稀土改性的Ni3P催化剂及其应用的制作方法
本发明属于多相催化
技术领域:
,涉及一类稀土元素改性的ni3p催化剂在生物质油加氢脱氧反应中的应用。
背景技术:
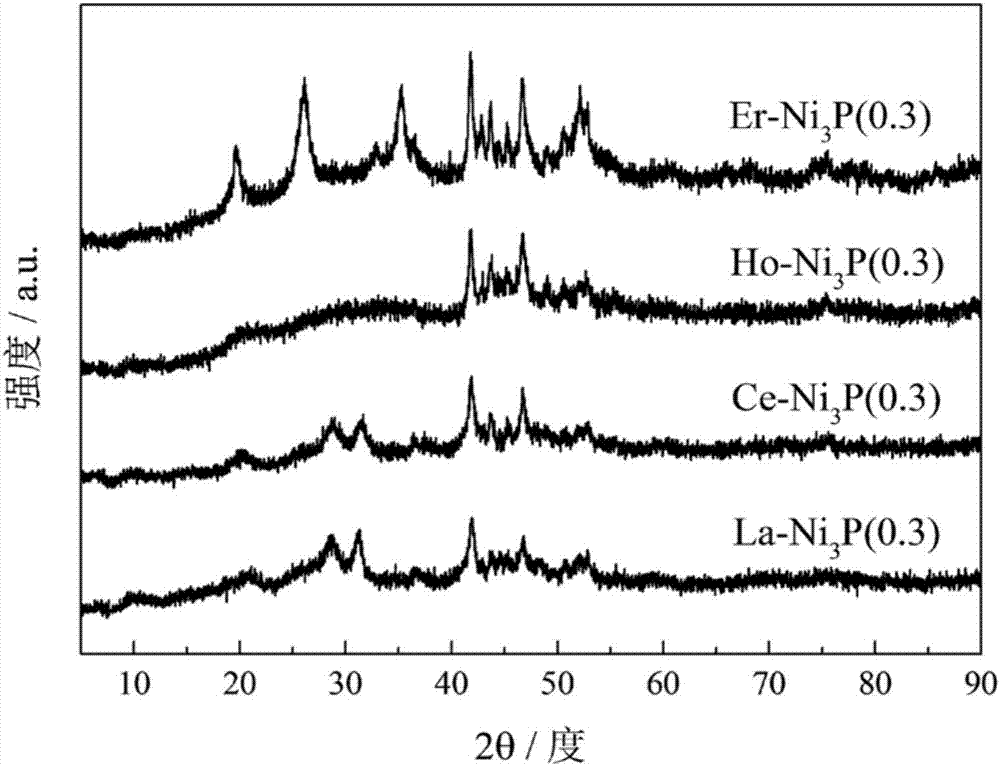
:近年来,能源消耗与环境污染日益加剧,以生物质油为代表的可再生能源的开发利用受到关注。生物质油具有可再生性、二氧化碳零排放、硫和氮含量较低、资源分布广、转化方式多等优点,但其中氧和水的含量较高。生物质油中大量的含氧化合物会导致一些严重的问题,如油品热值低、化学稳定性差、粘度大、腐蚀设备等,严重阻碍其作为汽柴油替代燃料的广泛使用,必须对其进行脱氧精制。在加氢脱氧(hdo)催化剂中,研究较多的是炼油工业用的ni(co)mos/γ-al2o3加氢脱硫(hds)催化剂。然而,在hdo反应过程中,硫化物催化剂会因硫原子被氧原子同晶取代或者被水氧化成相应的硫酸盐而导致失活,故而需要开发耐水的、高活性及稳定性的加氢脱氧催化剂。以ni2p、mop、wp、co2p以及cop为代表的一系列过渡金属磷化物以其高活性和稳定性在加氢精制(包括加氢脱氧)领域有广阔的应用前景。在这些过渡金属磷化物中,ni2p为常用的催化剂,但其稳定性较差(energ.fuel.,2011,25:854-863),有待提高。在前期的研究中发现,ni3p具有较高的催化苯酚水相及油相加氢脱氧活性及稳定性。但对于苯酚的衍生物(即取代苯酚),在低温条件下,加氢脱氧活性较低,亟待提高。稀土金属磷酸盐(如cepo4)作为一种功能材料在催化领域逐渐引起重视(appl.catal.a:gen.,2015,504:4-10;chem.commun.,2011,47:5307-5309)。在催化反应过程中,ce作为助剂可以通过ce3+和ce4+的相互转化形成氧空穴或者促进电子转移,从而提高主催化剂的反应活性。但目前多数采用稀土氧化物(如ceo2)作为助剂(top.catal.,2012,55:1010-1021.),未有稀土磷酸盐作为助催化剂在ni3p催化生物油中苯酚及其衍生物加氢脱氧中应用的报道。技术实现要素:本发明的目的首先在于提供一种改性的ni3p催化剂及其制备方法。所述的稀土改性的ni3p催化剂通过下述方法制备:(1)将硝酸镍和稀土元素硝酸盐共溶于去离子水中,不断搅拌下逐滴加入磷酸氢二铵溶液,蒸发掉水分后干燥,高温焙烧制得催化剂前体;(2)催化剂前体经氢气程序升温还原活化。显然,本发明提供的上述催化剂的制备方法包括如下步骤:(1)将硝酸镍和稀土元素硝酸盐共溶于去离子水中,不断搅拌下逐滴加入磷酸氢二铵溶液,蒸发掉水分后干燥,高温焙烧制得催化剂前体;(2)催化剂前体经氢气程序升温还原活化。在本发明上述研究中,我们发现稀土金属磷酸盐可以大幅度提高ni3p的低温加氢脱氧活性。采用该催化剂,在固定床反应器中以较温和的反应条件进行连续加氢脱氧反应,可以高效实现生物质油中苯酚及其衍生物的加氢脱除。因此,本发明另一方面的目的在于提供上述本发明的稀土金属改性ni3p催化剂在苯酚及其衍生物加氢脱氧反应中的应用。附图说明本发明附图2幅:图1是采用h2程序升温还原法制备ni3p和ce-ni3p(x)催化剂的xrd谱图;图2是采用h2程序升温还原法制备la-ni3p(0.3)、ce-ni3p(0.3)、ho-ni3p(0.3)和er-ni3p(0.3)催化剂的xrd谱图。具体实施方式本发明提供一种新的稀土改性的ni3p催化剂,及其制备方法和在苯酚及其衍生物加氢脱氧反应中的应用。本发明所述的稀土改性的ni3p催化剂,通过下述方法制备:(1)将硝酸镍和稀土元素硝酸盐共溶于去离子水中,不断搅拌下逐滴加入磷酸氢二铵溶液,蒸发掉水分后干燥,高温焙烧制得催化剂前体;(2)催化剂前体经氢气程序升温还原活化。其中所述的步骤(1)中,稀土元素与镍摩尔比为x,镍与磷的摩尔比为3/(1+3x)。其中所述的稀土元素与镍摩尔比x为0.01-1。优选0.1~0.5;最优选0.2-0.4。进一步具体的实施方式中,所述的稀土元素是镧、铈、钬或铒。优选金属铈。其中所述的步骤(2)中,氢气流量150ml/min,升温程序为从室温以2℃/min升至400℃后,再以1℃/min升至500℃并保持120min,随后降温。显而易见,上述通过制备方法定义稀土改性的ni3p催化剂的描述中,也详细地表述了该催化剂的制备过程及方法。在本发明上述研究中,我们发现稀土金属磷酸盐可以大幅度提高ni3p的低温加氢脱氧活性:从溶剂来看,在水相条件下,促进效果尤为明显;从反应底物来看,改性后的催化剂对于邻甲酚、苯甲醚和愈创木酚等空间位阻较大的反应底物促进效果更为明显。采用该催化剂,在固定床反应器中以较温和的反应条件进行连续加氢脱氧反应,可以高效实现生物质油中苯酚及其衍生物的加氢脱除。基于此,本发明进一步提供所述稀土改性的ni3p催化剂在苯酚及其衍生物加氢脱氧反应中的应用。具体实施方式中,所述的苯酚及其衍生物包括苯酚及取代苯酚,取代苯酚由下述基团任意取代:甲基、羟基或甲氧基。所述的取代苯酚优选一元取代化合物。所述的苯酚及其衍生物可具体举例但不限于苯酚、苯甲醚、邻甲酚、间甲酚和愈创木酚。其中,又以邻甲酚、苯甲醚和愈创木酚为优选。在所述的加氢脱氧反应的应用中,所述的加氢脱氧反应温度为100-400℃,反应压力为1.0-5.0mpa。所述反应为水相反应或油相反应。水相反应的反应介质为去离子水,反应底物的质量分数为1-10%,产物用二氯甲烷萃取后以无水硫酸镁干燥;油相反应的反应介质为十氢萘,反应底物的质量分数为0.1-5%。优选水相反应。以下结合具体实施例对本发明的内容作进一步说明。实施例1采用h2程序升温还原法制备ce改性的ni3p催化剂。按化学计量比称取定量的ni(no3)2·6h2o和ce(no3)3·6h2o共溶于10毫升去离子水中制成溶液a,再称取定量的(nh4)2hpo4溶于5毫升去离子水中制成溶液b,在不断搅拌下将b逐滴加入a中形成绿色悬浊液。蒸干后于120℃烘干12h,再置于马弗炉中以4℃/min的速率升温至500℃,焙烧3h,制得焙烧前体c。将c置于管式反应器中,于常压,氢气流量150ml/min的条件下,以2℃/min的程序升温至400℃再以1℃/min的程序升温至500℃,于500℃还原2h制得催化剂,分别记为ce-ni3p(x)(其中x代表ce/ni摩尔比)。通过将这些催化剂的xrd谱图与ni3p和cepo4的标准谱图对比(图1)可以看出,采用程序升温还原法成功制备ce改性的ni3p催化剂,且催化剂的晶粒尺寸随着ce含量的增加而减小。实施例2采用h2程序升温还原法制备la、ho和er改性的ni3p催化剂。制备方法与实施例1类似,区别仅在于将ce(no3)3·6h2o分别替换成la(no3)3·6h2o、ho(no3)3·5h2o和er(no3)3·5h2o,制得催化剂分别记为la-ni3p(0.3)、ho-ni3p(0.3)和er-ni3p(0.3)(其中0.3代表稀土元素与ni摩尔比)。通过将这些催化剂的xrd谱图与ni3p和稀土磷酸盐的标准谱图对比(图2)可以看出,采用程序升温还原法成功制备稀土元素改性的ni3p催化剂。实施例3苯酚在ni3p和ce-ni3p(x)上不同温度下水相加氢脱氧反应。反应在内径10毫米的高压固定床管式反应器中进行。按实施例1在反应器中制得相应的催化剂后,将反应器温度调至反应温度(300℃),总压增至4mpa,用高压恒流泵打入反应原料(质量浓度为5%的苯酚水溶液),重时空速30h-1,氢/油体积比1000,脱氧率以环己烷收率计算。反应稳定后取液体样品,用二氯甲烷萃取有机相产物,再用无水mgso4脱除残留的微量水,在aglient6890n型气相色谱分析,色谱柱为市售inno-wax毛细管柱,氢火焰检测器,反应结果列于表1(苯酚在ni3p和ce-ni3p(x)上300℃下水相加氢脱氧反应性能)。由表1可以看出,加入ce后催化剂苯酚的转化率以及脱氧率均大幅度提高,按如下顺序上升:ni3p<ce-ni3p(0.05)<ce-ni3p(0.1)<ce-ni3p(0.2)<ce-ni3p(0.4)<ce-ni3p(0.3),故而优选ce/ni为0.3的催化剂。表1:苯酚在ni3p和ce-ni3p(x)上300℃下水相加氢脱氧反应性能催化剂转化率(%)脱氧率(%)ni3p85.624.2ce-ni3p(0.05)92.725.3ce-ni3p(0.1)93.528.3ce-ni3p(0.2)94.343.3ce-ni3p(0.3)99.068.4ce-ni3p(0.4)93.851.8实施例4苯酚在la-ni3p(0.3)、ho-ni3p(0.3)和er-ni3p(0.3)上300℃下水相加氢脱氧反应。反应条件与实施例3类似,不同之处是催化剂为实施例2中的la-ni3p(0.3)、ho-ni3p(0.3)和er-ni3p(0.3),反应结果列于表2(苯酚在la-ni3p(0.3)、ho-ni3p(0.3)和er-ni3p(0.3)上300℃下水相加氢脱氧反应性能)。由表2可以看出,稀土元素改性后的ni3p催化剂催化苯酚水相加氢脱氧性能提高,促进幅度按如下顺序上升:ho-ni3p(0.3)<la-ni3p(0.3)<ce-ni3p(0.3)≈er-ni3p(0.3),考虑到ce的储量相对丰富,优选ce改性催化剂。表2:苯酚在la-ni3p(0.3)、ho-ni3p(0.3)和er-ni3p(0.3)上300℃下水相加氢脱氧反应性能催化剂转化率(%)脱氧率(%)ni3p85.624.2la-ni3p(0.3)99.664.2ce-ni3p(0.3)99.068.4ho-ni3p(0.3)99.554.5er-ni3p(0.3)99.369.2实施例5邻甲酚在ni3p和ce-ni3p(0.3)上300℃下水相加氢脱氧反应。反应条件与实施例3类似,区别之处是原料为质量浓度为5%的邻甲酚水溶液,反应结果列于表3(邻甲酚在ni3p和ce-ni3p(0.3)上300℃下水相加氢脱氧反应性能)。可以看出,加入ce后邻甲酚的转化率以及脱氧率均大幅度提高,说明ce的引入促进ni3p催化邻甲酚水相加氢脱氧性能。表3:邻甲酚在ni3p和ce-ni3p(0.3)上300℃下水相加氢脱氧反应性能催化剂转化率(%)脱氧率(%)ni3p72.722.6ce-ni3p(0.3)93.830.4实施例6间甲酚在ni3p和ce-ni3p(0.3)上300℃下水相加氢脱氧反应。反应条件与实施例3类似,区别之处是原料为质量浓度为5%的间甲酚水溶液,反应结果列于表4(间甲酚在ni3p和ce-ni3p(0.3)上300℃下水相加氢脱氧反应性能)。可以看出,加入ce后间甲酚的转化率以及脱氧率均大幅度提高,说明ce的引入促进ni3p催化邻间甲酚水相加氢脱氧性能。表4:间甲酚在ni3p和ce-ni3p(0.3)上300℃下水相加氢脱氧反应性能催化剂转化率(%)脱氧率(%)ni3p78.249.4ce-ni3p(0.3)96.366.0实施例7愈创木酚在ni3p和ce-ni3p(0.3)上300℃下水相加氢脱氧反应。反应条件与实施例3类似,区别之处是原料为质量浓度为5%的愈创木酚水溶液,反应结果列于表5(愈创木酚在ni3p和ce-ni3p(0.3)上300℃下水相加氢脱氧反应性能)。可以看出,加入ce后愈创木酚的转化率以及脱氧率均大幅度提高,说明ce的引入促进ni3p催化愈创木酚水相加氢脱氧性能。表5:愈创木酚在ni3p和ce-ni3p(0.3)上300℃下水相加氢脱氧反应性能催化剂转化率(%)脱氧率(%)ni3p45.628.5ce-ni3p(0.3)77.830.8实施例8苯酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应。反应在内径10毫米的高压固定床管式反应器中进行。按实施例1在反应器中制得相应的催化剂后,将反应器温度调至反应温度(200℃),总压增至4mpa,用高压恒流泵打入反应原料(质量浓度为1%的苯酚十氢萘溶液),重时空速26.7h-1,氢/油体积比1000,脱氧率以相应的烃收率计算。反应稳定后取液体样品,在aglient6890n型气相色谱分析,色谱柱为市售inno-wax毛细管柱,氢火焰检测器,反应结果列于表6(苯酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应性能)。可以看出,加入ce后,苯酚的转化率以及脱氧率略有增高,说明引入ce增加ni3p催化苯酚油相加氢脱氧活性。通过对比表1和表6,ce改性对于催化苯酚水相加氢脱氧效果更为明显。表6:苯酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应性能催化剂转化率(%)脱氧率(%)ni3p98.440.1ce-ni3p(0.3)99.445.0实施例9苯甲醚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应。反应条件与实施例8类似,区别之处是原料为质量浓度为1%的苯甲醚十氢萘溶液,反应结果列于表7(苯甲醚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应性能)。可以看出,加入ce后苯甲醚的转化率以及脱氧率均大幅度提高,说明ce的引入促进ni3p催化苯甲醚油相加氢脱氧性能。表7:苯甲醚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应性能催化剂转化率(%)脱氧率(%)ni3p52.223.2ce-ni3p(0.3)99.735.6实施例10邻甲酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应。反应条件与实施例8类似,区别之处是原料为质量浓度为1%的邻甲酚十氢萘溶液,反应结果列于表8(邻甲酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应性能)。同样可以看出,相比于ni3p催化剂,在ce-ni3p(0.3)上,邻甲酚的转化率以及脱氧率均提高,说明ce的改性促进ni3p催化邻甲酚加氢脱氧性能。表8:邻甲酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应性能催化剂转化率(%)脱氧率(%)ni3p68.99.3ce-ni3p(0.3)97.010.7实施例11间甲酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应。反应条件与实施例8类似,区别之处是原料为质量浓度为1%的间甲酚十氢萘溶液,反应结果列于表9(间甲酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应性能)。同样可以看出,相比于ni3p催化剂,在ce-ni3p(0.3)上,间甲酚的转化率以及脱氧率均提高,说明ce的改性促进ni3p催化间甲酚加氢脱氧性能。表9:间甲酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应性能催化剂转化率(%)脱氧率(%)ni3p82.754.5ce-ni3p(0.3)99.255.1实施例12愈创木酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应。反应条件与实施例8类似,区别之处是原料为质量浓度为1%的愈创木酚十氢萘溶液,反应结果列于表10(愈创木酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应性能)。同样可以看出,相比于ni3p催化剂,在ce-ni3p(0.3)上,愈创木酚的转化率以及脱氧率均提高,说明ce的改性促进ni3p催化愈创木酚加氢脱氧性能。通过对比表6-表10,在ni3p催化剂上,苯酚及取代物反应活性依次下降:苯酚>间甲酚>邻甲酚>苯甲醚>愈创木酚;经过ce改性后苯酚及其取代物的反应活性均大幅增加,尤其是对于ni3p催化活性较低的邻甲酚、苯甲醚和愈创木酚。表10:愈创木酚在ni3p和ce-ni3p(0.3)上200℃下油相加氢脱氧反应性能催化剂转化率(%)脱氧率(%)ni3p14.50.3ce-ni3p(0.3)96.20.5本发明经过上述的描述,已清楚地公开了本发明催化剂组成和制备条件。但是,本领域内的技术人员十分清楚,对本发明可以进行一些修改和改进。所以,只要不离开本发明的精神,对本发明所进行的任何修改和改进都应在本发明的范围内。本发明的范围在附属的权利要求书中提出。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1