四六面体纳米颗粒
四六面体纳米颗粒
1.美国政府支持声明
2.本发明是在由能源部(department of energy)授予的授权号de
‑
sc0000989
‑
0002下由政府支持进行的。政府拥有本发明的某些权利。
背景技术:
3.具有高指数小平面的纳米结构在催化中极为重要,但用于规模化生产所述纳米结构的方式有限(zhang等人,《今日纳米(nano today)》11,661
‑
677(2016),tian等人,《物理化学期刊c(j.phys.chem.c)》112,19801
‑
19817(2008))。通常使用依赖于小平面稳定配体的相对低通量的电化学方法或相对高通量的液相法来制备所述纳米结构。重要的是,无论粒径和组成如何,此类小平面的原子结构都可以直接影响催化活性(xia等人,《美国国家科学院院刊(proc.natl.acad.sci.u.s.a)》110,6669
‑
6673(2013),lee等人,《应用化学国际版(angew.chem.int.ed.)》118,7988
‑
7992(2006))。然而,出于两个原因,限制使用配体来控制颗粒形状。第一,此类药剂的作用尚不十分清楚,并且根据金属类型和期望的颗粒形状需要不同的配体(wang等人,《美国化学会志(j.am.chem.soc.)》133,1106
‑
1111(2010),personick等人,《纳米通讯(nano lett.)》11,3394
‑
3398(2011))。第二,稳定的配体通常难以去除,并且通过阻断活性位点而对催化活性产生不利影响(niu等人,《材料化学(chem.mater.)》26,72
‑
83(2013))。需要开发一种制备具有高指数小平面的纳米颗粒的直接、无配体且可通用的方法、用于如甲酸氧化等氧化反应的高效电催化剂以及回收废催化剂的方法。
技术实现要素:
4.本文提供了制备四六面体(“thh”)纳米颗粒的方法,所述方法包括:在存在第二金属的情况下,在500℃到1300℃下将包括第一金属的颗粒加热约0.5小时到约12小时以形成所述thh纳米颗粒;其中所述第一金属包括铂、钯、铑、镍、钴或其组合,并且所述第二金属包括sb、bi、pb、te或其组合。在实施例中,所述thh纳米颗粒包括高指数小平面。在实施例中,所述thh纳米颗粒包括{210}个小平面、{310}个小平面、其邻近平面或其组合中的一个或多个。在实施例中,所述第一金属是双金属的。在实施例中,所述第一金属包括ptni、ptco、ptcu、pdpt、pdau、pdni、pdco、pdcu、rhpt、rhco、rhni或其组合。
5.在实施例中,本文公开的方法是在反应器中执行的,其中所述第二金属定向在所述颗粒的上游并且通过气流携带到所述颗粒。在实施例中,其中气体包括氩气、氮气、氦气、氢气、一氧化碳、二氧化碳或其组合。
6.在实施例中,将包括所述第一金属的所述颗粒掺入到载体上。在实施例中,所述载体包括二氧化硅、二氧化钛、二氧化铈、氧化铝、氧化锆、氧化铌、氧化锌、氧化铁、氧化钒或其组合。在实施例中,所述载体是导电的。在实施例中,所述导电载体包括炭黑、石墨烯、石墨、碳纳米管、碳纤维、碳化钨或其组合。在实施例中,所述颗粒是通过分解和/或还原所述第一金属的盐而形成的,或所述颗粒是由所述第一金属的金属合金形成的。在实施例中,所
述第二金属是通过分解和/或还原所述第二金属的盐而形成的。在实施例中,所述第一金属包括铂。在实施例中,包括所述第一金属的所述颗粒是非thh颗粒。在实施例中,所述第二金属包括sb。在实施例中,所述第二金属包括bi。在实施例中,本文公开的方法在不存在有机配体的情况下执行。
7.本文还提供了将本文公开的所述thh纳米颗粒用作氧化催化剂的方法。在实施例中,所述thh纳米颗粒催化甲酸到co和/或co2的氧化。
附图说明
8.图1示出了用于通过与异金属(sb、bi、pb和te)合金化/去合金化来合成thh颗粒的实验装置和一些实验结果。(a)用于利用cvd设置合成thh颗粒的方案。将大约1mg的异金属粉末装入燃烧舟中,然后将燃烧舟转移到管式炉。将涂覆有所关注金属盐前体或形状不规则的颗粒的硅晶片放置在管中,在异金属粉末的下游。在ar(或ar/h2)气氛中进行热处理之后,将管在空气中淬火,并且然后冷却到室温。通过蒸发将异金属粉末完全转移到具有金属前体的硅晶片上,并且thh颗粒形成。(b)在900℃下反应后合成颗粒的作为时间的函数的eds光谱。cu信号来自tem样品架。在900℃下反应持续以下时间后同一区域中的合成颗粒的stem图像和eds元素图c:(c)0分钟(26%pt,74%sb),(d)20分钟(38%pt,62%sb)以及(e)40分钟(85%pt,<15%sb)。在(e)中,圈出颗粒的stem图像和对应eds元素图(右)提供了颗粒形态和元素分布的清晰视图。(i和ii)中的比例尺为50nm。剩余部分为300nm。
9.图2在(a
‑
c)中示出了从[100]晶体朝向、[110]晶体朝向和[111]晶体朝向观察到的截短的thh pt颗粒的代表性sem图像。pt颗粒的(d
‑
e)sem图像分散在硅晶片上。(f)沿[001]方向记录的截短的thh pt纳米颗粒的tem图像。白线用于突出纳米颗粒的作为眼睛的引导的小平面。(g)(f)中纳米颗粒的对应衍射图。(h)沿[001]方向投影的{210}小平面包围的thh形状的纳米颗粒的理想模型。仔细测量(f)中纳米颗粒的表面平面之间的角度指示暴露的高指数小平面的密勒指数(miller index)为{210}。(i)从(f)中标记了方框区域记录的hrtem图像。绘制红线以突出作为眼睛的引导的表面(210)平面。(j)(210)平面的原子模型。通过图2在硅晶片上合成的pt颗粒的sem图像。在(k)中,在不使用sb粉末的情况下,通过在700℃下将h2ptcl6热分解30分钟来合成颗粒,在(l)中,使用1mg sb粉末通过在900℃下将(k)中的样品加热30分钟来合成颗粒,并且在(m)中,在不使用sb粉末的情况下,通过在900℃下将(l)中的样品加热30分钟来合成颗粒。比例尺:在(f)中为20nm,并且在(k到m)中为300nm。
[0010]
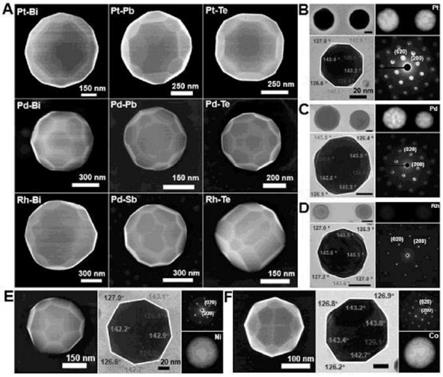
图3在(a)中示出了通过sb、bi、pb和te修饰合成的pt、pd和rh颗粒的sem图像。通过bi修饰的图3(b)中的pt颗粒、(c)中的pd颗粒和(d)中的rh颗粒的stem图像、eds元素图、tem图像和对应衍射图。示出了通过bi修饰的图3(e)中的ni颗粒和(f)中的co颗粒的sem图像、tem图像、对应衍射图和eds元素图。比例尺:50nm,除非另有说明。
[0011]
图4在(a)中示出了不同催化剂在0.5v甲酸氧化下的比活性的极化曲线并且在(b)中示出了所述比活性的直方图。
[0012]
图5示出了在以下不同条件下用约1mg sb粉末作为来源合成的pt颗粒的sem图像:(a)600℃持续12h,(b)800℃持续0分钟,(c)800℃持续30分钟,(d)800℃持续1小时,(e)800℃持续12小时,(f)900℃持续0分钟,(g)900℃持续30分钟,(h)900℃持续1小时,(i)900℃
持续4小时,(j)1000℃持续30分钟,并且(k)1000℃持续1小时。比例尺:500nm。
[0013]
图6示出了在900℃下在(a)30分钟内用0.1mg sb粉末作为来源合成的pt颗粒的sem图像以及在(b)1小时内用作为来源供应的10mg sb粉末每30分钟合成的pt颗粒的sem图像。比例尺:500nm。
[0014]
图7示出了在不添加sb的情况下直接分解pt前体而在硅晶片上合成的pt纳米颗粒的sem图像。比例尺:500nm。
[0015]
图8示出了在以下时间内在900℃下反应之后的合成颗粒的stem图像和eds元素图,其中1mg sb粉末用作来源:(a)0分钟(26%pt,74%sb),(b)10分钟(32%pt,68%sb),(c)20分钟(38%pt,62%sb),(d)30分钟(69%pt,31%sb)以及(e)40分钟(85%pt,15%sb)。在图(d)和(e)中,右侧示出了圈出颗粒的stem图像和对应eds图以获得形态和元素分布的清晰视图。(i
‑
iv)中的比例尺为50nm。剩余部分为300nm。
[0016]
图9示出了在以下时间后在900℃下合成的颗粒的eds光谱:(a)0分钟(26%pt,74%sb),(b)10分钟(32%pt,68%sb),(c)20分钟(38%pt,62%sb),(d)30分钟(69%pt,31%sb)以及(e)40分钟(85%pt,15%sb)。
[0017]
图10示出了通过以下利用1mg bi粉末来源合成的pt颗粒的sem图像:将旋涂有h2ptcl6水溶液的硅晶片加热到800℃(上升速率为10℃/min),随后在冰冷的水中淬灭管。比例尺:200nm。
[0018]
图11在(a)中示出了硅晶片上的合成后的截短的thh pt颗粒的sem图像,在(b)中示出了利用1mg sb粉末来源的在900℃下将样品(a)退火30分钟之后合成的pt颗粒的sem图像,以及(c)在没有sb粉末来源的情况下在900℃下将样品(b)退火30分钟之后合成的截短的thh pt纳米颗粒的sem图像。每列都是前一列的放大视图。(d)和(e)分别是(b)和(c)中方框区域的放大视图。比例尺:在(a
‑
c)中为500nm(第二列)和250nm(第三列),在(d
‑
e)中为250nm。
[0019]
图12在(a)中示出了在不使用额外的sb粉末来源的情况下合成后的thh形状的pt纳米颗粒的sem图像以及在(b)中示出了在900℃下退火30分钟后的纳米颗粒的sem图像。在退火处理之后,颗粒形状保持完整。比例尺:500nm。
[0020]
图13在(a)中示出了在不使用任何外部sb粉末来源的情况下通过在700℃下直接将旋涂有h2ptcl6水溶液的硅晶片退火30分钟而获得的合成后的pt颗粒的sem图像,在(b)中示出了利用1mg sb粉末来源通过将样品(a)在900℃下退火30分钟合成的pt颗粒的sem图像,以及在(c)中示出了在没有sb粉末来源的情况下通过将样品(b)在900℃下退火30分钟合成的截短的thh pt颗粒的sem图像。(d)是(c)中的方框区域的放大图。将每个图的右下角处的二氧化硅残留物用作颗粒的位置参考。
[0021]
图14在(a)中示出了将1mg sb粉末用作来源通过在900℃下将前体加热1小时,随后以5℃/min的速度将管缓慢冷却到室温而合成的pt颗粒的sem图像,并且在(b)中示出了在不使用外部sb粉末的情况下通过在900℃下对样品重新加热30分钟获得的具有光滑表面的pt颗粒的sem图像。比例尺:500nm。
[0022]
图15示出了在600℃下进行热退火以下不同持续时间的pt颗粒的sem图像:(i)0分钟,(ii)2小时,和(iii)12小时。(a)和(b)中的(iv)是在不使用额外的sb粉末来源的情况下在900℃下退火30分钟后的相同颗粒的sem图像。比例尺:300nm。
[0023]
图16(在a,b中)示出了合成后的thh形状的颗粒并且(在c,d中)在600℃下进行12小时的热退火之后的thh形状的颗粒的stem图像、eds元素图和对应光谱。颗粒的原子组成(在a,b中)为pt
76
sb
24
,并且(在c,d中)为pt
79
sb
21
。cu信号来自tem样品架。比例尺:50nm。
[0024]
图17示出了通过bi修饰合成的pt、pd、rh、ni、co和双金属纳米颗粒(ptrh、pdau、ptcu、pdcu、ptco、pdco、rhco、ptni、pdni和rhni)的文库的eds光谱。
[0025]
图18示出了thh形状的(a)ptcu纳米颗粒、(b)ptni纳米颗粒、(c)ptco纳米颗粒、(d)pdcu纳米颗粒、(e)pdau和(f)rhpt纳米颗粒的tem图像、对应衍射图和eds元素图。白线用于突出作为眼睛的引导的颗粒小平面。比例尺:20nm。
[0026]
图19示出了在900℃下加热(在a,b中)0分钟、(在c,d中)10分钟、(在e,f中)20分钟、(在g,h中)30分钟和(在i,j中)40分钟之后合成的颗粒的sem图像(左列)对应xrd图(中间列)。在(d)、(f)和(h)中,插入图是方框区域的放大视图,并且pt3sb金属间相的峰位置由参考线标记。右栏中的xrd图来自(k)中硅晶片、(l)中硅晶片上的通过在900℃下直接分解h2ptcl6盐持续10分钟而合成的纯pt颗粒,以及(m)中用于这项工作的纯sb粉末。比例尺:500nm。
[0027]
图20示出了装饰有异金属(sb、bi、pb和te)的pt小平面的板结构。银色原子和棕色原子分别代表pt和异金属(sb、bi、pb和te)。
[0028]
图21示出了装饰有异金属(sb、bi、pb和te)的pt小平面的板结构。银色原子和棕色原子分别代表pt和异金属(sb、bi、pb和te)。
[0029]
图22示出了不同的经修饰pt小平面的作为异金属(sb)的表面覆盖率的函数的比表面能。使用每种异金属的最低化学势计算比表面能。
[0030]
图23示出了不同的经修饰pt小平面的作为异金属(te)的表面覆盖率的函数的比表面能。使用每种异金属的最低化学势计算比表面能。
[0031]
图24示出了不同的经修饰pt小平面的作为异金属(bi)的表面覆盖率的函数的比表面能。使用每种异金属的最低化学势计算比表面能。
[0032]
图25示出了不同的经修饰pt小平面的作为异金属(pb)的表面覆盖率的函数的比表面能。使用每种异金属的最低化学势计算比表面能。
[0033]
图26示出了不同的经修饰pt小平面的作为异金属(sb、te、bi和pb)的化学势的函数的比表面能。对于每个小平面,在图22
‑
25中考虑了给出最低表面能的异金属的表面覆盖率。
[0034]
图27示出了在ar饱和的0.5m h2so4溶液中的thh形状的pt
‑
m(m=sb、bi、pb和te)催化剂、pt对照样品和商业pt/c催化剂的循环伏安图。扫描速率:50mv s
‑1。
[0035]
图28示出了在0.5v下在3600秒内ar饱和的0.5m hcooh+0.5m h2so4溶液中的pt
‑
m(m=sb、bi、pb和te)催化剂、pt对照样品和商业pt/c催化剂的安培分析曲线。插入图示出了方框区域的放大视图。
[0036]
图29在(a)中示出了不同催化剂在0.5v甲酸氧化下的比活性的极化曲线并且在(b)中示出了所述比活性的直方图。
[0037]
图30示出了炭黑上的合成催化剂的stem图像。可以从偶尔观察到的大颗粒中清楚地鉴别处截短的thh的形状。比例尺:50nm。
[0038]
图31示出了在用于在0.5v下进行甲酸电氧化3600秒之前(a)和之后(b)通过bi修
饰合成的pt thh形状的纳米颗粒的sem图像。比例尺:400nm。
[0039]
图32示出了将1mg bi粉末用作来源的(a)失活的商业pt/c催化剂的stem图像和(b)通过在900℃下将(a)中的商业催化剂加热1小时而形成的thh形状的pt颗粒的stem图像。(a)和(b)中成像的区域相同。每列都是前一列的放大视图。(c)商业pt/c催化剂在回收之前和之后的极化曲线。
[0040]
图33示出了通过bi修饰合成的thh形状的双金属颗粒。图33在(a)中示出了在硅晶片上合成的thh形状的ptpd颗粒的sem图像。图33在(b)中示出了thh形状的ptpd颗粒的tem图像、对应衍射图和eds元素图。图33在(c)中一系列thh形状的双金属颗粒的stem图像(第一列)和eds元素图(第二列和第三列)。比例尺:在(a)中为500nm并且在(b和c)中为20nm。
[0041]
图34示出了用于合成thh颗粒的替代性方法。(a)用于通过直接在硅晶片上加热异金属粉末和所关注的金属前体的混合物来合成thh颗粒的方案。(b)通过sb修饰合成的thh pt颗粒的sem图像。(c)用于通过直接加热异金属粉末、金属前体和炭黑粉末的混合物来在高比表面积炭黑粉末上制备thh颗粒的方案。(d)通过bi修饰的约1.3g thh pt催化剂颗粒(在碳xc
‑
72上装载20%的pt)的照片。(e)炭黑粉末上的thh pt颗粒的stem图像。(f)用于通过在硅晶片上直接加热异金属粉末并且预先合成均匀颗粒来产生单分散的thh颗粒的方案。(g)通过液相法合成的pd纳米立方体的stem图像。(h)直方图,其示出了pd纳米立方体的大小部分(样品大小:100)。(i)从(g)中的pd纳米立方体转换的pd thh的stem图像。(j)直方图,其示出了pd thh颗粒的大小部分(样品大小:100)。pd thh颗粒的体积和分散性与初始pd纳米立方体的体积和分散性类似。对于纳米立方体和{210}小平面封闭的等体积的thh颗粒,b=1.31*a。(g)和(i)中插入图像的比例尺分别为20和30nm。
[0042]
图35示出了在0.5v下利用商业pt/c催化剂、thh pt
‑
m(m=sb、bi、pb、te)催化剂以及通过bi修饰从商业pt/c催化剂合成的thh pt催化剂(pt/c
‑
bi thh)进行甲酸氧化的质量活性的(a)极化曲线和(b)直方图。在0.5v下利用商业pd/c催化剂和thh pd
‑
bi催化剂进行甲酸氧化的质量活性的(c)极化曲线和(d)直方图。
具体实施方式
[0043]
本文提供了由在存在异金属的情况下加热的金属颗粒混合物制备四六面体(“thh”)纳米颗粒的方法。在一些情况下,所述方法包括在存在第二金属的情况下,在800℃到1300℃下将包括第一金属的颗粒加热约30分钟到约120分钟以形成所述thh纳米颗粒;其中所述第一金属包括铂、钯、铑、镍、钴或其组合,并且所述第二金属包括sb、bi、pb、te或其组合。这些方法可以提供可以在工业规模上合成催化活性的thh纳米颗粒的一步式无配体的可通用的方法。这些方法还可以提供用于回收如工业中的废催化剂等非thh形状的纳米颗粒并形成本文公开的催化活性的thh纳米颗粒的方法。
[0044]
本文的方法可以用于在适当的热处理之后制备四面体(thh)形状的pt、pd、rh、ni、co和双金属纳米颗粒,无论异金属如何。在一些情况下,所述方法提供了thh形状的pt、pd和rh纳米颗粒。密度泛函理论(dft)和电子显微镜研究两者均用于确定此过程的发生原因和方式。
[0045]
thh纳米颗粒可以例如在电化学氧化反应中用作催化剂。例如,thh纳米颗粒可以用于例如将甲酸氧化为co和/或co2。在一些情况下,所述方法包括向thh纳米颗粒、h2so4和
甲酸的混合物施加电流以形成co和/或co2。将经sb修饰、经bi修饰、经pb修饰和经te修饰的thh形状的纳米颗粒作为甲酸氧化的电催化剂进行评估,并确定这些纳米颗粒优于商业pt/c催化剂,由此提供可以在下一代燃料电池开发中变得重要的有希望的替代品。
[0046]
除非另有所指,否则在描述本文的公开内容的上下文中(特别是在权利要求的上下文中)使用的术语“一个/一种(a/an)”和“所述(the)”以及类似的指代词应被解释为涵盖单数和复数两者。除非本文中另外指明,否则本文中对值范围的叙述仅旨在充当单独地提及落入所述范围内的每个单独值的速记方法,并且每个单独值并入本说明书中,如同在本文中单独叙述一样。除非另外声明,否则本文提供的任何和所有实例或示例性语言(例如,“如”)的使用旨在更好地阐明本文的公开内容,并且不是对本发明的范围的限制。本说明书中的任何语言都不应被解释为将任何未要求保护的要素指示为是实践本文的公开内容所必不可少的。如本文所使用的,除非上下文另外明确指出,否则单数形式“一个(a)”、“一种(an)”、和“所述(the)”包含复数指示物。因此,例如,术语“材料”旨在意指一种或多种材料或其组合。
[0047]
本文所使用的术语“约”和“大约”通常意指所述值的正负10%。例如,约0.5将会包含0.45和0.55,约10将会包含9到11,约1000将会包含900到1100。
[0048]
本文公开的方法和其单独步骤的实践可以手动地和/或借助于电子装备或由电子装备提供的自动化来执行。尽管已经参考特定实施例描述了各过程,但本领域的普通技术人员将容易理解,可以使用执行与所述方法相关联的行为的其它方式。例如,除非以其它方式描述,否则可以在不背离所述方法的范围或精神的情况下,改变步骤中的若干步骤的顺序。另外,可以将单独步骤中的一些组合步骤、省略或进一步细分为另外的步骤。
[0049]
制备thh纳米颗粒的方法
[0050]
本文的thh纳米颗粒可以通过在存在第二金属的情况下,在500℃到1300℃下将包括第一金属的颗粒加热约0.5小时到约12小时来制备。第一金属可以包括铂、钯、铑、镍、钴或其组合。在一些情况下,第一金属可以包括pt、pd、rh或其组合。在一些情况下,第一金属可以是双金属的。在实施例中,双金属的第一金属可以包括pt、ni、co、cu、pd、au、rh或其组合。例如,双金属的第一金属可以包括ptni、ptco、ptcu、pdpt、pdau、pdco、pdcu、rhpt、rhco、rhni或其组合。第二金属可以包括sb、bi、pb、te或其组合。
[0051]
固态前体的热解是工业上用于大规模生产贵金属纳米颗粒的广泛使用的技术(m.v.twigg,《催化剂手册(catalyst handbook)》(crc,1989));然而,通过所述方法制备的颗粒通常具有低指数小平面和热力学上有利的催化非理想形状(barnard,《美国化学学会纳米(acs nano)》3,1431
‑
1436(2009))。如本文所述的痕量的形状调节金属元素或第二金属的欠电位沉积(personick等人,《纳米通讯》11,3394
‑
3398(2011))可以用于合成如本文所述的高指数小平面纳米颗粒。本文中的方法用于以痕量的形状调节异金属元素控制各向异性生长。
[0052]
在一些情况下,将第一金属前体和第二金属粉末的混合物在硅晶片上加热(图34,a中)。在反应完成并随后冷却后,所得纳米颗粒的形态为thh并且可以通过扫描电子显微镜(sem)来确认(例如,图34中,b中)。通过用具有高比表面积(如炭黑)的与纳米颗粒具有强相互作用的典型催化剂载体代替平坦的晶片,所述合成可以容易地按比例缩放到克量(例如在图34中,c和d)。在实施例中,这种情况导致合成后的thh颗粒的大小从平均大于约200nm
骤减到小于或等于约20nm(例如图34中,e中)。尽管直接从金属盐前体转化的thh颗粒的大小在实施例中可以是多分散的(沿[100]方向的边缘长度:206
±
144nm,样品大小:100),但是可以通过将单分散的立方体形状的纳米颗粒(边缘长度:46
±
3.7nm,样品大小:100)用作起始材料并且然后用相同的方法将所述纳米颗粒转化到单分散的thh颗粒中来获得单分散的thh颗粒(沿[100]方向的边缘长度:63
±
6.0nm,样品大小:100)(图34,f到j中)。
[0053]
为了更好地了解thh形成工艺,可以使用化学气相沉积(cvd)室,在所述室中将第二金属放置在管式炉的上游并且在热处理时通过氩气/氢气流携带到第一金属前体(图1)。反应器可以用于确定对如经sb修饰的pt等thh纳米颗粒的结构和形态演变。sem图像确认通过cvd形成thh颗粒的高产率(1000个中的1000个)(图2中,a到e中)。主要暴露的小平面为高指数{210}平面,所述平面由透射电子显微镜(tem)图像及其对应的选定区域电子衍射(saed)图案索引。由{210}小平面包围的理想thh模型与tem成像结构密切相关。纳米颗粒的高分辨率tem(hrtem)图像表明了(210)平面上的原子的原子排列,所述原子排列还与(210)小平面的原子模型密切相关(图2,f到j中)。
[0054]
系统地探索反应温度、时间和sb粉末的量,以确定其相对重要性(图5
‑
10)。较高温度导致更快地形成thh形状的颗粒,但也导致减少生成高质量thh的时间窗口。在较高温度下,sb去合金化以更快的速率发生,并且需要等于或高于900℃的温度来产生具有光滑表面的thh形状的pt颗粒。有趣的是,当使用较大量的sb时,thh形状的形成被延迟了,因为去合金化工艺的发生需要更长的时间(图7)。实际上,扫描透射电子显微镜(stem)和能量色散x射线光谱学(eds)支持此结论(图1,b到e中和图8
‑
9)。具体地,所述两种技术表明随着从初始富含sb的pt
‑
sb合金颗粒中去除sb,形成thh形状的pt颗粒。这种新颖的合金化/去合金化形状调节工艺不同于限定常规湿化学方法的经典添加剂生长工艺。最后,如果从thh颗粒开始并使用相同的设置将sb转移到所述颗粒上,则合金化工艺会产生不规则形状的颗粒,这表明sb去合金化工艺对于产生近乎完美的thh颗粒至关重要(图11
‑
12)。
[0055]
由于pt纳米颗粒利用sb蒸气进行的均与初始纳米颗粒形状无关的合金化和去合金化,所述方法可以用作用于回收形状不良的pt废催化剂的有效方法,这对于贵金属催化剂的大规模应用是重要的工业关注点。在实施例中,可以通过在700℃下热分解h2ptcl6在硅晶片上合成pt纳米颗粒。(图2,k中和图13,a中)。所得纳米颗粒表现出类似于用废催化剂观察到的随机形状的不规则形状,由此建立用于测试合金化
‑
去合金化工艺的形状修复能力的模型系统。在实施例中,然后可以将1mg sb用作第二金属源将样品加热到900℃持续30分钟。所得pt
‑
sb合金纳米颗粒主要表现出低指数立方体形状(图2,l中和图13,b中)。去除sb来源,并将纳米颗粒在900℃下进一步加热另外的30分钟。所得纳米颗粒的sem图像(图2,m中以及图13,c和d中)确认>99%的纳米颗粒表现出截短的thh形态,这表明本文的方法可以用于从已经失活的催化剂重新建立催化重要的thh颗粒。在完成热处理之后,通过从炉中去除含有thh纳米颗粒的管来将所述纳米颗粒淬火到室温。如果相反,使用缓慢冷却,则获得与从淬火产生的具有光滑表面的颗粒相反的具有粗糙表面的thh纳米颗粒(图14)。可以在高指数平面中用sb稳定光滑的表面,并且在低于900℃的温度下加热时,所述表面是不稳定的并且通过纳米级台面和台阶结构(nanoscopic terrace
‑
and
‑
step structure)重组到小平面中。实际上,如果高质量thh结构在600℃下退火12小时,则所述过程可以通过电子显微镜而可视化(图15和16)。光滑的{210}小平面可以通过在900℃下重新加热颗粒来重新产
生,这启动了sb去合金化工艺(图14和15)。如pt等第一金属{210}小平面的由sb的内部重新分布引起的这种可逆表面重构是新型的形状恢复工艺。
[0056]
除了sb之外,痕量元素,即bi、pb和te还可以诱导形成截短的thh形状的pt纳米颗粒(图3,a中)。此策略证明可用于合成贵金属pd和rh以及ni和co的thh形状的纳米颗粒,其中尚未报告形状控制(图3和图17)。确认这些颗粒的暴露平面的密勒指数为{210},这与经sb修饰的pt thh形状的纳米颗粒的暴露平面的密勒指数相同。此方法的通用性通过以下进一步证实:通过bi修饰合成双金属纳米颗粒的文库(图33以及图17和18)。
[0057]
对于具有fcc结构的pt纳米颗粒,对不同晶体小平面的比表面能(按每面积计)按等级排序:σ
(111)
<σ
(100)
<σ
(110)
<σ
(210)
(zhang等人,《应用表面科学(appl.surf.sci.)》229,34
‑
42(2004)),并且单结晶pt颗粒的平衡形状为截角八面体。合成pt颗粒中{210}小平面的主要存在指示sb修饰通过降低其比表面能来稳定{210}小平面。x射线衍射数据(图19)证明了颗粒内的pt和sb的原子顺序不能成为形成thh形状的pt颗粒的原因。实际上,时程研究表明,在较早时间点形成的金属间相(pt3sb)颗粒不呈现thh形态。直到去除了绝大多数的sb并且大部分是纯pt才观察到thh形态。因此,可以得出结论,此类颗粒中的sb修饰主要发生在表面上。为了测试此假设,使用dft计算(210)平面、三个低指数小平面(100)、(111)和(110)以及sb修饰后不同类型的高指数平面的比表面能(图20
‑
25,表1
‑
4)。重要的是,计算结果确认,在sb修饰后,这些小平面的比表面能发生了急剧的变化,并且pt(210)
‑
sb的比表面能在所考虑的小平面中最低,这与以下结论一致:颗粒表面处的sb在稳定thh形态中发挥核心作用。
[0058]
在各个实施例中,制备thh纳米颗粒的方法可以包括具有任何高指数小平面的thh纳米颗粒。在一些情况下,制备thh纳米颗粒的方法可以包括thh纳米颗粒,所述纳米颗粒包括{210}小平面、{310}小平面、其邻近平面或其组合。
[0059]
在实施例中,第一金属可以包括适于本领域的技术人员的任何贵金属。在实施例中,第一金属可以包括pt、pd、rh、ni、co或其组合,或者第一金属可以是双金属的。在实施例中,第一金属可以包括pt、pd、rh或其组合。在实施例中,双金属的第一金属可以包括pt、ni、co、cu、pd、au、rh或其组合。例如,双金属的第一金属可以包括ptni、ptco、ptcu、pdpt、pdau、pdco、pdcu、rhpt、rhco、rhni或其组合。在实施例中,第一金属可以包括铂。在实施例中,制备thh纳米颗粒的方法可以包括由第一金属的金属合金形成通过分解和/或还原第一金属的盐而形成的颗粒。在实施例中,制备thh纳米颗粒的方法可以包括通过分解和/或还原h2ptcl6·
6h2o、na2pdcl4、na3rhcl6、ni(no3)2·
6h2o、co(no3)2·
6h2o、cu(no3)2·
xh2o(其中x为1到10)、haucl4·
3h2o或其组合而形成的颗粒。在实施例中,制备thh纳米颗粒的方法可以包括通过分解和/或还原h2ptcl6·
6h2o、na2pdcl4、na3rhcl6或其组合而形成的颗粒。
[0060]
在实施例中,制备thh纳米颗粒的方法可以包括第二金属,其中第二金属是适合本领域的技术人员的任何形状指示的金属。本文描述了术语“形状指示”,并且所述术语在personick等人.《纳米快报》11,3394
‑
3398(2011)中进一步描述。在一些情况下,第二金属可以包括bi、pb、sb、te或其组合。在一些实施例中,第二金属可以包括bi。在一些情况下,第二金属可以包括sb。在一些情况下,制备thh纳米颗粒的方法可以包括通过分解和/或还原第二金属的盐而形成的第二金属。在实施例中,第二金属以相对于起始第一金属颗粒和起始第二金属的总量的量存在。例如,第二金属可以相对于所使用的第一金属颗粒和第二金
属的总量以0.01wt%到25wt%的量存在。在实施例中,第二金属可以以0.01wt%到15wt%,或0.01wt%到10wt%,或0.01wt%到5wt%,或0.01wt%到1wt%,如0.1wt%、1wt%、2wt%、3wt%、4wt%、5wt%、6wt%、7wt%、8wt%、9wt%、10wt%、11wt%、12wt%、13wt%、14wt%或15wt%的量存在。
[0061]
由于颗粒利用第二金属蒸气进行的合金化和去合金化与初始颗粒形状无关,因此本文中的方法可以用作用于回收非thh(“不规则形状”)第一金属纳米颗粒(“废催化剂”)的有效方法,这对于贵金属催化剂的大规模应用是重要的工业关注点。(morgan等人,《美国化学学会催化剂(acs catal.)》5,3430
‑
3445(2015))。在实施例中,可以通过在700℃下热分解h2ptcl6在si晶片上合成pt颗粒。(图13,a中)。所得颗粒表现出类似于用废催化剂观察到的随机形状的不规则形状,由此建立用于测试合金化/去合金化工艺的形状修复能力的模型系统。因此,然后将1mg sb用作异金属源将样品加热到900℃持续30分钟。所得sb
‑
pt合金颗粒主要表现出低指数立方体形状(图13,b中)。去除sb来源,并将颗粒在900℃下进一步加热另外30分钟。所得颗粒的sem图像(图13,c
‑
d中)确认颗粒中的基本上所有颗粒都表现出截短的thh形态,从而确认此方法可以用于从已经失活的催化剂中重新建立催化重要的thh颗粒。
[0062]
在实施例中,在完成热处理之后,通过从炉中去除含有颗粒的管来将所述颗粒淬火到室温。如果相反,使用缓慢冷却,则获得与从淬火产生的具有光滑表面的颗粒相反的具有粗糙表面的thh颗粒(图14)。在高指数平面中用sb稳定光滑的表面,并且在低于900℃的温度下加热时,所述表面是不稳定的并且通过纳米级台面和台阶结构重组到小平面中。实际上,如果高质量thh结构在600℃下退火12小时,则此过程可以通过电子显微镜而清晰地可视化(图15
‑
16)。光滑的{210}小平面可以通过在900℃下重新加热相同的颗粒来重新产生,这启动了sb去合金化工艺(图14
‑
15)。pt{210}小平面的由sb的内部重新分布引起的这种可逆表面重构是形状恢复工艺。
[0063]
在实施例中,制备thh纳米颗粒的方法可以在反应器中执行,其中第二金属定向在颗粒的上游并且通过气流携带到颗粒。对于本领域的技术人员而言,本文所述的气体可以是任何合适的气体。在实施例中,气体可以包括氩气、氮气、氦气、氢气、一氧化碳、二氧化碳或其组合。
[0064]
在实施例中,可以将包括所述第一金属的所述颗粒掺入到载体上。本文所述的载体可以是适于本领域的技术人员的任何载体。在实施例中,载体可以包括适于本领域的技术人员的任何氧化物。在实施例中,载体可以包括二氧化硅、二氧化钛、二氧化铈、氧化铝、氧化锆、氧化铌、氧化锌、氧化铁、氧化钒或其组合。在一些情况下,载体可以是导电的。在实施例中,导电载体可以包括碳。在实施例中,导电载体可以包括炭黑、石墨烯、石墨、碳纳米管、碳纤维、碳化钨或其组合。
[0065]
在实施例中,制备thh纳米颗粒的方法可以在不存在适合本领域的技术人员的任何配体的情况下执行。在实施例中,制备thh纳米颗粒的方法可以在不存在有机配体的情况下执行。
[0066]
氧化催化剂
[0067]
本文提供了将本文所述的thh纳米颗粒用作氧化催化剂的方法。在实施例中,本文所述的thh纳米颗粒可以催化合适的合适小分子的氧化,所述小分子如甲酸、甲醇、乙醇、一
氧化碳或铵。在实施例中,本文所述的thh纳米颗粒可以催化甲酸到co和/或co2的氧化。
[0068]
元素sb、bi、pb和te可有利于促进催化剂对甲酸的电氧化的效率和稳定性(l.an等人,《纳米能源(nano energy)》)15,24
‑
32(2015)),这对于燃料电池中的化学燃料是一个有吸引力的选择。此外,{210}小平面在[001]区中具有最高台阶原子密度并且是fcc晶体的最“开放”的平面。最后,已经报道具有{210}小平面的pt纳米颗粒对甲酸电氧化表现出极高的催化活性(sun等人,《电分析化学杂志(j.electroanal.chem.)》370,273
‑
280(1994))。研究了在炭黑上大规模合成的pt
‑
m(m=sb、bi、pb、te)和pd
‑
bi thh np对甲酸电氧化的催化活性并确认相比而言,如本文所述的pt
‑
m(m=sb、bi、pb、te)thh np比商业pt/c和pd/c催化剂更具活性(图4,图27
‑
31,图34,c到e中和图35)。测试了与商业pt/c催化剂的催化活性相比,在炭黑上大规模合成的pt
‑
m(m=sb、bi、pb、te)纳米颗粒对甲酸电氧化的催化活性(图4,图26
‑
28)。如众所周知,催化剂表面上的甲酸电氧化通常遵循双重路径:1)直接脱氢路径和2)间接脱水路径(yu等人,《电源杂志(j.power sources)》182,124
‑
132(2008),rice等人,《电源杂志》111,83
‑
89(2002))。在约0.5v下的峰i对应于通过脱氢路径的甲酸氧化,并且在约0.9v下的峰ii对应于通过脱水路径形成的co
ads
氧化(图4,a中)。间接路径在商业pt/c催化剂的主导阻碍了所述催化剂在直接甲酸燃料电池中的应用(yu等人,《电源杂志》182,124
‑
132(2008),rice等人,《电源杂志》111,83
‑
89(2002))。然而,合成的thh形状的pt
‑
bi催化剂有利于直接路径,几乎没有证据证明存在通过间接路径的贡献。与商业pt/c催化剂(r=0.5)相比,thh形状的pt
‑
pb(r=2.1)、pt
‑
sb(r=1.8)和pt
‑
te(r=1.1)催化剂的较高i
peak i
对i
peak ii
比(r)还指示直接脱水路径对这些催化剂更有利。具体地,在0.5v下,pt
‑
bi(13.25ma/cm2)、pt
‑
pb(3.43ma/cm2)、pt
‑
sb(2.71ma/cm2)和pt
‑
te(2.26ma/cm2)催化剂的电流密度分别是商业pt/c催化剂(0.21ma/cm2)的电流密度的63倍、16倍、13倍和11倍。重要的是,不使用异金属元素合成的pt纳米颗粒表现出较差的催化活性(pt对照样品,在0.5v下为0.28ma/cm2),从而强调了源自形状控制和异金属修饰的催化效益。另外,为了区分形状控制与异金属修饰的重要性,通过重复性电化学沉积制备用bi原子进行的经修饰的商业pt/c催化剂,并评估所述催化剂的催化性质。在相同条件下,此材料在0.5v下表现出的电流密度为3.65ma/cm2,这是thh形状的pt
‑
bi催化剂的电流密度的四分之一(图29)。综上所述,必须得出的结论以下结论:除了异金属修饰之外,高指数小平面也是催化性能的主要因素。最后,安培i
‑
t曲线(图28)表明这些thh形状的催化剂是坚固的。在0.5v的固定电势下连续操作1小时后,所测得的电流密度为:对于thh形状的pt
‑
bi为0.44ma/cm2、对于thh形状的pt
‑
pb为0.34ma/cm2、对于thh形状的pt
‑
sb为0.30ma/cm2并且对于thh形状的pt
‑
te为0.21ma/cm2(分别是商业pt/c催化剂的电流密度(0.02ma/cm2)的22倍、17倍、15倍和11倍)。
[0069]
与常规高指数小平面纳米颗粒合成方法不同,本文描述的方法非常易于采用、可扩展且对工业生产有效。另外,由于可以通过本文描述的方法回收已经失去其高指数小平面的失活结构,因此所述方法对于催化剂的回收和重新活化(这是已经用商业催化剂样品进行进一步研究和表征的过程)特别具有吸引力(图32)。
[0070]
实例
[0071]
材料和方法
[0072]
sb粉末(99.5%)、bi粉末(99.99+%)、pb粉末(99.95%)、te粉末(99.997%)、溴化十六烷基三甲铵(ctab,>99%)、l
‑
抗坏血酸(>99%)、金属化合物(h2ptcl6·
6h2o、na2pdcl4、
na3rhcl6、haucl4·
3h2o、co(no3)2·
6h2o、ni(no3)2·
6h2o和cu(no3)2·
xh2o)、甲酸(≥95%)、硫酸(99.999%)、pt/c催化剂(20wt.%pt装载)、pd/c催化剂(10wt.%pd装载)并且全氟磺酸(nafion)溶液(5wt.%)购自西格玛奥德里奇(sigma
‑
aldrich)。炭黑粉末(vulcan xc
‑
72)购自卡博特公司(cabot)。硅晶片购自nova电子材料(nova electronic materials)。以上材料按原样使用。实验中使用的所有水都是密理博(millipore)超纯水(18.2mω
·
cm)。
[0073]
在硅晶片上合成pt
‑
m(m=sb、bi、pb或te)、pd
‑
m(m=sb、bi、pb或te)、rh
‑
m(m=bi或te)、ni
‑
bi、co
‑
bi和双金属
‑
bi thh形状的颗粒:首先在60w下用氧等离子体处理一块硅晶片持续5分钟,随后以1000rpm(上升速率为500rpm/s)旋涂200μl所关注的金属前体的水溶液(混合物)(对于pt,10.75mg/ml h2ptcl6·
6h2o;对于pd,6.11mg/ml na2pdcl4;对于rh,7.97mg/ml na3rhcl6;对于ni,12.08mg/ml ni(no3)2·
6h2o;对于co,12.07mg/ml co(no3)2·
6h2o;对于cu,7.78mg/ml cu(no3)2·
xh2o;并且对于au,16.33mg/ml haucl4·
3h2o)持续60秒。在空气中干燥之后,将硅晶片放置在管式炉的中心。将装入燃烧舟中的大约1mg sb(或bi、pb和te)粉末放置在炉中上游的硅晶片附近。为方便起见,还可以将异金属粉末直接装载到si晶片上。对热处理进行如下编程:在h2气流下,在12分钟内上升到600℃,在600℃下保持10分钟,在1小时内冷却到25℃,将气氛切换到ar(或ar/h2混合物)。对于不同的组合,高温处理有所不同。(1)为了合成pt
‑
m(m=sb、bi、pb或te)、ni
‑
bi、co
‑
bi、ptni
‑
bi、ptco
‑
bi和ptcu
‑
bi颗粒:在20分钟内上升到900℃,在900℃下保持1小时。(2)为了合成pd
‑
bi、rh
‑
bi、pdpt
‑
bi、pdau
‑
bi、pdni
‑
bi、pdco
‑
bi、pdcu
‑
bi、rhpt
‑
bi、rhco
‑
bi和rhni
‑
bi颗粒:在20分钟内上升到1000℃,在1000℃下保持1小时。(3)为了合成pd
‑
m(m=sb、pb或te)和rh
‑
te颗粒:在4小时内上升到1200℃,在1200℃下保持1小时。对于所有组合,在完成热处理后将管淬火到室温(样品仍处于ar或ar/h2气氛中)。ar(或ar/h2混合物)的流速为200sccm。
[0074]
在硅晶片上替代性合成pt
‑
m(m=sb、bi、pb和te)、pd
‑
m(m=sb、bi、pb和te)和rh
‑
m(m=bi和te)截短的thh形状的颗粒:将装入燃烧舟中的大约1mg sb(或bi、pb和te)粉末放置在管式炉的中心处作为来源。首先在60w下用氧等离子体处理一块硅晶片持续5分钟,随后以1000rpm(上升速率为500rpm/s)旋涂h2ptcl6·
6h2o的水溶液(对于pd,na2pdcl4,并且对于rh,na3rhcl6)持续60秒。在空气中干燥之后,将硅晶片放置在炉的下游的金属粉末来源附近。对热处理进行如下编程:在h2气流下,在12分钟内上升到600℃,在600℃下保持10分钟,在1小时内冷却到25℃,将气氛切换到ar。高温处理彼此不同。(1)对于合成pt
‑
m(m= sb、bi、pb和te)颗粒:在20分钟内上升到900℃,在900℃下保持1小时。 (2)对于合成pd
‑
bi和rh
‑
bi颗粒:在20分钟内上升到1000℃,在1000℃下保持1小时。(3)对于合成pd
‑
m(m=sb、pb和te)和rh
‑
te颗粒:在4小时内上升到1200℃,在1200℃下保持1小时。在完成热处理后将管淬火到室温(样品仍处于ar气氛中)。
[0075]
合成均匀pd纳米立方体。基于文献方法但通过很小的改变来合成单分散的pd纳米立方体(niu等人,《晶体生长与设计(cryst.growth des.)》8,4440
‑
4444(2008))。(1)通过在搅拌的同时向20ml的12.5mm ctab溶液添加一等分试样的1ml的10mm h2pdcl4溶液来合成22nm pd边缘长度纳米立方体。在95℃下将所得溶液在油浴中加热5分钟。然后向混合物中添加一等分试样的160μl的100mm抗坏血酸溶液。在另外搅拌并在95℃下加热30分钟后,将溶液冷却到室温并且在不纯化的情况下用作用于生长较大pd纳米立方体的种子溶液。(2)
合成约50nm边缘长度的pd纳米立方体。将125μl的10mm h2pdcl4溶液和80μl的制备的种子溶液添加到5ml的100mm ctab中,随后添加50μl的100mm抗坏血酸溶液。通过涡旋混合器将所得溶液充分混合,并置于40℃的水浴中持续24小时。通过离心(在8000rpm下两个循环)收集纳米立方体,将其重悬于水中,并且然后分散在衬底上。在空气中干燥后,将所述纳米立方体用作thh形状调节实验的起始材料。形状调节处理与用于合成pd
‑
bi thh的处理相同。
[0076]
工作电极的制备。为了在炭黑上合成thh形状的pt
‑
m(m=sb、bi、pb或te)颗粒,对炭黑粉末和h2ptcl6·
6h2o(20wt.%pt装载)的5mg/ml水性混合物进行超声处理60分钟。在燃烧舟中在空气中干燥之后,将样品放置在管式炉的中心。将1mg m粉末装入燃烧舟中并放置在管中,在炭黑粉末和金属前体的混合物的上游。为方便起见,异金属粉末还可以与炭黑粉末和金属前体一起混合在燃烧舟中。热处理与在硅晶片上合成thh形状的pt
‑
m(m=sb、bi、pb或te)颗粒的处理相同。收集制备的样品并将其分散在水中(5mg/ml)。将5μl分散液转移到旋转盘电极(rde)(直径为3mm)上。在室温下干燥之后,将2μl的全氟磺酸溶液(0.5wt.%)沉积在电极表面上并使其干燥。
[0077]
工作电极的替代性制备。为了在炭黑上合成thh形状的pt
‑
m(m=sb、bi、pb和te)颗粒,将装在燃烧舟中的大约1mg sb(或bi、pb和te)粉末放置在管式炉的中心处作为来源。对炭黑粉末和h2ptcl6·
6h2o(20%pt装载)的5mg/ml水性混合物进行超声处理60分钟。在燃烧舟中在空气中干燥之后,将样品放置在管的中心,粉末来源的下游。热处理与在硅晶片上合成thh形状的pt
‑
m(m=sb、bi、pb和te)颗粒的处理相同。收集制备的样品并将其分散在水中(5mg/ml)。将5μl分散液转移到旋转盘电极(rde)(直径为3mm)上。在室温下干燥之后,将2μl的全氟磺酸溶液(0.5wt.%)覆盖在电极表面上并使其干燥。
[0078]
测量不同催化剂的电化学表面积(ecsa)和甲酸的电氧化反应活性。使用epsilon eclipse工作站在298k下的三电极玻璃槽中执行电化学测量。将盘绕的铂丝和ag/agcl电极分别用作对电极和参考电极。相对于可逆氢电极(rhe)校准所有电势。通过0.05v与0.4v之间的氢的吸附
‑
脱附来电化学测定ecsa,并假设pt表面上的单层吸附氢为210μc/cm2。在以50mv/s的扫描速率的ar气流下,在0.5m h2so4中执行循环伏安法(cv)测量。以50mv/s的扫描速率在0.5m h2so4+0.5m hcooh中执行对甲酸电氧化的所有测量。记录第二次扫描。
[0079]
替代性测量不同催化剂的电化学表面积(ecsa)和甲酸的电氧化反应活性。使用epsilon eclipse工作站在298k下的三电极玻璃槽中执行电化学测量。将盘绕的铂丝和ag/agcl电极分别用作对电极和参考电极。相对于可逆氢电极(rhe)校准所有电势。通过0.05v与0.4v之间的氢的吸附
‑
脱附来电化学测定ecsa,并假设pt表面上的单层吸附氢为210μc/cm2。在以50mv/s的扫描速率的ar气流下,在0.5m h2so4中执行循环伏安法(cv)测量。以50mv/s的扫描速率在0.5m h2so4+0.5m hcooh中执行对甲酸电氧化的所有测量。记录第二次扫描。
[0080]
表征。使用日立(hitachi)su
‑
8030场发射sem拍摄扫描电子显微镜(sem)图像。使用日立hd
‑
2300stem以200kv的加速电压拍摄扫描透射电子显微镜(stem)。使用赛默科技(thermo scientific)nss 2.3获得能量分散x射线光谱(eds)光谱和元素图。使用jeol arm 300f grandarm tem以300kv的加速电压拍摄hrtem图像。使用cu kα来源在理学(rigaku)ultima上收集x射线衍射(xrd)光谱。
[0081]
计算细节。使用利用投影增强波(paw)电势的维也纳从头算模拟包(vienna ab
‑
initio simulation package,vasp)和广义梯度近似(gga)的perdew
‑
burke
‑
ernzerhof(pbe)公式来执行所有密度函数理论(dft)计算(32
‑
34)。在此研究中使用了超晶胞近似法,其中通过沿垂直于表面的方向周期性排列的晶体板和真空区域对表面进行建模。为了将板的厚度保持在不小于为不同的表面选择不同数量的原子层(表1)。对于所有表面,真空区域的厚度为将中间三层固定,并且使剩余层放松。对于用于表示电子波函数的平面波基组,使用400ev的能量截止值。使用γ中心的k点网格对布里渊区(brillouin
‑
zone)积分进行采样(表1)。通过计算比表面能,其中为板的总能量,n
i
为板中的i原子数,μ
i
为元素i的化学势,并且a为表面积。
[0082]
实验结果
[0083]
综合参数的影响:thh形态的形成受反应温度、时间和sb粉末来源的量的影响(图5
‑
6)。当将1mg sb粉末用作来源时,在600℃下,甚至在反应12小时后,pt颗粒的形状也保持不规则。在800℃下反应开始时,颗粒表现出不规则形状。在反应30分钟后,观察到立方体颗粒。在热处理1
‑
12小时后,pt颗粒可能呈现thh形状,但这些颗粒的表面相当粗糙,其中在高指数平面上形成纳米级台面和台阶结构。在900℃下,反应开始时颗粒的形态也是不规则的。然而,在热处理1小时后,产生了具有光滑表面的thh形状的颗粒。这些观察结果表明,需要大约900℃的高温来产生具有光滑表面的thh形状的颗粒。经过另外3小时的热处理,原始的尖锐thh颗粒被破坏,其中边缘被截短并倾向于表现出更加球形的形状。在1000℃反应1小时后合成的thh形状的颗粒已经被轻微破坏,这表明用于在1000℃下产生高质量thh的时间窗口比在900℃下产生高质量thh的时间窗口减少。
[0084]
sb粉末来源的量还对thh形态的形成有重要影响。当sb量减少到0.1mg时,将形成thh形状的pt颗粒所需的时间缩短到30分钟。虽然如果每30分钟供应10mg sb粉末,但是甚至在反应1小时后,也没有观察到thh形状的pt颗粒。上述结果表明,当使用过量的sb粉末来源时,thh形状的形成将被延迟。然而据发现,在合成期间,sb是诱导pt thh形状形成所必需的:发现在不使用sb粉末的情况下在900℃下反应1小时之后合成的pt颗粒表现出由低指数小平面包围的不规则形状,并且未发现thh形状的pt颗粒(图7)。
[0085]
stem和eds结果(图8
‑
9)确认在反应开始时在900℃下观察到的不规则形状的颗粒为富含sb的ptsb合金。在到900℃的升温期间,熔化并蒸发sb,然后流动的ar气体将其携带到pt纳米颗粒位点。sb与pt之间的强相互作用导致形成ptsb合金化颗粒。随着反应的进行,颗粒保持经历形状重构,并且由于sb粉末来源的消耗,颗粒中的平均sb含量连续下降。在处理30分钟后,发现sb在颗粒表面上更加富集,并且开始形成thh形状。经过另外10分钟的退火,sb进一步从颗粒中释放,但维持thh形态。这些结果表明thh形状的pt颗粒的形成是通过从颗粒内提取过量的sb而引发的。
[0086]
本文合成的pt颗粒的形状并非完全尖锐的thh,但是被截角和截顶,其中{100}平面和{111}平面暴露。为了排除这些低指数小平面的存在是由于过度退火造成的可能性,通过将1mg bi粉末用作来源并将旋涂的样品加热到800℃(上升速率为10℃/min),随后将管在冰冷的水中淬灭来合成pt颗粒。根据文献结果,直径为大约200nm的thh形状的pt颗粒甚至被加热到约815℃(上升速率为7℃/min)也可以保持其形状(tian等人,《科学(science)》316,732
‑
735(2007))。然而,与文献报道相比,本文提供了展现出thh形状的pt颗粒已经被
截角和截顶,即使以较短的时间和较低的完工温度温和地进行热处理(图10)。此结果可以证明在合成过程期间没有产生完全尖锐的thh形状的颗粒。
[0087]
外部sb渗透对thh形态的影响:为了检查外部sb渗透对thh形态的影响,首先在硅晶片上合成截短的thh形状的pt颗粒(图11,a中)。在用1mg sb粉末来源在900℃下加热30分钟后,这些颗粒的thh形状被破坏(图11,b中)。另一个对照实验表明,如果没有另外的sb粉末来源,则在以其它方式进行相同处理后,截短的thh形状的pt颗粒截图保留其形态(图12)。这些观察结果证明与外部sb的合金化组织pt颗粒形成thh形状。根据前述时间序列的eds表征结果(图8
‑
9),预期在具有其thh形状的颗粒内过量的sb被破坏。因此,如果可以在900℃下提取sb,则颗粒仍应能够形成thh形态。为了证明这一点,在不使用额外的sb粉末来源的情况下,将同一样品在900℃下进一步加热30分钟,并且sem图像确认重新形成pt颗粒的thh形状(图11,c中)。处理后这些颗粒的移动和粗化不如在第一步中使用1mg sb粉末来源进行退火的颗粒的移动和粗化那样强烈。然而,在提取sb期间,颗粒的朝向稍微改变(图11,d
‑
e中)。
[0088]
{210}高指数平面的可逆重组:在合成thh形状的pt颗粒时,至关重要的是,在完成热处理后,通过从炉中去除管来将颗粒淬火到室温,以减少在较低温度范围内的停留时间。如果以5℃/min的速度将颗粒缓慢冷却到室温,则形成具有粗糙表面的thh形状的颗粒(图14)。此结果表明,在低于900℃的温度下退火之后,sb稳定的高指数平面不稳定并且将重组到纳米级台面和台阶结构中。为了进一步了解此现象,通过sem追踪在600℃下进行热退火后具有原子光滑{210}平面的thh形状的pt颗粒的形态演化(图15)。在退火处理后,出现了纳米级台面,并且{111}平面的大小缩小。pt族金属的高指数平面的吸附剂诱导的微刻面在表面科学中是众所周知的(ermanoski等人,《表面科学(surf.sci.)》596,89
‑
97(2005))。然而,报道的研究主要集中在单结晶贵金属表面与如o2、co等非金属气体之间的相互作用。本文公开了由金属元素sb诱导的pt纳米颗粒的形态变化。由于没有供应外部sb来源,因此在不旨在受理论的束缚的情况下,pt颗粒的表面重构主要是由于sb在pt表面上的重新分布。eds图还确认,在600℃下进行12小时的热退火后,颗粒内几乎没有组成变化(从pt
76
sb
24
到pt
79
sb
21
,原子组成)(图16)。可以通过在900℃下重新加热颗粒来重新形成原子光滑的{210}小平面(图14
‑
15)。还已经报道了pt(557)和pt(332)平面上的pt高指数小平面的这种可逆的表面重建(39),所述平面属于与在此项工作中的pt{210}平面的[001]区不同的[110]的结晶区。而且,报道的表面重构是由外部co气体诱导的,而在本研究中,可逆的pt颗粒表面结构是由内部sb的重新分布,即一种新颖的颗粒形状恢复过程导致的。
[0089]
dft计算:使用原子模拟环境(ase)构建表面板结构(图19),并且使用vesta(momma等人,《应用结晶学杂志(j.appl.crystallogr.)》44,1272
‑
1276(2011))可视化这些表面。异金属(sb、bi、pb和te)原子仅出现在所有表面结构中最顶部表面层上。在本文中,选择每原子总pt的总能量作为pt的化学势,即对于吸附在pt表面上的异金属(sb、bi、pb和te),需要限定其化学势。在本文中,以sb为例说明如何确定其化学势。在不旨在受理论束缚的情况下,为了研究表面稳定性对环境的依赖性,假设μ
sb
在热力学允许的化学势与之间变化,其中为每原子元素总sb的总能量,并且通过(为每原子化合物sb2pt3的总能量)与μ
pt
相关。根据来自开
放式量子材料数据库(oqmd)的凸包(saal等人,《金属杂志(jom)》65,1501
‑
1509(2013),kirklin等人,《npj计算材料学(npj comput.mater.)》1,15010(2015)),化合物sb2pt3在pt
‑
sb合金区域内具有最大百分比的sb。因此,将是sb的化学势的下限。类似地,获得了其它三种金属bi、pb和te的化学势范围(表2)。
[0090]
为了确认sb在稳定{210}小平面方面的能力,使用dft计算在sb修饰之前和之后的(210)平面和三个低指数小平面(100)、(111)和(110)的比表面能(图19
‑
20,表1
‑
4)。在sb修饰之前,这些小平面能量的计算结果与先前报道(nicholas等人,《今日物理(phys.today)》18,67(1965))一致:(111)平面具有最低的比表面能(1.519j/m2)并且(210)平面的能量在四个考虑的小平面中最高(1.900j/m2)。然而,在sb修饰之后,这些小平面的比表面能发生了急剧的变化,并且其顺序也发生了变化,其中pt(111)
‑
sb的能量现在最高(4.357j/m2),而pt(210)
‑
sb的能量最低(0.392j/m2)。
[0091]
在不旨在受理论束缚的情况下,优先形成pt{210}小平面还意味着这些小平面的比表面能在所有类型的高指数小平面中是最低的。在广泛用于说明晶体平面坐标的单位立体三角形中,(210)小平面定位于[001]晶体区中并且可以表示为2(100)
×
(110),这指示由原子宽度为(100)对称的台面构成的台阶表面由(110)对称的单原子台阶分离。使用dft,还计算了经sb修饰的pt的比表面能,(310)(=3(100)
×
(110))和pt(320)(=3(110)
×
(100))小平面,这些表示在[001]区具有过多(110)或(100)子表面的台阶平面。此外,选择经sb修饰的pt(221)和pt(211)的比表面能以进行比较,所述比表面能表示其它两个晶体区中的台阶平面。对于所有考虑的小平面,用异金属sb的不同表面覆盖率计算所述小平面的比表面能(图22
‑
25)。计算结果验证,被1单层(ml)的表面sb原子覆盖的pt(210)的比表面能最低,这支持了实验观察结果。此外,探索了sb化学势对这些小平面的比表面能的影响并确认在sb修饰后,pt(210)小平面始终具有最低比表面能(图26)。最后,以相同的方式对其它三种异金属(bi、pb和te)执行dft计算并确认在改性后,pt(210)小平面的比表面能在所考虑的小平面中仍然是最低的,无论异金属的类型如何。
[0092]
表1.每个表面板模型的原子层数和k点网格大小
[0093][0094]
表2:sb、te、bi和pb的化学势范围(ev/原子)
[0095][0096]
表3:不同pt小平面的比表面能
[0097][0098]
表4:不同经修饰的pt小平面的比表面能(j/m2)。每个元素的最低化学势用于计算比表面能。
[0099][0100]
电化学表征:催化剂表面上的甲酸电氧化通常遵循双重路径:1)直接脱氢路径和2)间接脱水路径(d.等人,《表面科学》59,195
‑
204(1976)和t.avanesian等人,《美国化学会志》139,4551
‑
4558(2017))。在约0.5v下的峰i对应于通过脱氢路径的甲酸氧化,并且在约0.9v下的峰ii对应于通过脱水路径形成的co
ads
氧化(图4,a中)。间接路径在商业pt/c催化剂的主导阻碍了所述催化剂在直接甲酸燃料电池中的应用。然而,合成的thh形状的pt
‑
bi催化剂有利于直接路径,几乎没有来自间接路径的贡献。与商业pt/c催化剂(r=0.5)相比,thh形状的pt
‑
pb(r=2.1)、pt
‑
sb(r=1.8)和pt
‑
te(r=1.1)催化剂的较高i
peak i
对i
peak ii
比率(r)还指示直接脱水路径对这些催化剂更有利。具体地,在0.5v下,pt
‑
bi(13.25ma/cm2)、pt
‑
pb(3.43ma/cm2)、pt
‑
sb(2.71ma/cm2)和pt
‑
te(2.26ma/cm2)催化剂的电流密度分别是商业pt/c催化剂(0.21ma/cm2)的电流密度的63倍、16倍、13倍和11倍。重要的是,不使用异金属元素合成的pt纳米颗粒表现出较差的催化活性(pt对照样品,在0.5v下为0.28ma/cm2),从而强调了源自形状控制和异金属修饰的催化效益。另外,为了区分形状控制与异金属修饰的重要性,通过重复性电化学沉积(q.s.chen等人,《美国化学会志》133,12930
‑
12933(2011))来修饰商业pt/c催化剂并评估所述催化剂的催化性质。在相同条件下,此材料在0.5v下表现出的电流密度为3.65ma/cm2,这是thh形状的pt
‑
bi催化剂的电流密度的四分之一(图29)。综上所述,除了异金属修饰之外,高指数小平面也是催化性能的主要因素。最后,在操作几分钟到一小时后,通常通过将稳态电流密度与来自商业pt/c催化剂的电流密度进行比较来评估催化剂的稳定性。安培i
‑
t曲线(图28)表明这些thh形状的催化剂是坚固的。在0.5v的固定电势下连续操作3600秒后,所测得的电流密度为:对于thh形状的pt
‑
bi为0.44ma/cm2、对于thh形状的pt
‑
pb为0.34ma/cm2、对于thh形状的pt
‑
sb为0.30ma/cm2并且对于thh形状的pt
‑
te为0.21ma/cm2(分别是商业pt/c催化剂的电流密度(0.02ma/cm2)的22倍、17倍、15倍和11倍)。在电化学测试后,合成后的thh形状的pt
‑
bi未观察到明显的形态变化,从而进一步确认催化剂的固有稳定性(图31)。为了进一步评估本文中thh催化剂的实际适用性,比较了其质量活性(图35,a和b中)。在0.5v的超电势下,thh pt
‑
bi(1.49a/mg)、pt
‑
pb(0.42a/mg)、pt
‑
te(0.22a/mg)和pt
‑
sb(0.21a/mg)催化剂的电流密度分
别是商业pt/c催化剂(0.07a/mg)的电流密度的21倍、6倍、3倍和3倍。重要的是,通过bi修饰从商业pt/c催化剂合成的thh pt催化剂(pt/c
‑
bi thh)在0.5v下表现出的电流密度为1.37a/mg,这是商业pt/c催化剂的电流密度的20倍。除pt之外,pd还被广泛用作甲酸氧化的电催化剂。通过bi修饰合成的pd thh颗粒在0.5v下表现出的电流密度为1.44a/mg,这是商业pd/c催化剂(0.47a/mg)的电流密度的3倍(图35,c和d中)。
[0101]
废催化剂:失活的商业pt/c催化剂样品在用作甲酸氧化的电催化剂后,通过将bi用作痕量元素形状指示金属利用此策略进行回收。同一区域中的stem图像确认,已经将颗粒转化到thh中(图32,a中和b中)。此外,在回收之后,催化剂的性能显著提高;在0.5v的超电势下,催化剂的电流密度在回收前和回收后为0.09ma/cm2(回收之前)和12.65ma/cm2(回收之后)(图32,c中)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1