一种电化学选择性脱氯制备4-氨基-3,6-二氯吡啶甲酸的方法与流程
本发明涉及一种电化学选择性脱氯制备4-氨基-3,6-二氯吡啶甲酸的方法。
(二)
背景技术:
4-氨基-3,6-二氯吡啶甲酸,商品名为胺草定、氯氨吡啶酸、二氯氨基吡啶酸,是一种吡啶羧酸类除草剂,它能迅速进入植物体内,从而导致植物生长中断并迅速死亡,主要用于牧场、种植园和非农作物区的杂草控制。另外,4-氨基-3,6-二氯吡啶甲酸还是氟氯吡啶酯和氯氟吡啶酯合成的关键中间体。氟氯吡啶酯和氯氟吡啶酯是陶氏益农公司开发的新型芳基吡啶甲酸酯类除草剂,是激素类除草剂中的新品种,具有药量更低,杀草谱更广的特点。
美国专利us7666293、us8685222公布了4-氨基-3,5,6-三氯吡啶甲酸电化学选择性脱氯制备4-氨基-3,6-二氯吡啶甲酸的方法。该方法以无隔膜电解槽为反应器,hastelloyc为阳极材料,活化银网为阴极材料,碱性水溶液为电解液,可以得到较高收率的4-氨基-3,6-二氯吡啶甲酸。该方法的问题是,部分原料和产物会在hastelloyc电极上或者银电极活化过程中氧化,从而使产品纯度下降,色泽变红。为了解决该问题,中国专利201611135958公布了4-氨基-3,5,6-三氯吡啶甲酸电化学选择性脱氯制备4-氨基-3,6-二氯吡啶甲酸的方法。该方法以隔膜电解槽为反应器,316不锈钢为阳极材料,银为阴极材料,碱性水溶液为电解液,可以得到较高收率的4-氨基-3,6-二氯吡啶甲酸。该方法能够避免原料和产物与阳极材料接触从而避免产品纯度下降,色泽变红。该方法的问题是,银电极的脱氯活性和选择性很难保证和维持。
(三)
技术实现要素:
本发明目的是提供一种4-氨基-3,5,6-三氯吡啶甲酸(i)电化学高选择性脱氯制备4-氨基-3,6-二氯吡啶甲酸(ii)的方法,采用电子级超纯水配置脱氯用电解液、以高纯的银网为阴极(银的纯度≥99.95%)、银电极活化前用盐酸浸泡可以显著改善活性银电极的脱氯活性和选择性。
本发明采用的技术方案是:
本发明提供一种电化学选择性脱氯制备4-氨基-3,6-二氯吡啶甲酸的方法,所述方法为:采用隔膜电解槽,以电子级超纯水配制的4-氨基-3,5,6-三氯吡啶甲酸(i)盐水溶液为阴极液,以电子级超纯水配制的碱金属氢氧化物水溶液为阳极液,以镍基材料为阳极,以纯度99.95%-99.99%的银为阴极;在电流密度为1~20a/dm2、反应温度为0~90℃的条件下进行电解反应,电流从阳极穿过阳极液、隔膜和阴极液最后到达阴极,达到所需通电量后停止通电,电解反应完全后,阴极液分离纯化得到4-氨基-3,6-二氯吡啶甲酸(ii);所述配制阴极液的电子级超纯水为达到gb/tii446.1-2013(ew-ii)规定的水,并且其中钴、锡、铋、镉、汞离子的单一含量不超过1微克/升;所述配制阳极液的电子级超纯水为达到gb/tii446.1-2013(ew-iii)规定的水,并且其中钴、锡、铋、镉、汞离子的单一含量不超过5微克/升。
所述阳极液中碱金属氢氧化物为naoh、koh,优选naoh。所述阴极液中4-氨基-3,5,6-三氯吡啶甲酸浓度为0.2~0.8m;所述阳极液中碱金属氢氧化物浓度为1.0~2.0m。
所述镍基材料,包括哈氏合金、314不锈钢、316不锈钢、纯镍等,优选哈氏合金(hastalloyc-276)。所述的隔膜为阳离子膜,优选全氟磺酸阳离子膜。
所述电流为直流电,电流密度优选5~10a/dm2,反应温度优选35~50℃,更优选45-50℃。
所述阴极为活化银,所述的活化银是将银先用盐酸水溶液浸泡,然后用氧化还原法制得,具体如下:(1)浸泡:在质量浓度5~35wt%盐酸水溶液中将新鲜的银或者使用过的银浸泡5~300min,浸泡温度为20~50℃,获得浸泡后的银;(2)氧化:在以nafion117阳离子膜为隔膜的h型电解槽(如图1所示)中,以含有氯离子或溴离子的水溶液为工作电极液,以1mnaoh水溶液为对电极液,以步骤(1)浸泡后的银为工作电极,相同面积的铂片为对电极,银/氯化银为参比电极,在电流密度为0.1~5a/dm2(优选0.5~2a/dm2)、温度为0~50℃(优选20~25℃)条件下氧化至有氧气析出为止(优选电极电位到达+0.5vs.ag/agcl);(3)还原:以步骤(2)银为对电极,铂片为工作电极,其他条件相同,还原至有氢气析出为止(优选电极电位到达-0.5vs.ag/agcl),获得活化银。
更优选步骤(1)所述银为新的银网或使用过的银网,本体银含量≥99.95wt%,优选≥99.99wt%。步骤(1)所述的盐酸水溶液的浓度为10~15wt%,温度为20~25℃,浸泡时间为20~60min;步骤(2)所述含有氯离子或溴离子的水溶液为0.1~1.0mnacl+0~1.0mnaoh的水溶液,更优选0.1nacl+1.0mnaoh水溶液,或1nacl水溶液。如果是新的银网,上述氧化还原过程可以重复2次。所需产品通过常规技术回收。例如,通过酸化然后过滤或用不与水混溶的有机溶剂萃取。
与现有技术相比,本发明的有益效果主要体现在:(1)本发明采用电子级超纯水配制的阴极液和阳极液,能够高选择性脱氯制备4-氨基-3,6-二氯吡啶甲酸,4-氨基-3,6-二氯吡啶甲酸产品收率(上升4-10%)和纯度更高(上升2-5%);(2)采用活化银电极为阴极,脱氯活性(使用400分钟后,转化率高出6%)和选择性维持更久(使用400分钟后,选择性高出4.5%)。
(四)附图说明
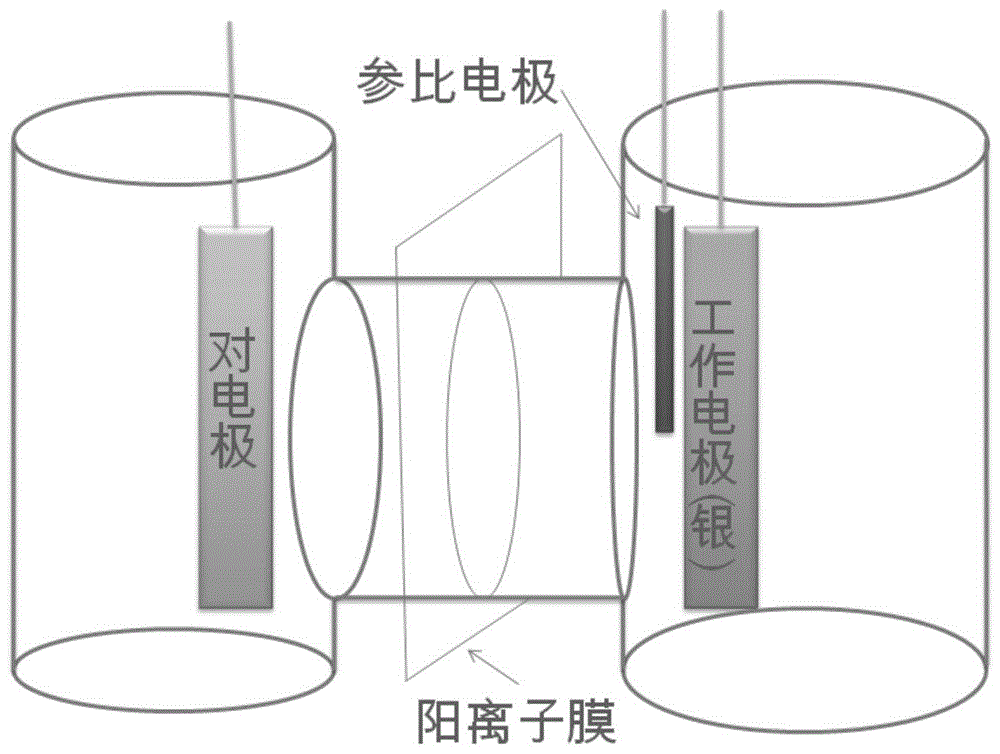
图1为以nafion117阳离子膜为隔膜的h型电解槽示意图,阴阳极之间距离约为8cm,离子膜居中放置,离子膜的面积3.14×2×2=12.56cm2。
(五)具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
下述所有实施例和对比例中,所述配制阴极液的电子级超纯水为达到gb/tii446.1-2013(ew-ii)规定要求的水,并且其中钴、锡、铋、镉、汞离子的单一含量不超过1微克/升;所述配制阳极液的电子级超纯水为达到gb/tii446.1-2013(ew-iii)规定要求的水,并且其中钴、锡、铋、镉、汞离子的单一含量不超过5微克/升;所述去离子水为达到gb/tii446.1-2013(ew-iv)规定要求的水,并且其中钴、锡、铋、镉、汞离子的单一含量不超过200微克/升。
实施例1:活化银电极的制备
(1)浸泡:将银网(纯度为99.99wt%,尺寸为0.3cm×4.0cm×6.0cm)浸泡在静止的10wt%盐酸水溶液(用去离子水配制),20℃浸泡20min,获得浸泡后的银网;
(2)氧化:在以nafion117阳离子膜为隔膜的h型电解槽(如图1所示)中,步骤(1)银网为工作电极,相同面积的铂片为对电极;银/氯化银为参比电极。工作电极室为0.1mnacl+1.0mnaoh水溶液(用去离子水配制),对电极室为1.0m氢氧化钠水溶液(用去离子水配制)。控制工作电极室水溶液的温度为20~25℃,首先对银电极施加0.5a/dm2的阳极氧化电流直至电极电位到达+0.5vs.vs.ag/agcl;
(3)还原:以步骤(2)中银网为对电极,铂片为工作电极,其他同步骤(2),然后对银网施加0.5a/dm2的阴极还原电流直至电极电位到达-0.5vs.ag/agcl。取出银网即为银电极,置于去离子水中备用。
实施例2:活化银电极的制备
(1)浸泡:将纯度分别为99.95wt%(记为银网1#)和99.0wt%(记为银网2#),尺寸均为0.3cm×4.0cm×6.0cm的银网,分别浸泡在15wt%盐酸水溶液(用去离子水配制)中,25℃静置60min;
(2)氧化:在以nafion117阳离子膜为隔膜的h型电解槽中(图1),分别以步骤(1)银网1#和银网2#为工作电极,相同面积的铂片为对电极;银/氯化银为参比电极。工作电极室为1.0mnacl水溶液(用去离子水配制),对电极室为1.0m氢氧化钠水溶液(用去离子水配制)。控制工作电极室水溶液的温度为20~25℃,首先对银网施加2a/dm2的阳极氧化电流直至电极电位到达+0.5vs.ag/agcl;
(3)将步骤(2)中银网作为对电极,铂片作为工作电极,其他条件相同,然后对银网施加2a/dm2的阴极还原电流直至电极电位到达-0.5vs.ag/agcl。取出银网,即为活化银网,置于去离子水中备用。
实施例3:活化银电极的制备(盐酸不浸泡)
(1)氧化:在以nafion117阳离子膜为隔膜的h型电解槽(图1)中,银网(纯度为99.95wt%,尺寸为0.3cm×4.0cm×6.0cm)为工作电极;相同面积的铂片为对电极;银/氯化银为参比电极。工作电极室为0.1mnacl+1.0mnaoh水溶液(用去离子水配制),对电极室为1.0m氢氧化钠水溶液(用去离子水配制)。控制工作电极室的水溶液的温度为20~25℃,首先对银网施加0.5a/dm2的阳极氧化电流直至电极电位到达+0.5vs.vs.ag/agcl;
(2)还原:将步骤(1)中银网作为对电极,铂片作为工作电极,其他条件相同,然后对银网施加0.5a/dm2的阴极还原电流直至电极电位到达-0.5vs.ag/agcl。取出银网,即为活化银网,置于去离子水中备用。
实施例4:电化学脱氯合成4-氨基-3,6-二氯吡啶甲酸(电子级超纯水配制溶液)
在以nafion117阳离子膜为隔膜的h型电解槽(图1)中,以实施例1方法制备的活化银网为阴极(工作电极),相同面积的hastalloyc-276片(几何尺寸为0.5cm×4.0cm×6.0cm)为阳极(对电极)。300ml1.0mnaoh水溶液为阳极液(用电子级超纯水配制),300ml0.2m4-氨基-3,5,6-三氯吡啶甲酸钠水溶液(用电子级超纯水配制)为阴极液,ph=12.5。在35-40℃下,搅拌阴极液,通入5a/dm2的电流,反应200分钟后停止电解。用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为98.5%,4-氨基-3,6-二氯吡啶甲酸收率为96.4%。首先,在85~90℃下用质量浓度36%浓盐酸将阴极液ph调到1,过夜自然冷却结晶;然后,对析出晶体进行过滤、去离子水水洗、80℃下烘干处理,得到雪白色晶体;最后,用高效液相色谱测定白色晶体中4-氨基-3,6-二氯吡啶甲酸的纯度为98.6%。
所述高效液相色谱测定条件为:c18对称柱(250mmlength_4.6mmi.d.,5mmparticlesize)为分离柱;含有30mm磷酸的乙腈/甲醇/水(体积比1:3:6)混合溶液为流动相;流速为:1ml/min;检测波长为230nm;waters2996pda为检测器。
实施例5:电化学脱氯合成4-氨基-3,6-二氯吡啶甲酸(电子级超纯水配制溶液)
在以nafion117阳离子膜为隔膜的h型电解槽(图1)中,以实施例2方法制备的1#活化银网为阴极,相同面积的hastalloyc-276片(几何尺寸为0.5cm×4.0cm×6.0cm)为阳极。300ml2.0mkoh水溶液为阳极液(用电子级超纯水配制),300ml0.8m4-氨基-3,5,6-三氯吡啶甲酸钾水溶液(用电子级超纯水配制)为阴极液,ph=12.7。在45-50℃下,搅拌阴极液,通入10a/dm2的电流,反应400分钟后停止电解。采用实施例4方法用高效液相色谱测定4-氨基-3,5,6-三氯吡啶甲酸的转化率为98.2%,4-氨基-3,6-二氯吡啶甲酸收率为94.1%。首先,在85~90℃下用质量浓度36%浓盐酸将阴极液ph调到1,过夜自然冷却结晶;然后,对析出晶体进行过滤、去离子水水洗、80℃下烘干处理,得到雪白色晶体;最后,用高效液相色谱测定白色晶体中4-氨基-3,6-二氯吡啶甲酸的纯度为98.3%。
实施例6:电化学脱氯合成4-氨基-3,6-二氯吡啶甲酸(重复使用实施例5的银电极)
在以nafion117阳离子膜为隔膜的h型电解槽(图1)中,以实施例5使用完的银网为阴极,相同面积的hastalloyc-276片(几何尺寸为0.5cm×4.0cm×6.0cm)为阳极。300ml2.0mkoh水溶液为阳极液(用电子级超纯水配制),300ml0.8m4-氨基-3,5,6-三氯吡啶甲酸钾水溶液(用电子级超纯水配制)为阴极液。在45-50℃下,搅拌阴极液,通入10a/dm2的电流,反应400分钟后停止电解。用高效液相色谱测定4-氨基-3,5,6-三氯吡啶甲酸的转化率为98.2%,4-氨基-3,6-二氯吡啶甲酸收率为93.2%。首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却结晶;然后,对析出晶体进行过滤、去离子水水洗、80℃下烘干处理,得到雪白色晶体;最后,用高效液相色谱测定白色晶体中4-氨基-3,6-二氯吡啶甲酸的纯度为98.1%。
比较例1(与实施例4对比):电化学脱氯合成4-氨基-3,6-二氯吡啶甲酸(去离子水配制溶液)
在以nafion117阳离子膜为隔膜的h型电解槽(图1)中,以实施例1方法制备的活化银网为阴极,相同面积的hastalloyc-276片(几何尺寸为0.5cm×4.0cm×6.0cm)为阳极。300ml1.0mnaoh水溶液为阳极液(用去离子水配制),300ml0.2m4-氨基-3,5,6-三氯吡啶甲酸钠水溶液(用去离子水配制)为阴极液。在25-30℃下,搅拌阴极液,通入5a/dm2的电流,反应200分钟后停止电解。用高效液相色谱测定4-氨基-3,5,6-三氯吡啶甲酸的转化率为96.5%,4-氨基-3,6-二氯吡啶甲酸收率为88.4%。首先,在85~90℃下用浓盐酸将阴极液ph调到1,过夜自然冷却结晶;然后,对析出晶体进行过滤、去离子水水洗、80℃下烘干处理,得到白色粉末;最后,用高效液相色谱测定白色晶体中4-氨基-3,6-二氯吡啶甲酸的纯度为96.5%。
与实施例4相比,采用去离子水配制电解液,转化率下降了2.0%、产品收率下降了8.0%、产品纯度下降了2.1%。
比较例2(与实施例5对比):电化学脱氯合成4-氨基-3,6-二氯吡啶甲酸(去离子水配制溶液)
在以nafion117阳离子膜为隔膜的h型电解槽(图1)中,以实施例2方法制备的1#活化银网为阴极,相同面积的hastalloyc-276片(几何尺寸为0.5cm×4.0cm×6.0cm)为阳极。300ml2.0mkoh水溶液为阳极液(用去离子水配制),300ml0.8m4-氨基-3,5,6-三氯吡啶甲酸钾水溶液(用去离子水配制)为阴极液。在45-50℃下,搅拌阴极液,通入10a/dm2的电流,反应400分钟后停止电解。用高效液相色谱测定4-氨基-3,5,6-三氯吡啶甲酸的转化率为95.2%,4-氨基-3,6-二氯吡啶甲酸收率为87.4%。首先,在85~90℃下用浓盐酸将阴极液ph调到1,过夜自然冷却结晶;然后,对析出晶体进行过滤、去离子水水洗、80℃下烘干处理,得到白色粉末;最后,用高效液相色谱测定白色晶体中4-氨基-3,6-二氯吡啶甲酸的纯度为96.4%。
与实施例5相比,采用去离子水配制电解液连续第一次使用银网电极时,转化率下降了3.0%、产品收率下降了6.7%、产品纯度下降了1.9%。
比较例3(与实施例6对比):电化学脱氯合成4-氨基-3,6-二氯吡啶甲酸(重复使用比较例2的银电极)
在以nafion117阳离子膜为隔膜的h型电解槽(图1)中,以比较例2使用过的银网为阴极,相同面积的hastalloyc-276片(几何尺寸为0.5cm×4.0cm×6.0cm)为阳极。300ml2.0mkoh水溶液为阳极液(用去离子水配制),300ml0.8m4-氨基-3,5,6-三氯吡啶甲酸钾水溶液(用去离子水配制)为阴极液。在45-50℃下,搅拌阴极液,通入10a/dm2的电流,反应400分钟后停止电解。用高效液相色谱测定4-氨基-3,5,6-三氯吡啶甲酸的转化率为92.2%,4-氨基-3,6-二氯吡啶甲酸收率为83.4%。首先,在85~90℃下用浓盐酸将阴极液ph调到1,过夜自然冷却结晶;然后,对析出晶体进行过滤、去离子水水洗、80℃下烘干处理,得到白色粉末;最后,用高效液相色谱测定白色晶体中4-氨基-3,6-二氯吡啶甲酸的纯度为93.4%。
与实施例6对比,采用去离子水配制电解液,原料转化率、产品收率和纯度分别下降了6.0%、9.8%和4.7%。
比较例4(与实施例4对比):电化学脱氯合成4-氨基-3,6-二氯吡啶甲酸(盐酸不浸泡的银电极)
在以nafion117阳离子膜为隔膜的h型电解槽(图1)中,以实施例3方法制备的活化银网为阴极,相同面积的hastalloyc-276片(几何尺寸为0.5cm×4.0cm×6.0cm)为阳极;300ml1.0mnaoh水溶液为阳极液(用去离子水配制),300ml0.2m4-氨基-3,5,6-三氯吡啶甲酸钠水溶液(用去离子水配制)为阴极液。在25-30℃下,搅拌阴极液,通入5a/dm2的电流,反应200分钟后停止电解。用实施例4所述高效液相色谱测定4-氨基-3,5,6-三氯吡啶甲酸的转化率为96.5%,4-氨基-3,6-二氯吡啶甲酸收率为90.2%。首先,在85~90℃下用浓盐酸将阴极液ph调到1,过夜自然冷却结晶;然后,对析出晶体进行过滤、去离子水水洗、80℃下烘干处理,得到雪白色晶体;最后,用高效液相色谱测定白色粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为96.3%。
与实施例4对比,采用盐酸不浸泡的银电极作为阴极,原料转化率、产品收率和产品纯度分别下降了2.0%,6.2%和2.3%。
比较例5(与实施例5对比):电化学脱氯合成4-氨基-3,6-二氯吡啶甲酸(银网的纯度降到99.0%)
在以nafion117阳离子膜为隔膜的h型电解槽(图1)中,以实施例2方法制备的2#活化银网为阴极,相同面积的hastalloyc-276片(几何尺寸为0.5cm×4.0cm×6.0cm)为阳极;银/氯化银为参比电极。300ml2.0mkoh水溶液为阳极液(用电子级超纯水配制),300ml0.8m4-氨基-3,5,6-三氯吡啶甲酸钾水溶液(用电子级超纯水配制)为阴极液,ph=12.7。在45-50℃下,搅拌阴极液,通入10a/dm2的电流,反应400分钟后停止电解。采用实施例4方法用高效液相色谱测定4-氨基-3,5,6-三氯吡啶甲酸的转化率为97.1%,4-氨基-3,6-二氯吡啶甲酸收率为84.8%。首先,在85~90℃下用质量浓度36%浓盐酸将阴极液ph调到1,过夜自然冷却结晶;然后,对析出晶体进行过滤、去离子水水洗、80℃下烘干处理,得到雪白色晶体;最后,用高效液相色谱测定白色晶体中4-氨基-3,6-二氯吡啶甲酸的纯度为91.1%。
与实施例5相比,采用纯度为99%的银网作为阴极后,原料转化率、产品收率和产品纯度分别下降了1.1%,9.3%和7.2%。
比较例6(与实施例6对比):电化学脱氯合成4-氨基-3,6-二氯吡啶甲酸(重复使用比较例5的银电极)
在以nafion117阳离子膜为隔膜的h型电解槽(图1)中,以比较例5使用完的银网为阴极,相同面积的hastalloyc-276片(几何尺寸为0.5cm×4.0cm×6.0cm)为阳极;银/氯化银为参比电极。300ml2.0mkoh水溶液为阳极液(用电子级超纯水配制),300ml0.8m4-氨基-3,5,6-三氯吡啶甲酸钾水溶液(用电子级超纯水配制)为阴极液。在45-50℃下,搅拌阴极液,通入10a/dm2的电流,反应400分钟后停止电解。用实施例4高效液相色谱测定4-氨基-3,5,6-三氯吡啶甲酸的转化率为95.1%,4-氨基-3,6-二氯吡啶甲酸收率为81.1%。首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却结晶;然后,对析出晶体进行过滤、去离子水水洗、80℃下烘干处理,得到雪白色晶体;最后,用高效液相色谱测定白色晶体中4-氨基-3,6-二氯吡啶甲酸的纯度为88.8%。
与实施例6相比,采用纯度为99%的银网作为阴极连续第二次使用时,原料转化率、产品收率和产品纯度分别下降了3.1%,12.1%和9.3%。
- 还没有人留言评论。精彩留言会获得点赞!