一种苝酰亚胺及其衍生物的还原态离子盐及制备方法与流程
本发明涉及一种有机物及其电化学制备方法。
背景技术:
:
还原态苝酰亚胺由于具有近红外吸收特性和高的电子迁移率在半导体材料和光电子应用方面具有潜在的价值和光明的前景。
目前制备环境稳定的还原态苝酰亚胺及其衍生物的资料相对较少,而环境不稳定的苝酰亚胺的离子盐的制备工艺仅仅局限于,用硫代硫酸钠的还原、电化学还原和C/Pd-H2还原、电化学还原,并且这些制备工艺对原料和合成条件都有苛刻的要求,且在真空氮气保护条件下才能应用于科学研究;空气稳定的苝酰亚胺的离子盐的制备工艺仅仅局限于80℃条件下的碳酸钾还原。
技术实现要素:
:
本发明的目的在于提供一种产品性能稳定且制备工艺条件温和、简单、成本低、适于工业化的苝酰亚胺及其衍生物的还原态离子盐及制备方法。本发明主要是采用3,4,9,10-苝四酸二酐为原料,通过溴化反应、酰胺化反应、亲核取代反应合成了具有低LUMO能级、优异光电性能的苝酰亚胺类衍生物;然后依据电化学还原法较容易地合成空气稳定的苝酰亚胺自由基阴离子盐。
一、本发明的化合物苝酰亚胺及其衍生物的还原态离子盐具有如下结构通式(1)
其中:R1为烷基,R2、R3为溴或苯氧基或芳香基,R4是代表四丁基六氟磷酸铵,作稳定剂.
二、上述苝酰亚胺及其衍生物的还原态离子盐的制备方法具体如下:
(1)溴化反应合成中间体M1-1,2:
采用3,4,9,10-苝四酸二酐为原料,将1当量3,4,9,10-苝四羧酸基二酐(PTDA)加入到盛有40-80当量浓H2SO4(1.836g/mL)的容器中,经35Hz超声波震荡10min溶解,再将其置于集热式恒温磁力搅拌器上并加冷凝装置,室温下搅拌6h后,将温度调至80℃,回流反应,其间加入0.5-10当量I2作为催化剂并用恒压滴液漏斗向其内缓慢滴加1.2-12当量液溴。待反应24h后,用蒸馏水将浓硫酸反应液稀释到60%后抽滤得到滤饼,将滤饼放在90℃真空干燥箱中干燥24h。滤饼中主要含有a,b,c三种化合物其中a:b:c=7:2:1。此产物M1由于溶解性的问题,可不需要进一步提纯,直接进行合成反应下一步。
(2)酰胺化反应合成中间体M2:
上述反应方程式是以含量多的中间体M1为代表的。
a R1为支链烷基
分别称取步骤(1)获得的1当量(物质的量)溴代苝四酸酐,1-10当量催化剂冰醋酸,2-5当量异辛胺加入到盛有30-60当量N-甲基吡咯烷酮(NMP)的容器中,在氩气保护下,在80℃反应约6小时。并且反应过程中,用CH2C12点板观察反应程度。待反应结束后,放置至室温,然后将产物倒入装有200mL去离子水的烧杯中,析出暗红色絮状沉淀,然后抽滤,用甲醇洗涤,将得到的滤饼于80℃真空干燥得深红色固体,最后用二氯甲烷:石油醚(v:v)=5:1或4:1作洗脱剂硅胶柱层析纯化得红色粉末M2-1。
b R1为环形烷基
分别称取步骤(1)获得的1当量(物质的量)溴代苝四酸酐,1-12当量催化剂冰醋酸,2-10当量环己胺加入到盛有30-60当量N-甲基吡咯烷酮(NMP)容器中,在氩气保护下,在80℃反应约4.5小时。并且反应过程中,用CH2C12点板观察反应程度。待反应结束后,放置至室温,然后将产物倒入装有200mL去离子水的烧杯中,析出暗红色絮状沉淀,然后抽滤,用甲醇洗涤,将得到的滤饼于80℃真空干燥得深红色固体,最后用乙酸乙酯:石油醚(v:v)=5:1或1:0作洗脱剂硅胶柱层析纯化得红色粉末M2-2。
(3)亲核取代反应合成苝酰亚胺类衍生物中间体M3:
a M2-1及M2-2与五氟苯酚反应
取步骤(2)获得的1当量的中间体M2,0.8-5当量的三乙胺和1-3当量五氟苯酚加入到盛有30-60当量除水的氮氮二甲基甲酰胺(DMF)的反应容器中。氩气保护下缓慢升温至80℃。TLC检测反应进程,至走板原料点消失停止反应。一般反应4.5h结束,减压(真空度小于133Pa)蒸馏,在80℃时除去DMF,然后用二氯甲烷:石油醚(v:v)=3:1或4:1溶解,并用其作洗脱剂,硅胶柱层析分离提纯,得到最终粉红色产物M3-1,M3-2.
b M2-1及M2-2与硼酸酯反应
取步骤(2)获得的1当量的中间体M2,1-6当量的无水碳酸钾和2-5当量硼酸酯,2-10当量四三苯基磷钯作为Suzuki反应的催化剂加入到盛有30-60当量除水的THF(四氢呋喃)的反应容器中。氩气保护下缓慢升温至80℃。TLC检测反应进程,至走板原料点消失停止反应。一般反应8h结束,减压(真空度小于133Pa)蒸馏,在45℃时除去THF(四氢呋喃),然后用乙酸乙酯:石油醚(v:v)=3:1溶解,并用其作洗脱剂,硅胶柱层析分离提纯,得到最终粉红色产物M3-3,M-4.
(4)终产物M的制备:M3-1,2,3,4的电化学还原
将1当量的中间体M3,1-10当量的四丁基六氟磷酸铵加入到盛有DMF的容器中。常温条件下,将恒压点位的两个电极插入容器中。当施加电位为中间体的循环伏安曲线显示的第一还原电位时,在阴极电位处的溶液由粉红色全部变成绿色,并用紫外吸收光谱检测本征态吸收峰消失时停止电化学还原,减压蒸馏除去DMF,然后用四氢呋喃:石油醚(v:v)=3:1溶解,并用其作洗脱剂,进行柱层析分离提纯,最终得到暗红色产物苝酰亚胺自由基阴离子盐M-1。
本发明与现有技术相比具有如下优点:
1、本发明的化合物具有相当好的空气稳定性,如图3所示,并且在光激发过程中,在近红外区域具有低能量的电子跃迁以及荧光淬灭。
2、本发明的化合物虽处于还原态,但对空气具有很强的稳定性。
3、本发明设计了一种简单、新颖的制备方法,即电化学还原法对在机溶剂体系中的苝酰亚胺进行还原得到环境稳定的苝酰亚胺离子盐。此方法,对氧气和有机溶剂中的水分无严格要求,后处理提纯简单,并且制备出的离子盐的溶液在暴露空气中的条件下至少能稳定存在一个月。
4、本发明中制备的还原态化合物所用的原料易得,价格便宜,反应产率高且可调控,范围为10.7%-52.9%,具有工业化价值,
附图说明
图1是本发明的苝酰亚胺及其衍生物的自由基阴离子的紫外-可见光吸收光谱图。
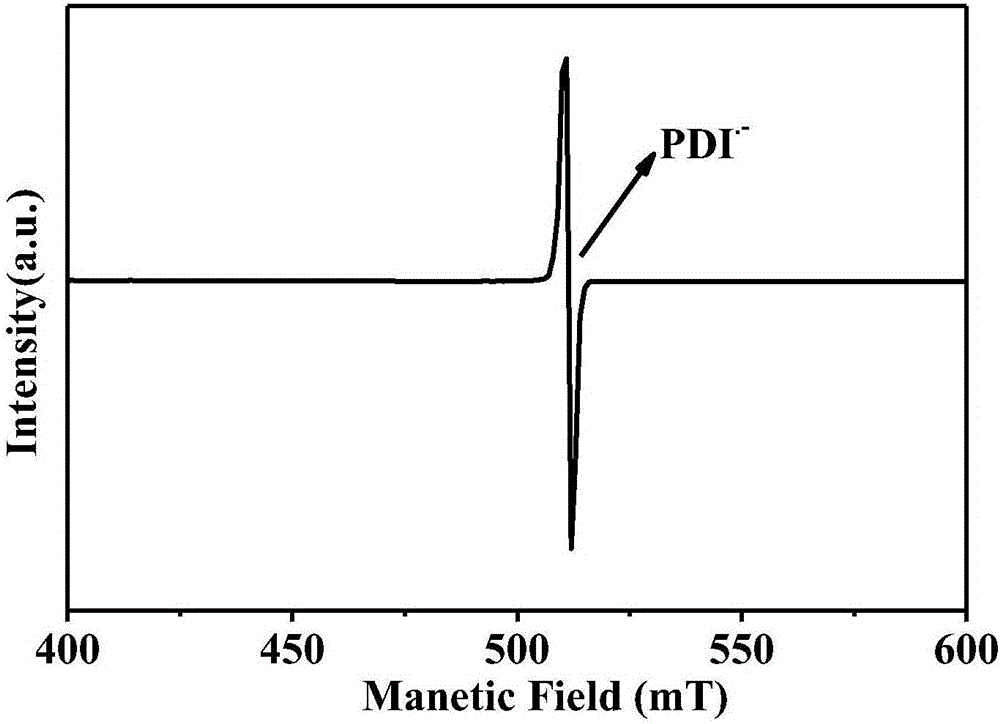
图2是本发明的苝酰亚胺及其衍生物自由基阴离子的顺磁共振谱图。
图3是本发明的苝酰亚胺及其衍生物自由基阴离子的紫外吸收光谱图。
从图1所示的本发明的苝酰亚胺及其衍生物自由基阴离子的紫外-可见光吸收光谱图中可以看出,苝酰亚胺被还原后,本征态特征吸收峰500nm-600nm消失,苝酰亚胺还原态一价离子盐的特征吸收峰700-800nm出现。
从图2所示的本发明的苝酰亚胺及其衍生物自由基阴离子的顺磁共振谱图中可以看出,很明显的峰表明顺磁物种的产生。
从图3所示的本发明的苝酰亚胺及其衍生物自由基阴离子的在空气中放置后的紫外吸收光谱,从图可以看出,其具有很强的空气稳定性。
具体实施方式
下面给出实施例以对本发明进行具体描述,有必要在此指出的是以下实施例只用于对本发明进行进一步的说明,不能理解为对本发明保护范围的限制,本领域的技术熟练人员根据上述本发明的内容对本发明做出的一些非本质的改进和调整仍属于本发明的保护范围。
实施例1
将2.14g的3,4,9,10-苝四羧酸基二酐(PTDA)(M=392)加入到盛有18mL浓(98%)H2SO4(M=98,1.836g/mL)的三口烧瓶中,经35Hz超声波震荡10min,再将其置于集热式恒温磁力搅拌器上并加冷凝装置,室温下搅拌6h后,将温度调至80℃,回流反应,其间加入0.053g I2(M=127)作为催化剂并用恒压滴液漏斗向其内缓慢滴加液溴(M=160)约0.577mL(3.12g/mL)。待反应24h后,用蒸馏水将反应液浓硫酸稀释到60%后抽滤得到滤饼,将滤饼放在90℃真空干燥箱中干燥24h。滤饼中主要含有前面所述的a,b,c三种化合物其中a:b:c=7:2:1。
在100ml三口反应烧瓶中,分别加入0.89g由上述步骤制得的溴代苝四酸酐(M=550),0.42mL 17.5mol/L的冰醋酸(M=60)和0.63g异辛胺(M=129)加入到盛有13mL N-甲基吡咯烷酮(M=99,1.028g/mL)的三口反应烧瓶中,在氩气保护下,在80℃反应约6小时。并且反应过程中,用CH2C12点板观察反应程度。待反应结束后,放置至室温,然后将产物倒入装有去离子水的200mL的烧杯中,析出暗红色絮状沉淀,然后抽滤,用甲醇洗涤,将得到的滤饼于80℃真空干燥得深红色固体,最后用二氯甲烷:石油醚(v:v)=5:1作洗脱剂硅胶柱层析纯化得红色粉末M2。
将0.718g的中间体M2,0.03mL的三乙胺(0.73g/mL)和0.58g五氟苯酚(M=184)加入到14mL盛有除水的DMF(M=73)的三口圆底反应瓶中。氩气保护下缓慢升温至80℃。TLC检测反应进程,至走板原料点消失停止反应。反应4.5h结束,80℃减压(真空度小于133Pa)蒸馏除去DMF,然后用二氯甲烷:石油醚(v:v)=3:1溶解,并用其作洗脱剂,柱层析分离提纯,得到粉红色产物M3。
将0.38g的中间体M3,0.59g的四丁基六氟磷酸铵加入到盛有13mL DMF的容器中。常温条件下,将恒压点位的两个电极插入容器中。当施加电位为中间体的循环伏安曲线显示的第一还原电位时,在阴极电位处的溶液由粉红色全部变成绿色,并用紫外吸收光谱检测本征态吸收峰消失时停止电化学还原,减压蒸馏除去DMF,然后用四氢呋喃:石油醚(v:v)=3:1溶解,并用其作洗脱剂,进行柱层析分离提纯,得到最终暗红色产物得到终产物苝酰亚胺自由基阴离子M-1,得到暗红色产物,最终产率36.3%.
实施例2
将2.09g的3,4,9,10-苝四羧酸基二酐(PTDA)(M=392)加入到盛有18mL浓(98%)H2SO4(M=98,1.836g/mL)的三口烧瓶中,经35Hz超声波震荡10min,再将其置于集热式恒温磁力搅拌器上并加冷凝装置,室温下搅拌6h后,将温度调至80℃,回流反应,其间加入0.052g I2(M=127)作为催化剂并用恒压滴液漏斗向其内缓慢滴加液溴(M=160)约0.58mL(3.12g/mL)。待反应24h后,用蒸馏水将反应液浓硫酸稀释到60%后抽滤得到滤饼,将滤饼放在90℃真空干燥箱中干燥24h。滤饼中主要含有前面所述的a,b,c三种化合物其中a:b:c=7:2:1。
在100ml三口反应烧瓶中,分别加入0.91g由上述步骤制得的溴代苝四酸酐(M=550),0.42mL 17.5mol/L的冰醋酸(M=60)和0.633g异辛胺(M=129)加入到盛有13mL N-甲基吡咯烷酮(M=99,1.028g/mL)的三口反应烧瓶中,在氩气保护下,在80℃反应约6小时。并且反应过程中,用CH2C12点板观察反应程度。待反应结束后,放置至室温,然后将产物倒入装有去离子水的200mL的烧杯中,析出暗红色絮状沉淀,然后抽滤,用甲醇洗涤,将得到的滤饼于80℃真空干燥得深红色固体,最后用二氯甲烷:石油醚(v:v)=4:1作洗脱剂硅胶柱层析纯化得红色粉末M2。
将0.681g的中间体M2,0.03mL的三乙胺(0.73g/mL)和0.703g五氟苯酚(M=184)加入到14mL盛有除水的DMF(M=73)的三口圆底反应瓶中。氩气保护下缓慢升温至80℃。TLC检测反应进程,至走板原料点消失停止反应。反应4.5h结束,80℃减压(真空度小于133Pa)蒸馏除去DMF,然后用二氯甲烷:石油醚(v:v)=3:1溶解,并用其作洗脱剂,柱层析分离提纯,得到粉红色产物M3。
将0.365g的中间体M3,0.512g的四丁基六氟磷酸铵加入到盛有12mL DMF的容器中。常温条件下,将恒压点位的两个电极插入容器中。当施加电位为中间体的循环伏安曲线显示的第一还原电位时,在阴极电位处的溶液由粉红色全部变成绿色,并用紫外吸收光谱检测本征态吸收峰消失时停止电化学还原,减压蒸馏除去DMF,然后用四氢呋喃:石油醚(v:v)=3:1溶解,并用其作洗脱剂,进行柱层析分离提纯,得到最终暗红色产物得到终产物苝酰亚胺自由基阴离子M-1,得到暗红色产物,最终产率37.3%.
实施例3
将1.98g的3,4,9,10-苝四羧酸基二酐(PTDA)加入到盛有18mL浓(98%)H2SO4的三口烧瓶中,经35Hz超声波震荡10min,再将其置于集热式恒温磁力搅拌器上并加冷凝装置,室温下搅拌6h后,将温度调至80℃,回流反应,其间加入0.052g I2作为催化剂并用恒压滴液漏斗向其内缓慢滴加液溴约0.58mL。待反应24h后,用蒸馏水将反应液浓硫酸稀释到60%后抽滤得到滤饼,将滤饼放在90℃真空干燥箱中干燥24h。滤饼中主要含有前面所述的a,b,c三种化合物其中a:b:c=7:2:1。
在100ml三口反应烧瓶中,分别加入0.95g由上述步骤制得的溴代苝四酸酐,0.52mL17.5mol/L的催化剂冰醋酸,0.59g异辛胺加入到盛有15mL N-甲基吡咯烷酮的三口反应烧瓶中,在氩气保护下,在80℃反应约6小时。并且反应过程中,用CH2C12点板观察反应程度。待反应结束后,放置至室温,然后将产物倒入装有去离子水的200mL的烧杯中,析出暗红色絮状沉淀,然后抽滤,用甲醇洗涤,将得到的滤饼于80℃真空干燥得深红色固体,最后用二氯甲烷:石油醚(v:v)=5:1作洗脱剂硅胶柱层析纯化得红色粉末M2。
将0.83g的中间体M2,0.41g硼酸酯和0.46g四三苯基磷钯,0.69g无水碳酸钾加入到盛有除水的12mL THF(四氢呋喃)的三口圆底反应瓶中。氩气保护下缓慢升温至80℃。TLC检测反应进程,至走板原料点消失停止反应。反应8h结束,45℃减压(真空度小于133Pa)蒸馏除去THF(四氢呋喃),然后用乙酸乙酯:石油醚(v:v)=3:1溶解,并用其作洗脱剂,柱层析分离提纯,得到粉红色产物M3。
将0.41g的中间体M3,0.51g的四丁基六氟磷酸铵加入到盛有14mL DMF的容器中。常温条件下,将恒压点位的两个电极插入容器中。当施加电位为中间体的循环伏安曲线显示的第一还原电位时,在阴极电位处的溶液由粉红色全部变成绿色,并用紫外吸收光谱检测本征态吸收峰消失时停止电化学还原,减压蒸馏除去DMF,然后用四氢呋喃:石油醚(v:v)=3:1溶解,并用其作洗脱剂,进行柱层析分离提纯,得到最终暗红色产物得到终产物苝酰亚胺自由基阴离子M-1,得到暗红色产物,最终产率38.3%.
实施例4
将2.021g的3,4,9,10-苝四羧酸基二酐(PTDA)加入到盛有17mL浓(98%)H2SO4的三口烧瓶中,经35Hz超声波震荡10min,再将其置于集热式恒温磁力搅拌器上并加冷凝装置,室温下搅拌6h后,将温度调至80℃,回流反应,其间加入0.052g I2作为催化剂并用恒压滴液漏斗向其内缓慢滴加液溴约0.59mL。待反应24h后,用蒸馏水将反应液-浓硫酸-稀释到60%后抽滤得到滤饼,将滤饼放在90℃真空干燥箱中干燥24h。滤饼中主要含有前面所述的a,b,c三种化合物其中a:b:c=7:2:1。
称取上面步骤获得的0.943g中间体M1,0.46mL冰醋酸和0.515g环己胺加入到盛有41mL N-甲基吡咯烷酮三口烧瓶中,在氩气保护下,在80℃反应4.5小时。并且反应过程中,用CH2C12点板观察反应程度。待反应结束后,放置至室温,然后将产物倒入装有去离子水的200mL的烧杯中,析出暗红色絮状沉淀,然后抽滤,用甲醇洗涤,将得到的滤饼于80℃真空干燥得深红色固体,最后用乙酸乙酯作洗脱剂硅胶柱层析纯化得红色粉末M2。
称取上面步骤获得的0.859g中间体M2,0.05mL三乙胺和0.67g五氟苯酚加入到40mL盛有除水的DMF的三口圆底反应瓶中。氩气保护下缓慢升温至80℃。TLC检测反应进程,至走板原料点消失停止反应。反应4.5h结束,80℃减压(真空度小于133Pa)蒸馏除去DMF,然后用二氯甲烷:石油醚(v:v)=3:1溶解,并用其作洗脱剂,柱层析分离提纯,得到粉红色产物M3。
将0.358g的中间体M3,0.519g的四丁基六氟磷酸铵加入到盛有13mL DMF的容器中。常温条件下,将恒压点位的两个电极插入容器中。当施加电位为中间体的循环伏安曲线显示的第一还原电位时,在阴极电位处的溶液由粉红色全部变成绿色,并用紫外吸收光谱检测本征态吸收峰消失时停止电化学还原,减压蒸馏除去DMF,然后用四氢呋喃:石油醚(v:v)=3:1溶解,并用其作洗脱剂,进行柱层析分离提纯,得到最终暗红色产物得到终产物苝酰亚胺自由基阴离子M-1,得到暗红色产物,最终产率35.3%.
- 还没有人留言评论。精彩留言会获得点赞!