Co(L)(1,3,5-HBTC)]/ppy的带隙宽度测试:
如图8和图9所示,经过测试后计算出带隙能量为2.48ev。说明[Co(L)(1,3,5-HBTC)]/ppy为半导体材料。
对实施例的1~实施例3合成的[Co(L)(1,3,5-HBTC)]以及实施例的4~实施例6合成的[Co(L)(1,3,5-HBTC)]/ppy的光催化降解亚甲蓝性能测试实验的具体步骤如下:
将50mg的[Co(L)(1,3,5-HBTC)]/ppy复合材料加入150mL浓度为10.0mg·L–1的亚甲蓝水溶液中作为实验组,将50mg的配合物[Co(L)(1,3,5-HBTC)]加入150mL浓度为10.0mg·L–1的亚甲蓝水溶液中作为对照组,并取同体积的染料分子水溶液作为对照组,150mL浓度为10.0mg·L–1的亚甲蓝水溶液中作为空白组。
搅拌实验组、对照组和空白组的染料分子水溶液60min得到悬浮物,边搅拌边采用350W氙灯作为可见光光源进行照射(波长400nm以下光采用滤光片过滤)。每隔30min取出5mL亚甲蓝悬浮物进行离心分离得到澄清上层溶液进行UV测试,下层悬浮物倒回催化反应体系中。
如图13和图14所示,在经过180min可见光光照射后,在无催化材料的空白组和采用[Co(L)(1,3,5-HBTC)]配合物催化材料的对照组的情况下,上述对照组和空白的亚甲蓝染料分子溶液的紫外吸收峰几乎没有变化,这表明甲蓝染料分子几乎没有降解,证明在没有催化剂情况下,可见光无法降解亚甲蓝染料分子,也证明钴配合物[Co(L)(1,3,5-HBTC)]无法在可见光条件下催化降解亚甲蓝染料分子。
如图15所示,在存在[Co(L)(1,3,5-HBTC)]/ppy复合材料情况下,在15分钟下对亚甲蓝有机污染物的可见光催化降解率可达54.4%,仅用时75分钟,上述实验组亚甲蓝染料分子的特征吸收强度下降了85.7%,表明该染料分子已经发生分解。证明利用聚吡咯包裹的配合物复合材料[Co(L)(1,3,5-HBTC)]/ppy可作为水溶性染料分子的可见光快速催化降解催化剂。
以上仅为本发明的具体实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!