与αvβ3整联蛋白甲状腺拮抗剂缀合的不可裂解聚合物的制作方法
本申请要求2016年6月7日提交的题为“novelcompositionsandmothodsofusofnon-cleavablepolymerconjugatedwithnovelalpha-v-beta-3thyroidantagonists的美国申请号62/346,659的优先权和权益,其内容通过引用并入本文。
本公开大体上涉及甲状腺激素受体拮抗剂(称为“甲状腺整联蛋白(thyrointegrin)拮抗剂”)并且更具体的涉及α-v-β-3(αvβ3)整联蛋白-甲状腺激素受体拮抗剂,其通过具有或不具有短链peg的接头的不可裂解的键与一个或多个聚合物缀合(在实施方案中使用除peg之外的聚合物)。
背景技术:
整联蛋白是细胞表面粘附受体的超家族,其控制具有固体细胞外环境的细胞与细胞外基质(ecm)和其他细胞的附着。粘附对细胞至关重要;它提供了锚定、迁移的线索以及生长和分化的信号。整联蛋白直接参与许多正常和病理状况,因此是治疗性干预的主要靶标。整联蛋白是完整的跨膜蛋白,是异二聚体,其结合特异性取决于14个α-链中的哪个与8个β-链中的哪个组合。整联蛋白分为四个重叠的亚家族,包含β1、β2、β3或αv链。细胞可以表达来自每个亚家族的几种不同的整联蛋白。在过去的几十年中,已经显示整联蛋白是参与细胞粘附的主要受体,因此可能是治疗性干预的合适靶标。整联蛋白αvβ3调节细胞生长和存活,因为在某些情况下,该受体的连接可以诱导肿瘤细胞的细胞凋亡。已显示用抗αvβ3抗体、rgd肽和其他整联蛋白拮抗剂破坏细胞粘附可减缓肿瘤生长。
技术实现要素:
本公开的第一实施方案通常涉及一种组合物,其包含通式:
其中n5=1-5,并且a=ch或n,至少有一个a=n
本公开的第二实施方案通常涉及一种组合物,其包含通式:
本公开的第三实施方案通常涉及一种组合物,其包含甲状腺拮抗剂、不可生物降解的聚合物;和通过不可裂解的共价键与所述甲状腺拮抗剂和所述不可生物降解的聚合物共价键合的接头。其中,即使在某些情况下,一种或多种所列聚合物可能通过使用苛刻的环境条件而裂解,但是残留的聚合物链仍然可以共价键合到接头并且mat、dat或tat仍然能够限制缀合的甲状腺拮抗剂的细胞核摄取。
附图说明
一些实施方案将参考以下附图详细描述,其中相同的标记表示相同的构成部分,其中:
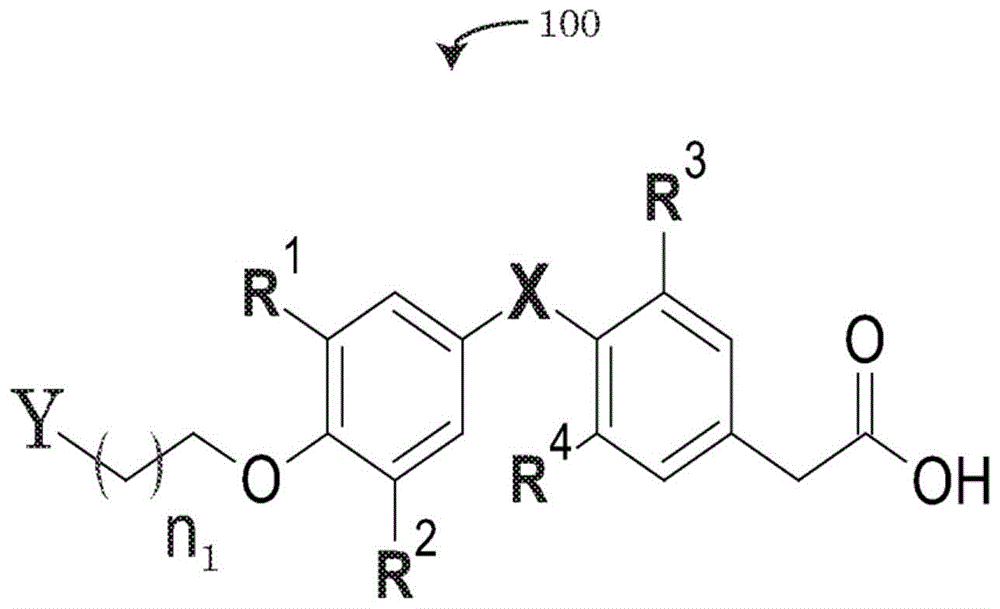
图1描绘了描述甲状腺拮抗剂及其衍生物的一般化学式的实施方案。
图2a描绘了四碘甲腺乙酸(tetrac)衍生物,单氨基丙基四碘甲腺乙酸(mat)的实施方案。
图2b描绘了四碘甲腺乙酸衍生物,二氨基丙基四碘甲腺乙酸(dat)的实施方案。
图2c描绘了四碘甲腺乙酸衍生物,炔丙基四碘甲腺乙酸(pgt)的实施方案。
图2d描绘了图2a的四碘甲腺乙酸衍生物的替代实施方案。
图2e描绘了图2b的四碘甲腺乙酸衍生物的替代实施方案。
图2f描绘了图2c的四碘甲腺乙酸衍生物的替代实施方案。
图3a描绘了三碘甲腺乙酸(triac)衍生物,单氨基丙基三碘甲腺乙酸(matri)的实施方案。
图3b描绘了三碘甲腺乙酸衍生物,二氨基丙基三碘甲腺乙酸(datri)的实施方案。
图3c描绘了三碘甲腺乙酸衍生物,炔丙基三碘甲腺乙酸(pgtri)的实施方案。
图4a描绘了甲状腺整联蛋白拮抗剂衍生物的实施方案。
图4b描绘了甲状腺整联蛋白受体拮抗剂衍生物的替代实施方案。
图4c描绘了甲状腺整联蛋白拮抗剂衍生物的另一个替代实施方案。
图5描绘了描述通过不可裂解键与聚合物缀合的甲状腺激素拮抗剂衍生物的一般化学式的实施方案。
图6描绘了通过不可裂解的键与聚乙二醇(peg)聚合物缀合的甲状腺激素拮抗剂衍生物的通式的实施方案。
图7a描绘了甲状腺激素拮抗剂通过不可裂解的单氨基键与peg聚合物缀合形成peg-单氨基丙基四碘甲腺乙酸(p-mat)的实施方案。
图7b描绘了甲状腺激素拮抗剂通过不可裂解的二氨基键与peg聚合物缀合形成peg-二氨基丙基四碘甲腺乙酸(p-dat)的实施方案。
图7c描绘了甲状腺激素拮抗剂通过不可裂解的n-c键与peg聚合物缀合形成peg-三唑四碘甲腺乙酸(p-tat)的实施方案。
图8描绘了使用甲氧基-聚乙二醇(mpeg)通过可裂解和不可裂解的接头合成αvβ3甲状腺激素拮抗剂的缀合途径的实施方案。
图9描绘了双官能甲状腺整联蛋白拮抗剂衍生物的通式的实施方案,该衍生物包含通过不可裂解键与peg聚合物的两个缀合。
图10a描绘了双官能甲状腺整联蛋白拮抗剂衍生物,聚乙二醇双-单氨基丙基四碘甲腺乙酸(p-双-mat)的实施方案。
图10b描绘了双官能甲状腺整联蛋白拮抗剂衍生物,聚乙二醇双-二氨基丙基四碘甲腺乙酸(p-双-dat)的实施方案。
图10c描绘了双官能甲状腺整联蛋白拮抗剂衍生物,聚乙二醇双-三唑四碘甲腺乙酸(p-双-tat)的实施方案。
图11描绘了用于合成p-双-mat、p-双-dat和p-双-tat的方法的实施方案。
图12描绘了使用四聚乙二醇以合成下列甲状腺整联蛋白拮抗剂的四官能衍生物的方法的实施方案:聚乙二醇四-单氨基丙基四碘甲腺乙酸(p-四-mat(p-四-mat)、聚乙二醇四-二氨基丙基四碘甲腺乙酸(p-四-dat)和聚乙二醇四三唑四碘甲腺乙酸(p-四-tat)。
图13a描绘了与环糊精缀合的甲状腺整联蛋白拮抗剂衍生物的通式的实施方案。
图13b描绘了环糊精单氨基丙基四碘甲腺乙酸(c-mat)的化学结构的实施方案。
图13c描绘了环糊精二氨基丙基四碘甲腺乙酸(c-dat)的化学结构的实施方案。
图13d描绘了环糊精三唑四碘甲腺乙酸(c-tat)的化学结构的实施方案。
图14a描绘了使用α、β或γ环糊精合成c-mat和c-dat的方法的实施方案。
图14b描绘了使用α、β或γ环糊精合成c-tat的方法的实施方案。
图15a描绘了甲状腺整联蛋白拮抗剂衍生物与海藻酸聚合物缀合形成海藻酸单氨基丙基四碘甲腺乙酸(a-mat)的实施方案。
图15b描绘了甲状腺整联蛋白拮抗剂衍生物与海藻酸聚合物缀合形成海藻酸二氨基丙基四碘甲腺乙酸(a-dat)的实施方案。
图15c描绘了甲状腺整联蛋白拮抗剂衍生物与海藻酸聚合物缀合形成海藻酸三唑四碘甲腺乙酸(a-tat)的实施方案。
图16a描绘了用于从海藻酸合成a-mat和a-dat的方法的实施方案。
图16b描绘了用于从海藻酸合成a-tat的方法的实施方案。
图17a描绘了甲状腺整联蛋白拮抗剂衍生物用不可裂解的单氨基键与透明质酸聚合物缀合形成透明质酸-单氨基丙基四碘甲腺乙酸(h-mat)的实施方案。
图17b描绘了甲状腺整联蛋白拮抗剂衍生物用不可裂解的二氨基键与透明质酸聚合物缀合形成透明质酸-二氨基丙基四碘甲腺乙酸(h-dat)的实施方案。
图17c描绘了甲状腺整联蛋白拮抗剂衍生物用不可裂解的三唑键与透明质酸聚合物缀合形成透明质酸-三唑丙基四碘甲腺乙酸(h-tat)的实施方案。
图18a描绘了用于从透明质酸合成h-mat和h-dat的方法的实施方案。
图18b描绘了用于从透明质酸合成h-tat的方法的实施方案。
图19描绘了用于合成具有包含炔丙基的接头的甲状腺整联蛋白拮抗剂的方法的实施方案。
图20描绘了用于合成炔丙基化四碘甲腺乙酸(pgt)的方法的实施方案。
图21描绘了p-双-tat对小鼠中基质胶生长因子植入物-介导的血管生成的影响,以及小鼠基质胶生长因子介导的血管生成中p-双-tat的剂量-依赖性抗血管生成功效。
图22描绘了p-双-tat(1mg/kg,scqd持续14天)对裸鼠中卵巢(ovcar3)-肿瘤血管生成的影响。
图23描绘了通过流式细胞术研究αvβ3蛋白的表达。
图24a-24d描绘了使用共聚焦显微镜观察gbm细胞中αvβ3蛋白的表达。
图25描绘了聚合物缀合的dat降低了在具有急性骨髓性白血病(aml)的转基因小鼠的骨髓和肝转移中的thp1-luc细胞的生物发光信号。
图26描绘了gbm肿瘤异种移植物的生物发光信号(接受治疗21天处死),对照和空白的平均生物发光信号强度为2-3×107个光子/秒并且治疗组(1mg/kg和3mg/kg)具有<1-2×104个光子/秒的生物发光信号强度。
图27描绘了gbm肿瘤异种移植物的生物发光信号(21天和脱离治疗22天处死),对照和空白的平均生物发光信号强度为2-3×107个光子/秒并且治疗组(1mg/kg和3mg/kg)具有<1-2×104个光子/秒的生物发光信号强度。
图28描绘了以每日1和3mg/kgdat或tat当量治疗21天后(接受治疗天数),以及另一组接受治疗21天随后脱离治疗22天后,聚合物缀合的dat或tat对裸鼠异种移植物中gbm肿瘤生长的影响。
图29描绘了以每日1和3mg/kgdat或tat当量治疗的21天后(接受治疗天数),以及另一组接受治疗21天随后脱离治疗22天后,聚合物缀合的c-tat和p-双-tat对裸鼠异种移植物中gbm肿瘤生长的影响。
图30描绘了以每日3和10mg/kg治疗(每日皮下(sc)(分别为1和3mg/kgtat当量))21天后(接受治疗天数),以及另一组接受治疗21天随后脱离治疗22天后,聚合物缀合的p-双-tat对裸鼠异种移植物中gbm肿瘤生长的影响。
图31描绘了由p-双-tat诱导的gbmu87mg小鼠异种移植物中肿瘤细胞活力的丧失。p-双-tat(3mg/kg)和p-双-tat(10mg/kg)分别是3mg和10mgp-双-tat/kg的皮下每日药物剂量,连续21天,细胞活力降低达到62%和72%(基于组织学分数)。误差标尺表示平均值的标准误差(s.e.m.)。
图32描绘了gbmu87mg-luc小鼠异种移植物的生物发光信号,在处死前接受治疗21天或接受治疗21天并脱离治疗22天。对照中的平均生物发光信号强度为2-3×107个光子/秒。在治疗组中,信号强度<1-2×104个光子/秒(检测限)。在这些ivis图像中,垂直发光彩色标尺(左边距)估计活力,范围从无活力(0p/秒/cm2/sr)到完全活力(6p/秒/cm2/sr)。
图33描绘了在施用p-双-tat和其他聚合物缀合的tat的组合物后30分钟和4小时体内原位脑(gbm)肿瘤中的cy5信号(上图)。下图显示了原位脑gbm肿瘤和皮下异种移植gbm肿瘤中cy5p-双-tat和其他聚合物缀合的tat的图像强度。
图34描绘了gbm脑肿瘤中p-双-tat的cy5信号强度的动力学的实施方案的图。
图35描绘了皮下异种移植gbm肿瘤中p-双-tat的cy5信号强度的动力学的实施方案的图。
图36描绘了描述在研究结束时(4小时)移除的脑和皮下gbm肿瘤中的cy5标记的p-双-tat信号强度的图的实施方案。
图37描绘了在1、3和10mg/kg,sc每日,持续7天的条件下,对照与p-双-tat治疗的动物的脑中gbm(u87-luc)原位肿瘤的生物发光信号。
图38描绘了描述p-双-tat和ntat(聚合物缀合的tat,1mg/kg,sc每日,持续7天)对脑中gbm(u87-luc)的生物发光信号的影响的图。
图39描绘了描述在一周治疗后p-双-tat对gbm皮下肿瘤生长的影响的结果的图。
图40描绘了描述使用ivis成像检测p-双-tat对gbm肿瘤活力的剂量效应的图。
图41a描绘了与对照相比,3mg/kg和10mg/kg的p-双-tat对胰腺癌suit2的抗癌功效的图,其中抗癌功效作为肿瘤重量的函数。
图41b描绘了与对照相比,3mg/kg和10mg/kg的p-双-tat对胰腺癌suit2的抗癌功效的图,其中抗癌功效作为细胞活力的函数。
图41c描绘了与对照相比,3mg/kg和10mg/kg的p-双-tat对胰腺癌suit2的抗癌功效的图,其中抗癌功效作为%细胞坏死的函数。
图42a描绘了描述单独的辐射/非辐射暴露(pbs),和与一定剂量的p-双-tat(3mg/kg和10mg/kg)组合的辐射或非/辐射暴露,对胰腺癌suit2的影响的图,该影响被描述为异种移植肿瘤重量的函数。
图42b描绘了描述单独的辐射/非辐射暴露(pbs),和与一定剂量的p-双-tat(3mg/kg和10mg/kg)组合的辐射或非/辐射暴露,对胰腺癌suit2的影响的图,该影响被描述为%细胞活力的函数。
图42c描绘了描述单独的辐射/非辐射暴露(pbs),和与一定剂量的p-双-tat(3mg/kg和10mg/kg)组合的辐射或非/辐射暴露,对胰腺癌suit2的影响的图,该影响被描述为%细胞坏死的函数。
图43描绘了描述单独的辐射暴露和与一定剂量p-双-tat组合的辐射暴露在1gy和5gy的辐射时对胰腺癌suit2的影响的图,该结果描绘为异种移植肿瘤重量的函数。
图44描绘了描述用p-双-tat治疗的雄性和雌性小鼠的血浆中肝脏、心脏和肾脏标记物水平的图。持续14天以所示浓度(ng/ml)的p-双-tat每日皮下治疗小鼠,测量该小鼠血浆中的肝损伤(天冬氨酸转氨酶(ast)和丙氨酸转氨酶(alt))、心肌损伤(心肌肌钙蛋白i(ctni))和肾功能(肌酐)的标记物。误差标尺表示平均值的标准误差(s.e.m.)。
具体实施方式
本文通过示例而非限制的方式参考附图给出了下文描述的所公开的装置和方法的实施方案的详细描述。尽管详细示出和描述了某些实施方案,但是应该理解的是,在不脱离所附权利要求的范围的情况下,可以进行各种改变和修改。本公开的范围绝不限于构成组件的数量、其材料、其形状、其颜色、其相对布置等,并且仅作为本公开的实施方案的例子公开。通过参考以下描述结合附图,可以获得对本实施方案及其优点的更完整的理解,其中相似的附图标记表示相似的特征。
作为详细描述的前言,应当注意,如在本说明书和所附权利要求中使用的,单数形式“一”、“一个”和“该”(“a”、“an”和“the”)包括复数指示物,除非上下文另有明确规定。
概述
本公开的实施方案描述了新的化学组合物及其合成方法。本文公开和描述的组合物可以针对并归类为抗血管生成剂,其可能能够与整联蛋白αvβ3受体家族的一种或多种细胞表面受体反应。本文所述的组合物可包括抗血管生成甲状腺激素或其衍生物,其通过不可裂解的接头与聚合物缀合,形成可被认为是微分子或大分子(取决于与甲状腺激素或其衍生物共价结合的聚合物的大小)的单一化学实体。单一化学实体的大小和不可裂解的共价键的强度可有利于防止甲状腺激素或其衍生物进入包含各种αvβ3整联蛋白的细胞表面受体的细胞。由于所连接的聚合物的大小,以及细胞周围环境不能从聚合物裂解甲状腺激素的强的、不可裂解的共价键,所述化学实体的甲状腺激素部分可能无法在甲状腺激素或其衍生物可能相互作用的细胞的核内内化。因此,所述化学实体的甲状腺激素部分可以以非基因组的方式(non-genomically)与细胞相互作用并避免可能由甲状腺激素或其衍生物进入细胞并与细胞核的核受体相互作用而引起的基因组相互作用。
本文公开的组合物的实施方案可以合成为包括但不限于实体,所述实体包含不可生物降解的聚合物例如聚乙二醇(peg)(1000-15000道尔顿,例如4000-8000道尔顿)、α、β或γ环糊精、壳聚糖、海藻酸或透明质酸,其通过不可裂解接头缀合到αvβ3甲状腺拮抗剂,所述不可裂解接头包含胺或三唑键,没有peg短链(100-800m.w.)。与聚合物缀合的甲状腺拮抗剂的实施方案可包括四碘甲腺乙酸(tetrac)、三碘甲腺乙酸(triac)、其衍生物及其变体。在一些实施方案中,包含四碘甲腺乙酸和三碘甲腺乙酸的甲状腺激素拮抗剂的一种或多种变体的实例可包括二氨基四碘甲腺乙酸(dat)或二氨基三碘甲腺乙酸(datri)(以下可以互换地称为“dat”)、单氨基四碘甲腺乙酸(mat)或单氨基三碘甲腺乙酸(matri)(以下可以互换地称为“mat”)、三唑四碘甲腺乙酸(tat)或三唑三碘甲腺乙酸(tatri)(以下可以互换地称为“tat”)、其衍生物或本领域技术人员已知的其他甲状腺拮抗剂。
已经进一步合成了本文所述组合物的实施方案,并将其表征为与不同分子量的聚乙二醇(1000至15000道尔顿)缀合的dat、mat或tat。我们已经扩大了相对最可溶的peg-dat(p-单-dat、p-双-dat)和peg-tat(p-单-tat、p-双-tat)的实施方案,用于各种体外和体内生物系统中的生物学表征。dat或tat和peg-dat或peg-tat以及c-dat和c-tat的化学标记用于成像和细胞动力学。数据显示,与dat或tat的强烈的细胞核摄取相比,聚合物与dat或tat的缀合导致限制了那些与聚合物缀合的dat或tat的细胞核摄取。这种独特的细胞分布的结果导致,与非缀合的dat、mat或tat相比,与聚合物缀合的dat、mat或tat缺乏基因组作用。描述了其他多聚物,例如与具有或不具有短链短链peg(100-1000道尔顿)的dat、mat或tat缀合的透明质酸、海藻酸、壳聚糖。使用双官能或四官能peg合成的与dat、mat或tat缀合的另外的聚合物可以包括,但也可以包括高达8个链的其它支链化的peg。
本申请中描述的每种化合物的实施方案可以具有多种类型的用途,用于治疗由血管生成或其抑制所调节的多种不同疾病。鉴于存在于所述组合物中的甲状腺拮抗剂的存在,本公开中描述的每种组合物可各自具有靶向位于遍及人体和各种动物体的多种类型细胞上的整联蛋白受体αvβ3的亲和力。
例如,本文公开的组合物的效用可用于治疗人或哺乳动物中的血管生成介导的疾病,例如癌症(实体瘤和液体肿瘤)。癌症可包括胶质母细胞瘤、胰腺癌、卵巢癌、乳腺癌、前列腺癌、膀胱癌、肺癌和肝癌。液体肿瘤还可以是急性骨髓性白血病、多发性骨髓瘤、淋巴瘤和慢性淋巴细胞白血病。本文所述的组合物可进一步治疗眼部病症(糖尿病性视网膜病和年龄相关性黄斑变性)、炎性病症(关节炎、骨关节炎)、动脉粥样硬化病变和皮肤病(红斑痤疮、牛皮癣、皮肤癌)其可以各自介导或依赖于通过血管生成产生新的血细胞以持续存在并且其治疗可以依赖于拮抗新血管的形成以减缓或消除血管生成途径。22
虽然本文描述的本公开的实施方案和实施例,为了说明的目的,基于所示的实施例,修改和改变对于本领域技术人员而言将变得显而易见。因此,所附实施例旨在包括落入本公开的真实精神和范围内的所有变化和此类修改和改变。
甲状腺整联蛋白拮抗剂组合物
参照附图,图1描述了通式100的实施方案,所述通式100描述了连接至接头和“y”的甲状腺激素拮抗剂,所述接头包含碳原子的重复连接,其可以由n1个碳亚基定义,所述“y”可以定义与通式100的甲状腺激素拮抗剂及其衍生物的接头相连的不可裂解的共价键。术语“甲状腺激素拮抗剂”可以描述通式100的分子抑制或拮抗本领域技术人员已知的一种或多种甲状腺激素受体的能力,例如甲状腺激素受体的整联蛋白家族,如甲状腺激素细胞表面受体αvβ3。由于甲状腺激素拮抗剂及其衍生物抑制整联蛋白受体的功能性,通式100定义的分子在本文中可进一步描述为甲状腺整联蛋白拮抗剂。
如图1的通式100的化学结构所示,该化学结构的实施方案可包括一个或多个定义图1的甲状腺整联蛋白拮抗剂的额外特征的变量。例如,在甲状腺整联蛋白拮抗剂的一些实施方案中,描述为r1、r2、r3和r4的变量可各自独立地由氢、碘、直链烷烃、支链烷烃和环状烷烃的分子取代。在一些实施方案中,变量“x”可以被定义为氧原子(o)或硫原子(s)。
与通式100的甲状腺拮抗剂连接的碳接头的实施方案可以在碳链的长度上是可变的。碳链的长度可以与氧分子和不可裂解的共价键“y”之间的一个碳原子一样小。在甲状腺整联蛋白拮抗剂的替代实施方案中,接头可以包含碳原子的重复连接,其可以由n1个重复定义。在一些实施方案中,n1可以≥0,而在通式100的替代实施方案中,n1的接头中碳原子的重复数可以≥0、≥1、≥2或≥3。
由变量“y”描绘的不可裂解的共价键的实施方案在某些情况下可以是胺键。例如,如图2a-2b、2d-2e、3a-3b、4a-4b的甲状腺整联蛋白拮抗剂210、215、220、225、310、320、410、420的实施例所示,在不可裂解的共价键中,通式的变量y可以是具有一个胺基的单胺或具有两个胺基的二胺。在替代实施方案中,如图2c、2f、3c和4c所示,被取代的变量y可以包括炔丙基。
如图2a-4c的实施方案所示,存在多种可由通式100形成的衍生物组合物。例如,在图2a中,组合物210可以包括碘取代的r1-r4,导致形成具有三碳接头和作为不可裂解共价键的单胺的四碘甲腺乙酸(tetrac)衍生物。组合物210可称为单胺-四碘甲腺乙酸(mat)。同样地,在图2b中,四碘甲腺乙酸分子还包含与接头连接的二氨基共价键。同样地,该组合物220可以称为二氨基四碘甲腺乙酸(dat)。在图2c的替代实施方案中,组合物230可包含如图所示与四碘甲腺乙酸分子和炔丙基之间的一碳连接相连的炔丙基。该衍生物组合物230可称为炔丙基四碘甲腺乙酸(pgt)。
由四碘甲腺乙酸合成炔丙基化的四碘甲腺乙酸(pgt)
以下实施例提供了根据图19中描述的通用化学式从四碘甲腺乙酸制备炔丙基四碘甲腺乙酸或其衍生物的样本方法,更具体地,如图20的合成图中所示应用于四碘甲腺乙酸。
步骤1:酯化
表1a提供了将四碘甲腺乙酸酯化成o-甲基四碘甲腺乙酸(mr-2)(甲基-2-(4-(4-羟基-3,5-二碘苯氧基)-3,5-二碘苯基)乙酸酯)的合成条件:
在用于合成mr-2的一个实施方案中,根据先前公开的保护方法合成组合物。将四碘甲腺乙酸(1g,1.33mmol,1当量)和三氟化硼二乙醚(bf3·et2o)(0.1ml)的甲醇(10ml)溶液在氮气下在环境温度下搅拌24小时。通过加入15ml饱和nahco3水溶液淬灭反应,将混合物搅拌10分钟,并将水相用乙酸乙酯(3×20ml)萃取。将合并的有机相用硫酸钠干燥,过滤,并在真空下浓缩,得到950mg粗品甲基-2-(4-(4-羟基-3,5-二碘苯氧基)-3,5-二碘苯基)乙酸酯2,该粗品然后用乙醇重结晶,得到纯化合物(630mg,0.82mmol),产率62%。重结晶溶剂:etoh;rf:0.62,tlc溶剂(正己烷:etoac/8:2);熔点=162-4℃;ftir(υcm-1):3371,3082,2943,1719cm-1(c=o),1586,1556,1539,1455,1430,1397,1283,1240,1220,1160,1238,912,847,823,782,700,597,527。1hnmr(cdcl3)δ(ppm):7.78(s,2h,arh),7.12(s,2h,arh),5.53(br,1h,oh),3.75(s,3h,-cooch3),3.58(s,2h,-ch2-coo);13cnmr(cdcl3)δ(ppm):171.0(-coome),152.8,150.2,149.6,141.3,135.2,126.1,90.9,81.8,52.7(-cooch3),39.8(-ch2-coo)。ms(esi+)m/z:785[m+na]+;(esi-)m/z:761[m-h]-。
步骤2:烷基化
表1b:mr-3(甲基{4-[3,5-二碘-4-(丙-2-炔-1-基氧基)苯氧基]-3,5-二碘苯基}乙酸酯)的合成条件
在一个实施方案中,可以进行烷基化步骤。将甲基保护的四碘甲腺乙酸(1当量)和炔丙基溴(3当量)和碳酸钾(5当量)在25ml丙酮中的混合物加热回流24小时。将反应物过滤、浓缩,然后使用正己烷和乙酸乙酯(9:1至7:3)在硅胶上通过快速柱层析进行粗品纯化,得到甲基{4-[3,5-二碘-4-(丙-2-炔-1-基氧基)苯氧基]-3,5-二碘苯基}乙酸酯,产率75-85%。1hnmr(cdcl3)δ(ppm):7.76(s,2h,arh),7.16(s,2h,arh),4.6(br,1h,oh),3.75(s,3h,-cooch3),3.56(s),2h,-ch2-coo);2.54(s,2h,-o-ch2-c-ch)。
步骤3:水解
表1c:mr-4{4-[3,5-二碘-4-(丙-2-炔-1-基氧基)苯氧基]-3,5-二碘苯基}乙酸的合成条件
化合物pgt230可以通过使用koh对化合物mr-3进行去保护来获得。简言之,将100mg化合物mr-3溶于6mlthf/甲醇(1:1)中。将混合物搅拌15分钟,然后加入6mlkoh2m并将反应物在室温下搅拌18小时。将有机溶剂完全蒸发并用1mhcl中和。沉淀物通过真空过滤收集,用水洗涤数次,并过夜干燥,得到83%的pgt230白色粉末。二氯甲烷/甲醇(9:1)用作tlc的溶剂。1hnmr(cdcl3)δ(ppm):7.85(s,2h,arh),7.16(s,2h,arh),4.6(br,1h),3.56(s,2h,-ch2-coo);2.96(s,2h,-o-ch2-c-ch)。
参考附图,图2d-2f的实施方案展示了图2a-2c的四碘甲腺乙酸衍生物的化学式。然而,衍生物组合物215、225和235各自包含硫取代的变量x,而不是包含氧的变量x。类似于图2d-2f中描绘的组合物215、225、235,衍生物310、320和330描述了通式100的另一种变体。在图3a-3c的实施方案中,描绘的甲状腺整联蛋白拮抗剂用氢取代r4,而r1-r3用碘取代。因此,由于三个碘和一个氢的取代,图3a-3c中所示的甲状腺拮抗剂可以是三碘甲腺乙酸(triac)而不是四碘甲腺乙酸。组合物310、320和330可以概括地标识为衍生物单氨基三碘甲腺乙酸310、二氨基三碘甲腺乙酸320和炔丙基三碘甲腺乙酸330。在甲状腺整联蛋白拮抗剂的实施方案中,甲状腺拮抗剂的衍生物可以不包括碘,这与图2a-3c中提供的实施例相反。图4a-4c的实施方案展示了甲状腺整联蛋白拮抗剂410、420、430,其中r1-r2各自被氢取代,r3-r4各自被异丙基取代。
下面提供的表2描述了可以制成通式100的变量的多个不同取代:
在本公开的甲状腺整联蛋白拮抗剂的一些实施方案中,如图5的实施方案500所公开的通式所示,通式100的甲状腺整联蛋白拮抗剂可以通过变量y的不可裂解的共价键与不可生物降解的聚合物(变量z)缀合。可以取代变量z的合适聚合物可以包括但不限于聚乙二醇(peg)、α-环糊精、β-环糊精、γ-环糊精、壳聚糖、海藻酸和透明质酸、其聚合物的组合或本领域技术人员已知或使用的任意其他不可裂解的聚合物。甲状腺整联蛋白拮抗剂的衍生物600描绘了实施方案500的一般实施方案中的变量z取代为peg聚合物的实施例,其中peg聚合物具有一个或多个由变量n2定义的重复单体亚单元。例如,变量n2可以是≥1的任意数量的重复单体。peg的大小可以根据peg链中单体的重复数量的数量而变化。例如,在一些实施方案中,在一些实施方案中peg的大小可以是1000-15000道尔顿,而在替代实施方案中,peg可以是1000-4000道尔顿或4000-6000道尔顿或更多。
在本申请的示例性实施方案中,与聚合物缀合的甲状腺激素可以是如上表1中所示的四碘甲腺乙酸、三碘甲腺乙酸或其衍生物。图7a-7c描绘了通过不可裂解的共价键与聚合物peg缀合的四碘甲腺乙酸的示例性实施方案的化学式。例如,在图7a中,附图描绘了甲状腺整联蛋白拮抗剂710,其包含通过接头与peg聚合物通过不可裂解的单胺键(n-c键)共价结合的四碘甲腺乙酸分子。所得组合物可描述为peg-单氨基丙基四碘甲腺乙酸(p-mat)。在图7b的替代实施方案720中,示例性组合物是通过接头与peg聚合物通过不可裂解的二胺键结合的四碘甲腺乙酸分子,得到可描述为peg-二氨基丙基四碘甲腺乙酸(p-dat)的组合物。在第三示例性实施方案730中,四碘甲腺乙酸可以通过不可裂解的三唑键与peg聚合物共价结合。所得组合物730可描述为peg-三唑四碘甲腺乙酸(p-tat)。
图8描绘了流程图800,其说明了通过与peg(1000-15000道尔顿)键合的不可裂解的接头将peg聚合物与各种活性αvβ3整联蛋白甲状腺拮抗剂缀合,导致形成p-mat、p-dat和p-tat的一个或多个步骤。在所提供的示例性方法中,本公开描绘的每种组合物,使用单甲氧基peg作为聚合物、接头和甲状腺拮抗剂的多种变体四碘甲腺乙酸、三碘甲腺乙酸和衍生物,在缀合步骤期间提供了如dat、mat和tat。
图8描绘了以单甲氧基peg(m-peg-oh)起始产生p-mat和p-dat的步骤。在步骤801下,可以将m-peg-oh甲苯磺酰化,形成单-甲苯磺酰化的peg807(m-peg-ots)和m-peg醛806。在步骤802中,可以在步骤802中将单-甲苯磺酰化peg引入mat-210或其衍生物,以通过点击化学形成p-mat710。类似地,不再将单-甲苯磺酰化的peg807引入mat310,而是在步骤803中,可以将单-甲苯磺酰化的peg807引入dat220,如所示通过点击化学形成p-dat720。
为了从单甲氧基-peg起始形成p-tat,可以通过步骤804将mpeg-oh转化为m-peg-叠氮化物,通过将m-peg-oh甲苯磺酰化为单-甲苯磺酰化的peg807并通过将单-甲苯磺酰化的peg807与nan3结合再将单-甲苯磺酰化的peg807转化为m-peg-叠氮化物808。m-peg-叠氮化物808可以在步骤805中与炔丙基四碘甲腺乙酸230结合,产生三唑键,得到p-tat730。
应该注意的是,对于附图中提供的和本申请全文中描述的每个实施例,均使用市售化学品制备,所述市售化学品在使用时未经进一步纯化。所有溶剂被干燥并使用活化分子筛(0.3或0.4nm,取决于溶剂的类型)以获得无水溶剂。所有反应(如果不特别含有水作为反应物、溶剂或共溶剂)在ar或n2气体环境下,在烘箱干燥过的玻璃器皿中进行。所有新化合物均得到令人满意的1hnmr和质谱结果。熔点在electrothermalmel-
在一些实施方案中,贯穿本公开内容描述的聚合物缀合的甲状腺整联蛋白拮抗剂可以是双官能或四官能组合物。术语“双官能”可以指聚合物缀合的甲状腺拮抗剂,其具有两个通过不可裂解的共价键与通式500的相同聚合物(z)缀合的甲状腺拮抗剂或其衍生物。双官能组合物之一能够在图9中看到,其中甲状腺整联蛋白拮抗剂900包含在实施方案900的两侧通过碳原子接头和由变量y表示的不可裂解的键与甲状腺激素拮抗剂缀合的peg聚合物。图10a-10c提供了图9的实施方案900中描绘的变量的一个或多个特定取代的实施例。
例如,在图10a中,具有一个或多个由变量n2表示的重复亚单元的peg聚合物可以使用两个不同的氨基键和与甲状腺激素拮抗剂(包括四碘甲腺乙酸或四碘甲腺乙酸衍生物)连接的碳接头缀合。如图10a所示,甲状腺整联蛋白受体拮抗剂可包含两个mat分子,其中每个mat分子可通过不可裂解的氨基连接与peg聚合物的最外侧peg分子缀合。因此,组合物1010可称为聚乙二醇-双-单胺丙基四碘甲腺乙酸(p-双-mat)1010。
类似于图10a的组合物1010,图10b的组合物可以用不可裂解的二氨基键替换双官能p-双-mat中的每个mat形成缀合,在peg聚合物和甲状腺激素拮抗剂之间形成连接。因此,包含二氨基键的组合物1020可以概括地描述为聚乙二醇-双-二氨基丙基四碘甲腺乙酸(p-双-dat)1020。在一个或多个替代实施方案中,通过不可裂解键与peg聚合物缀合的甲状腺整联蛋白拮抗剂可以是多个pgt230分子,而不是使用mat210或dat220。如图7c所示,pgt分子及其衍生物可在聚合物(peg)和pgt之间形成不可裂解的三唑键,导致形成p-tat730。类似于p-双-mat1010和p-双-dat1020,p-tat可以形成图10c中所示的其双官能变体,其包含将每个甲状腺激素拮抗剂或其衍生物与peg聚合物缀合的三唑键。该双-官能分子可称为聚乙二醇-双-三唑-四碘甲腺乙酸(p-双-tat)。
图11描述了使用peg(在实施方案1100中描述为oh-peg-oh)合成双-官能甲状腺整联蛋白拮抗剂p-双-mat1010、p-双-dat和p-双-tat的一种或多种方法。在方法1100的第一步中,peg可以在步骤1101中被甲苯磺酰化以形成双-甲苯磺酰化的peg。在下面描述的一个实施例中,可以如以下实施例中所述进行用于甲苯磺酰化peg的步骤:
将ho-peg-oh(1.5g,0.25mmol,当量=1)溶于50mldcm中并搅拌15分钟。将4-甲苯磺酰氯(0.38g,2.02mmol,当量=8)和1mltea加入混合物中。将反应在室温下搅拌过夜。将反应在dcm中稀释,并用hcl1n,(2x),盐水1x洗涤。将混合物浓缩并完全除去dcm,并将所得物通过乙酸乙酯重结晶过夜。过滤后收集白色粉末,得到1.4g残余物。
方法1100的第二步可以根据期望实现的所需最终产品而变化。如果所需最终甲状腺整联蛋白拮抗剂是p-双-mat1010,则双-甲苯磺酰化的peg可以通过步骤1102引入到已存在的mat210,导致双-甲苯磺酰化的peg中的甲苯磺酰基被替换为通过氨基键与peg聚合物的每一侧共价键合的mat210,生成p-双-mat。类似地,双-甲苯磺酰基-peg的甲苯磺酰基可以在步骤1103中被dat220基团替换。当在dat存在下引入双-甲苯磺酰基peg时,dat可以通过一个或多个共价二氨基键与peg结合,导致p-双-dat1020的形成。
然而,如果期望的目标是获得p-双-tat1030,则可以执行一个或多个附加步骤。首先,在步骤1104中,可以从步骤1101的双-甲苯磺酰化-peg形成双-叠氮基-peg。用于合成双-叠氮基-peg的步骤的实施方案的一个实施例可描述如下:
双-叠氮基-peg的合成(mw=4000)
将sto-peg-ots(2500mg,~0.5mmol,当量=1)溶解在20mldmf中,然后将3000mgnan3加入到溶液中,并将反应设定为80℃过夜。向溶液中加入200ml水,用dcm萃取三次。合并有机相并用盐水洗涤并用mgso4干燥。除去溶剂并在-20℃下用乙酸乙酯重结晶并过滤,得到1750mg的双-叠氮基修饰的peg。
在步骤1104中获得双-叠氮基修饰的peg后,双-叠氮基-peg可以在步骤1105中与存在的tat730反应,以形成如图11中所示的p-双-tat1030。以下实施例进一步详细说明了从双-叠氮基-peg到p-双-tat1030的反应的合成;p-双-tat(mw=4000)的合成
将双-叠氮基-peg(n3-peg-n3)(2000mg,0.5mmol,当量=1)溶解在8mldmf中,然后将1570mgtat(2mmol,当量=4)加入到反应中。将286mgcubr(2mmol,当量=4)和814μln,n,n',n”,n”-五甲基二亚乙基三胺(pmdeta)(4mmol,当量=8)溶于2mldmf中并加入溶液中并将反应设置在60-65℃过夜。将反应冷却至室温并将反应物在100mldcm中稀释。通过被羧酸负活化的氧化铝柱除去铜,并用dcm洗涤数次,然后浓缩混合物至100ml。用200ml水(3x)和盐水(2x)洗涤混合物。5)有机相经mgso4干燥,除去dcm,最后通过加入200ml乙酸乙酯重结晶最终产物。重复两次重结晶,过滤得到淡黄色粉末。
甲状腺类似物的酚羟基(-oh)是其修饰的重要位点,并且是将四碘甲腺乙酸转化为整联蛋白拮抗剂而不改变四碘甲腺乙酸的羧酸部分的靶位点。用炔丙基溴合成修饰分子结构四碘甲腺乙酸(tetrac)以制备炔烃修饰的四碘甲腺乙酸,炔丙基四碘甲腺乙酸(pgt),{4-[3,5-二碘-4-(丙-2-炔-1-基氧基)苯氧基]-3,5-二碘苯基}乙酸;分子量,785道尔顿)。pgt通过点击化学与o,o'-双(叠氮化物)聚乙二醇(分子量,4050道尔顿)缀合,得到分子量为5620道尔顿的p-双-tat,如图11和下述p-双-tat合成方案所示:
p-双-tat合成:
化学名称:o,o'-双({4-[3,5-二碘-4-(1-亚甲基-1,2,3-三唑-4-基甲氧基)苯氧基]-3,5-二碘苯基}乙酸)聚乙二醇。
物理外观:黄棕色粉末溶解性:在水中50mg/ml;在ph8.0磷酸盐缓冲液中,100mg/ml;在10%乙醇中,150mg/ml,以及在50%丙二醇中,200-300mg/ml。
在聚合物缀合的甲状腺整联蛋白拮抗剂的一些实施方案中,组合物不仅可以是如通式600所示的单官能的,或者如具有通式900的组合物所示的双官能的,而且可以进一步是如图12的实施例所证明的四官能的。例如,类似于如图11中和如上所示的制备p-双-mat1010、p-双-dat1020和p-双-tat的方法,可以合成p-四-mat1210、p-四-dat和p-四-tat的四官能衍生物。图12中所示的制备四官能甲状腺整联蛋白拮抗剂的合成步骤可以类似于图8和11的实施例方法中所示的制备单-缀合和双-缀合衍生物的步骤。
例如,可以使用四-peg而不使用m-peg或peg作为起始材料。在步骤1201中,四-peg可以被甲苯磺酰化成p-四-ots,随后在p-mat710或p-dat720的存在下反应,分别形成p-四-mat1210和p-四-dat1220。同样,类似于p-tat730和p-双-tat1030的合成,可以通过首先将甲苯磺酰化的p-四-ots转化为p-四-叠氮基来合成p-四-tat1230,如步骤1204所示。随后,在步骤1205中,p-四-叠氮基组合物可以在p-tat730的存在下反应以产生p-四-tat1230。
在甲状腺整联蛋白拮抗剂的替代实施方案中,通式500的聚合物z可以被α、β或γ环糊精取代,如图13a的通式1300所示。在式1300中,变量n3=1、2或3个重复的环糊精单体亚单元,其中重复亚单元的数目可以鉴定用作缀合的甲状腺整联蛋白拮抗剂中的聚合物的环糊精。例如,当n3=1时,环糊精是α-环糊精,n3=2,环糊精是β-环糊精,当或n3=3时,环糊精可以是γ-环糊精。与上述通式100和500描述的其他甲状腺整联蛋白拮抗剂衍生物类似,r1、r2、r3、r4、x、y和n1的取代可以在式1300中以类似的方式发生。图13b-13d提供了环糊精缀合的甲状腺整联蛋白拮抗剂的实施例。具体地,在图13b中,提供了环糊精-单氨基-丙基四碘甲腺乙酸(c-mat)1310,而在图13c中,例示了环糊精-二氨基-丙基四碘甲腺乙酸(c-dat)1320。此外,图13d描绘了环糊精-三唑四碘甲腺乙酸(c-tat)1330的化学结构的实施例。
合成c-mat1310、c-dat1320和c-tat1330的方法遵循与先前描述的p-mat710、p-dat720和p-tat730的替代缀合变体相似的合成步骤。如图14a和图14b所示,合成可以在步骤1401开始,通过将环糊精聚合物甲苯磺酰化为甲苯磺酰化-环糊精。为了产生c-mat1310或c-dat1320,可以分别在步骤1402或1403中在甲苯磺酰化-环糊精存在下使mat210或dat220反应。为了合成c-tat1330,由步骤1401得到的甲苯磺酰化-环糊精可以在步骤1404中进一步反应以产生环糊精-叠氮基组合物,其可以在步骤1405中在pgt230存在下进一步反应,导致c-tat1330的合成。
以下实施例更详细地描述了环糊精缀合的甲状腺整联蛋白拮抗剂的合成:
c-tat的合成-合成简介:甲状腺类似物的酚羟基(-oh)是其修饰的重要位点,并且是将四碘甲腺乙酸转化为整联蛋白拮抗剂而不改变四碘甲腺乙酸的羧酸部分的靶位点。用炔丙基溴合成修饰分子结构四碘甲腺乙酸(tetrac)以制备炔烃修饰的四碘甲腺乙酸,炔丙基四碘甲腺乙酸(pgt),{4-[3,5-二碘-4-(丙-2-炔-1-基氧基)苯氧基]-3,5-二碘苯基}乙酸;分子量,785道尔顿)。pgt通过点击化学与单-6-叠氮基-脱氧-6-β-环糊精缀合,得到β-c-tat。
tat的加载量为%42.41
βc-tat:分子量:1944.89道尔顿
化学名称:6-({4-[3,5-二碘-4-(1-亚甲基-1,2,3-三唑-4-基甲氧基)苯氧基]-3,5-二碘苯基}乙酸)-6-脱氧-β环糊精
γ-c-tat的合成-合成简介:
甲状腺类似物的酚羟基(-oh)是其修饰的重要位点,并且是将四碘甲腺乙酸转化为整联蛋白拮抗剂而不改变四碘甲腺乙酸的羧酸部分的靶位点。用炔丙基溴合成修饰分子结构四碘甲腺乙酸(tetrac)以制备炔烃修饰的四碘甲腺乙酸,炔丙基四碘甲腺乙酸(pgt),{4-[3,5-二碘-4-(丙-2-炔-1-基氧基)苯氧基]-3,5-二碘苯基}乙酸;分子量,785道尔顿)。pgt通过点击化学与单-6-叠氮基-脱氧-6-γ-环糊精缀合,得到分子量为2108道尔顿的γ-c-tat。
合成c-tat的详细示意图如下所示。
tat的加载量为%39.27
c-tat的结构
化学名称:6-({4-[3,5-二碘-4-(1-亚甲基-1,2,3-三唑-4-基甲氧基)苯氧基]-3,5-二碘苯基}乙酸)-6-脱氧-γ环糊精
分子量:2108道尔顿
在具有通式500的聚合物缀合的甲状腺整联蛋白拮抗剂的一些替代实施方案中,由变量z描述的聚合物可以用海藻酸聚合物取代,如图15a-15c中所描绘的实施例所示。例如,在图15a中,海藻酸聚合物可以与p-mat710缀合,导致形成海藻酸-单氨基-丙基四碘甲腺乙酸(a-mat)1510或其衍生物,其中变量n4定义了海藻酸的多个重复单体亚单元。变量n4可以是≥1的任意数量重复亚单元。在图15b中,可以使用利用dat720与海藻酸聚合物的替代缀合物或其衍生物。得到的海藻酸聚合物与dat720的缀合物可称为海藻酸-二氨基-丙基四碘甲腺乙酸(a-dat)1520,而tat730和海藻酸之间的缀合导致形成海藻酸-三唑四碘甲腺乙酸(a-tat)1530或其衍生物。
与先前上述peg和环糊精的聚合物缀合物的合成相比,a-mat1510、a-dat1520和a-tat1530的合成遵循一组略微改变的步骤。如图16a所示,合成方法从海藻酸聚合物开始。步骤1601不是将聚合物甲苯磺酰化,而是将海藻酸与n-羟基-琥珀酰亚胺反应,将n-羟基-琥珀酰亚胺(nhs)连接至海藻酸的羧基上。在步骤1602和1603中,nhs可以在取代反应中被除去,通过引入mat210(步骤1602)或dat220(步骤1603)以用mat210替代海藻酸的nhs,形成a-mat1510,或用dat220替代海藻酸的nhs,形成a-dat1520。
图16b描绘了由海藻酸起始材料制备a-tat1530的方法步骤。在步骤1601中,海藻酸与nhs反应形成nhs-海藻酸。在步骤1604中,nhs-海藻酸与叠氮化合物反应,用叠氮化合物替代连接在海藻酸的羧基上的nhs,得到海藻酸-叠氮化合物,如图16b所示。最后,由步骤1604形成的海藻酸-叠氮化合物可以在步骤1605中在pgt230存在下进一步反应以与叠氮基的n3形成三唑键,产生a-tat1530。
在具有通式500的聚合物缀合的甲状腺整联蛋白拮抗剂的一些替代实施方案中,由变量z描述的聚合物可以用透明质酸聚合物取代,如图17a-17c中所描绘的实施例所示。例如,在图17a中,透明质酸聚合物可以与p-mat710缀合,导致形成透明质酸-单氨基-丙基四碘甲腺乙酸(h-mat)1710或其衍生物,其中变量n4定义了透明质酸的多个重复单体亚单元。变量n4可以是≥1的任意数量重复亚单元。在图17b中,可以使用利用dat720与透明质酸聚合物的替代缀合物或其衍生物。得到的透明质酸聚合物与dat720的缀合物可称为透明质酸-二氨基-丙基四碘甲腺乙酸(a-dat)1720,而tat730和透明质酸之间的缀合导致形成透明质酸-三唑四碘甲腺乙酸(a-tat)1730或其衍生物。
h-mat1710、h-dat1720和h-tat1730的合成遵循与上面前述的图16a-16b的聚合物缀合的海藻酸方法类似的一组合成步骤。如图18a所示,合成方法始于透明质酸聚合物。步骤1801将透明质酸与nhs反应至透明质酸聚合物的羧基,而不是将聚合物甲苯磺酰化。在步骤1802和1803中,nhs可以在取代反应中被除去,通过引入mat210(步骤1802)或dat220(步骤1803)以用mat210替代海藻酸的nhs,形成h-mat1710,或用dat220替代海藻酸的nhs,形成h-dat1720。
图18b描绘了由透明质酸起始材料制备h-tat1730的方法步骤。在步骤1801中,透明质酸与nhs反应形成nhs-透明质酸。在步骤1804中,nhs-透明质酸与叠氮化合物反应,用叠氮化合物替代连接在透明质酸的羧基上的nhs,得到透明质酸-叠氮化合物,如图18b所示。最后,由步骤1804形成的透明质酸-叠氮化合物可以在步骤1805中在pgt230存在下进一步反应以与叠氮基的n3形成三唑键,产生h-tat1730。
聚合物-缀合的甲状腺整联蛋白拮抗剂使用方法
实施例1抗血管生成功效:
小鼠基质胶-生长因子植入血管生成模型:
根据动物安全和福利的机构指南进行小鼠基质胶模型。5-6周龄、体重20g的雌性小鼠c56/bl购自taconicfarms(hudson,ny,usa)。将动物维持在特定无病原体的条件下,并在温度(20-24℃)和湿度(60-70%)和12小时光照/黑暗循环的受控条件下每笼饲养4只动物。体内研究在退伍军人事务部(va)医疗中心的动物设施(albany,ny)中进行,并且实验方案由vaiacuc批准。在实验开始前使小鼠适应环境5天。准备含生长因子的基质胶matrixhighconcentration以促进血管生成并将该混合物以100μl/动物皮下注射四次。对照组中的动物仅注射100μl体积的基质胶。
将聚合物缀合的dat、tat或mat衍生物在三种不同剂量(10、30、100μg/10μl)下测试。所有组每组有3只小鼠,每组12次基质胶皮下注射。在植入植入物后第14天,处死所有动物并使用分光光度法定量血红蛋白含量。图21描绘了p-双-tat的代表性数据及其相对于生长因子的有效抗血管生成功效。如在图21中可见,在该研究期间测量的p-双-tat和其他甲状腺整联蛋白拮抗剂的抗血管生成作用证明了对血管生成的剂量依赖性反应,如通过测量的血红蛋白水平所证实的那样。
实施例2:血红蛋白(hb)水平的测定(血管生成指数的测量)
将基质胶植入物血红蛋白(hb)含量作为衡量新血管形成的指标。简言之,将基质胶植入物置于含有二重蒸馏水的0.5ml的试管中,然后均化5-10分钟。将样品以4000rpm离心10分钟,然后收集上清液。将50μl体积的上清液与50μldrabkin试剂混合,并使其在室温下静置15-30分钟,之后将该100μl置于96孔板中并用microplatemanagerelisa读取器在540nm处测量吸光度。基于与标准曲线的比较,hb浓度表示为mg/ml。
实施例3:小鼠皮下癌细胞植入物。
使用植入雌性裸鼠基质胶的卵巢癌细胞系(ovcar3)测试对抗肿瘤血管生成的抗血管生成功效。在每日用聚合物缀合的dat、mat和tat处理14天(1mg/kg,sc,qd)后,移除基质胶肿瘤植入物并分析血红蛋白。对于肿瘤介导的血管生成的抗血管生成作用的代表性数据被描绘为如图22的数据所示的血红蛋白测量的函数。
实施例4:不同癌细胞系中αvβ3的表达(流式细胞术分析)/共聚焦成像。
将细胞培养过夜,并在胰蛋白酶处理后收集细胞。然后将细胞与fitc缀合的抗αvβ3孵育30分钟,用pbs洗涤并使用流式细胞术研究αvβ3的表达,如图23和下表3所示。另外,在共聚焦显微镜下观察到αvβ3表达水平,如图24所示。
表3:各种癌细胞的αvβ3的表达(流式细胞术)
实施例5:急性骨髓性白血病模型:雄性转基因小鼠(nod.cg-prkdcscid)
将thp-1-luc细胞腹膜内注射(在0.1ml中5×106个细胞)到对照(载体)组中并且在聚合物缀合的二氨基四碘甲腺乙酸(p-dat720)或聚合物缀合的三唑四碘甲腺乙酸(p-tat730)中以3mg/kg,皮下注射,每天一次,持续3周。在注射细胞之前、在治疗之前收集血样,并在治疗之后每周收集一次。动物在三周后处死,通过四肢、肝脾和心脏和肺的ivis图像获得tph-1细胞的发光信号强度。另外,制备骨髓涂片并进行利什曼染色。图25描述了白血病模型的代表性数据,其中与聚合物缀合的p-dat720和p-tat730相比,在对照中观察到更多数量的母细胞。
实施例6:抗癌功效:聚合物缀合的dat和dat对裸鼠中u87mg异种移植物的影响:
胶质母细胞瘤的异种移植模型是用于研究gbm的标准模型。在目前的研究中,使用5-6周龄且体重18-20g的无胸腺、免疫缺陷的ncr裸鼠。给动物提供随意获取水和食物的途径。在研究之前允许动物适应该设施5天。收获培养的u87mg-luc细胞并在每个侧腹皮下(s.c.)植入(5×106个细胞在含有50%
所呈现的数据证明聚合物缀合的p-dat720和p-tat730在皮下u87mg胶质母细胞瘤异种移植物中是有效的。全身施用21天,该药物通过完全抑制血管生成、诱导广泛坏死并引起细胞凋亡来减少肿瘤体积。虽然聚合物缀合的p-dat720和p-tat730在αvβ3上具有单分子靶标,但是该靶差异调节这样的网络:细胞内信号传导途径和控制特定基因转录的质膜功能以及与癌症和癌症相关的血管生成高度相关的细胞表面血管生长因子受体功能的网络。小αvβ3在非分裂的非恶性细胞中表达或活化,从而限制了聚合物缀合的p-dat720和p-tat730对肿瘤细胞和肿瘤相关血管细胞的作用。图28-30中的数据描述了这些各种聚合物缀合的p-dat720和p-tat730对肿瘤进展和消退的影响。
图28-30中呈现的数据描述了3mg/kg和10mg/kg剂量的聚合物缀合的甲状腺整联蛋白拮抗剂显著降低了肿瘤重量。第二组治疗小鼠的异种移植物在没有进一步药物暴露的情况下观察另外22天(停止(off)治疗组),尺寸继续减小,在3mg/kg和10mg/kg剂量下,在43天的研究持续时间内都达到了肿瘤重量减少>95%(**p<0.01)。图28-30中呈现的观察结果进一步得到了在第21天收获的异种移植物中细胞活力的组织学评估(图31)的支持,其中p-双-tat1030作为代表性的聚合物缀合的p-tat730。
对石蜡包埋的组织切片染色(苏木精和伊红)的组织学评价,并对每个切片进行编码。用镜台测微器测量切片的面积,以及活肿瘤相对坏死肿瘤的百分比,表现为细胞密度的损失、质膜的溶解和视觉上估计的核结构的损失。计算了5个活肿瘤区域中每个高倍视野的有丝分裂和凋亡细胞的数量并每个组织切片取平均。存活区域的血管化程度不同,并且分为1-4级。在暴露于3mg/kg和10mg/kg剂量的p-双-tat1030的异种移植物中,第21天细胞存活率的降低达到60-70%(**p<0.001)。这些发现与导致坏死的肿瘤的系统性去血管化和聚合物p-dat720或p-tat730诱导的多种促凋亡机制一致。用于活细胞的ivis成像(图32)和组织学数据(图31)证实了p-双-tat1030作为代表性聚合物缀合的p-dat720或p-tat730在影响癌细胞存活中的影响。
实施例7:cy5标记的p-双-tat和其他聚合物缀合的甲状腺整联蛋白拮抗剂衍生物的动力学。
给每组动物提供原位植入物(gbm)和在脑中肿瘤生长后并皮下植入gbm异种移植物。将聚合物缀合的mat210、tat230或dat220衍生物与p-双-tat-cy5和其他聚合物缀合的tat230或dat220衍生物-cy5进行皮下注射。注射后立即进行ivis成像,并在30分钟、1小时、2小时、3小时、4小时进行ivis成像,以检测原位脑异种移植肿瘤部位(脑)和皮下异种移植肿瘤的荧光强度。提供图33作为代表性ivis成像,其显示向脑中gbm或皮下植入的肿瘤中的最佳和相当的递送。图34和35中描绘的图描述了脑中gbm肿瘤或皮下植入的肿瘤中甲状腺整联蛋白拮抗剂衍生物的摄入的动力学。图34-35描述了含有mat210、dat220和tat230的聚合物缀合的甲状腺整联蛋白拮抗剂衍生物随时间的影响,包括其双功能实施方案。
在30分钟时在脑中观察到p-双-tat1030和聚合物缀合的p-tat730衍生物-cy5的cy5信号,并用ivis监测4小时。4小时后动物处死并且在脑gbm肿瘤和皮下gbm肿瘤异种移植物中检测到cy5信号。cy5信号在脑gbm肿瘤和皮下gbm肿瘤异种移植物中是相当的。图36显示了在脑中gbm肿瘤和皮下gbm肿瘤异种移植物中相当的摄入。
实施例8:p-双-tat对gbm(u87-luc)异种移植物(原位和皮下)的影响:
胶质母细胞瘤细胞:将u87-luc细胞以20μl基质胶中的0.2×106个细胞原位植入无胸腺雌性小鼠中,并用基质胶以2×106个细胞/植入物皮下异种移植到无胸腺雌性小鼠中。在植入后1天后,用聚合物缀合的甲状腺整联蛋白拮抗剂,包括p-dat720、p-tat730的衍生物及其双功能衍生物治疗动物,持续7天,并进行脑肿瘤和皮下肿瘤异种移植物的ivis成像。图37描述了使用p-双-tat1030的肿瘤的剂量依赖性抑制,包括脑gbm肿瘤和皮下gbm肿瘤中的细胞活力的抑制。图38-40描绘了p-双-tat1030对脑gbm肿瘤和皮下gbm肿瘤的可测量的剂量依赖性影响,包括对脑中gbm的发光信号、平均肿瘤重量和生物发光强度的影响。
实施例9:p-双-tat对胰腺癌suit2异种移植物的影响。
用于体内研究的雌性ncr小鼠(taconicfarms,hudson,ny)在5-6周龄(20gm体重)时获得并维持在特定无病原体的条件下,并随意提供食物和水。允许动物适应环境5天。收获培养的人类suit2细胞,并在小鼠的每个侧腹皮下(s.c.)植入。制备包含100μl,50%基质胶并含有2x106个肿瘤细胞的接种物。使肿瘤生长7天,此时将动物随机分成对照组和p-双-tat组(6只动物/组,12-癌移植物/组)。从第0天开始,每天施用3mg或10mgp-双-tat/kg体重的药物,持续20天(至实验第19天)。对照动物每天接受载体(pbs)。在实验第20天处死动物。收获肿瘤并测量肿瘤的重量。肿瘤用福尔马林固定、用石蜡包埋并切片。
还评估了单独的和与p-双-tat结合的辐射疗法的有效性。辐射治疗是在第10天和第17天对右侧异种移植物以1个gy施用的。侧腹暴露的几何形状排除了对研究动物左侧的辐射暴露,因此在辐射研究中将左侧用作对照。
研究了代表性的聚合物-缀合的甲状腺整联蛋白拮抗剂(p-双-tat1030)对肿瘤重量、细胞活力和细胞坏死的影响。p-双-tat对胰腺癌suit2异种移植物重量、细胞活力和细胞坏死的抗癌功效如图41a-41c所示。p-双-tat以3mg/kg施用20天后肿瘤重量减少55%并且以10mg/kg施用减少65%(相对于对照,各自***p<0.001)(图41a)。如图41b所示,肿瘤细胞活力的降低百分比在3mg/kg的药物浓度下是不显著的,并且在10mg/kg时是43%(相对于对照,***p<0.001)。如图41c中的数据所示,组织切片中%细胞坏死的增加在3mg/kgp-双-tat时不显著,但在10mg/kg剂量下坏死增加大于一倍(相对于对照,***p<0.001)。因此,在这些使用p-双-tat的短期研究中,存在肿瘤大小和癌细胞活力和坏死的组织学评估的理想改变。
辐射暴露和p-双-tat施用的相互作用:
在图42a-42c中描绘了单独的辐射暴露和与p-双-tat结合的辐射对胰腺癌suit2异种移植物重量、细胞活力和细胞坏死的影响。相对于未暴露于辐射的左侧异种移植物,以1gy对右侧异种移植物的单独的辐射治疗导致了这些异种移植物的肿瘤重量减少了38%(图42a)。辐射和示例性实施方案聚合物缀合的甲状腺整联蛋白拮抗剂测试p-双-tat1030的组合导致肿瘤大小进一步降低71%(3mgp-双-tat/kg)和77%(10mgp-双-tat/kg)(相对于未被照射的对照,各自***p<0.001)。当药物和辐射结合时,不存在对肿瘤重量的p-双-tat剂量效应。
如图42b所示,在没有p-双-tat的1gy下显示的%细胞活力的降低是没有统计学意义的,但是,辐射和3mg/kg药物的组合导致54%的活力降低,并且在10mg/kg时观察到68%的降低(相对于未被照射的对照异种移植物,各自**p<0.05)。此外,如图42c中所示的图形数据所示,单独的辐射引起的坏死的上升趋势没有统计学意义,然而,辐射和3mg/kgp-双-tat的组合导致65%的坏死增加并且在10mg/kg时70%(相对于未被照射的对照,各自**p<0.05)。此外,图43进一步展示了数据,这些数据显示与对照相比,p-双-tat(3和10mg/kg,scqd)与1gy和5gy的胰腺肿瘤照射之间的相互作用。
因此,基于图42a-42c中提供的数据,可以得出结论,存在意外的组合疗法,其对肿瘤重量减少和细胞坏死和活力产生重要的累加效应。甲状腺整联蛋白拮抗剂、聚合物缀合的甲状腺整联蛋白拮抗剂及其双功能实施方案的抗肿瘤效力,包括p-双-tat的示例性组合物对肿瘤质量的影响在3mg/kg体重时对于药剂而言是接近最大的,即,剂量为3mg/kg和10mg/kg的结果是相当的。然而,在使用和不使用辐射的情况下获得的组织学变化的调查表明,10mgp-双-tat/kg比3mg/kg更有效。上述异种移植物观察组确定,聚合物缀合的甲状腺整联蛋白拮抗剂,例如p-bi-tat,是标准实验环境中胰腺癌异种移植物中的高效治疗干预。
实施例10:安全性研究,c57bl6小鼠。
p-双-tat的聚合物缀合的甲状腺整联蛋白拮抗剂抗血管生成剂的代表的临床前毒理学在5至6周龄的c57bl6小鼠中进行,用不同剂量的p-双-tat治疗14天。治疗组是对照(载体)、1mg/kg、3mg/kg、10mg/kg、30mg/kg、100mg/kg和330mg/kgp-双-tat,每天皮下给药,持续14天。每个治疗组由5只雄性动物和5只雌性动物组成,每周两次测量动物体重。小鼠在14天后处死并从眶后静脉丛收集血样。将血液样品离心,并将收获的血浆在-80℃存储直至进行下述分析。
通过测量在储存的血浆中的丙氨酸转氨酶(alt)和天冬氨酸转氨酶(ast)活性(比色试剂盒,biovision,inc.,milpitas,ca)来估计肝功能,其数据显示在图44中。通过elisa方法(lifediagnostics,westchester,pa)测量血浆中的心肌肌钙蛋白i(ctni)水平作为心肌损伤的指标(图44)。它是一种仅在心脏中而不是在其他形式的肌肉中发现的肌钙蛋白。通过血浆肌酸酐浓度的测量(比色试剂盒,biovision,inc.)估计肾功能(图44)。为每个肝脏和肾脏的试验构建令人满意的标准曲线。体重不受药剂影响(结果未显示)。没有证据表明,当暴露于高达实现最大(3-10mg/kg)抗癌治疗效果的p-双-tat剂量的100倍2周时,雄性或雌性动物中有肝脏、心脏或肾脏毒性。
已经出于说明的目的给出了对本发明的各种实施方案的描述,但是并不旨在穷举或限制于所公开的实施方案。在不脱离所描述的实施方案的范围和精神的情况下,许多修改和变化对于本领域普通技术人员来说是显而易见的。这里选择使用的术语是为了最好地解释实施方案的原理、实际应用或对市场中发现的技术的技术改进,或者使本领域普通技术人员能够理解本文公开的实施方案。
- 还没有人留言评论。精彩留言会获得点赞!