一种异质结丰富的CoNiP-P纳米催化剂的制备方法及其应用
本发明属于本发明属于催化剂制备领域,尤其是涉及一种异质结丰富的conip-p纳米催化剂的制备方法及其应用。
背景技术:
随着经济的迅速发展,化石能源的大量使用带来严重的环境问题,同时面临未来能源枯竭问题,寻找可代替的,清洁的可再生能源迫在眉睫。氢能是一种十分具有潜力的可再生能源,其燃烧产物为无污染的水,因此是一种十分清洁的能源,是未来新能源的重要发展方向,目前工业制氢主要为天然气蒸汽转化,轻油蒸汽转化和水煤气制氢,均依托于传统化石能源。与之形成对比的是电解水制氢,电解水制氢以水作为原料,通过电解法制备氢气,完全摆脱传统化石燃料的束缚,原料丰富且过程中不产生污染,并可充分与风能,地热能,潮汐能等可再生能源相结合,是未来大规模制氢最有前途的方式。
然而,基于电解水过程中缓慢的动力学过程,尤其是阳极析氧反应涉及复杂的电子-质子耦合反应,缓慢的速率控制步骤,造成过高的电解电位,成为电解水制氢商业化发展的巨大瓶颈。贵金属及其氧化物ruo2/iro2以及pt/c分别是性能优越的oer和her催化剂,令人遗憾的是,贵金属元素ru、ir和pt的稀缺性和高成本,以及不尽人意的电化学稳定性限制了其应用。为了减少贵金属元素的使用,降低催化剂成本,以及提高催化剂稳定性,近年来,过渡金属基催化剂在电解水中的应用受到人们的大量关注,如过渡金属的氧化物、氢氧化物、硫化物、氮化物、磷化物,以及层状双氢氧化物等。最近的研究表明,过渡金属磷化物是十分具有潜力的oer和her双功能催化剂,如何通过界面工程调控电子结构,从而进一步提高其双功能催化性能,成为过渡金属磷化物催化剂发展面临的挑战。
目前,现有国内外发明专利中关于过渡金属基催化剂大部分致力于单纯oer或her催化,关于双功能催化剂的研究和应用相对较少,此外,大部分过渡金属基双功能催化剂的设计和制备强调三维复合结构的构建,对于异质界面,尤其是晶态/非晶态异质界面的研究较少,本发明通过两步法构建了具有丰富晶态/非晶态异质界面的conip-p双功能电催化剂,基于异质界面的形成促使高活性位点的暴露,conip-p在oer和her催化中均表现出优异的性能,用于全解水时,在1.0mkoh溶液中,仅需1.565v即可驱动10macm-2的电流密度,优于大部分过渡金属基双功能电催化剂。
技术实现要素:
本发明要解决的第一个技术问题是提供一种异质结丰富的conip-p纳米催化剂的制备方法;本发明通过电沉积法和低温掺杂两步合成了conip-p纳米催化剂,基于两步磷化过程磷的不同掺杂方式,conip-p纳米催化剂形成了大量晶态/非晶态异质界面,赋予了催化剂优秀的oer、her双功能催化活性。
本发明要解决的第二个技术问题是上述方法制得的异质结丰富的conip-p纳米催化剂在电解水制氢中的应用。
为解决上述第一个技术问题,发明采用如下的技术方案:
一种异质结丰富的conip-p纳米催化剂的制备方法,包括如下步骤:
1)将钴源、镍源和磷源添加至去离子水中,配制成电解液,随后在室温下超声,将电解液倒入电解池中;
2)将泡沫镍裁切成片浸泡在稀盐酸溶液中并超声,随后分别用去离子水和无水乙醇清洗2-4次,烘干;
3)采用三电极体系,其中以饱和甘汞电极作为参比电极,碳棒为辅助电极,泡沫镍作为工作电极,泡沫镍完全浸入电解液面,采用循环伏安法进行电沉积制备,参数设定为:扫描次数20-60次,扫描速率100mv/s,扫描区域为0~-1.2v至0~-2.0vvssce;电沉积完成后取出泡沫镍,分别用去离子水和乙醇冲洗,随后置于干燥箱中室温真空干燥,得到生长有conip前驱体的泡沫镍;
4)将生长有conip前驱体的泡沫镍和次亚磷酸钠分开置于两个瓷舟内,将两个瓷舟顺序装入石英管中,其中装有次亚磷酸钠的瓷舟置于上游,装有泡沫镍的瓷舟置于下游,随后将石英管置于管式炉中;在常温下以90-110mlmin-1的气速通入氮气吹扫1h,随后升温至300℃,并保持2h;升温后将气速降低为15-25mlmin-1,自然降温,得到产物conip-p纳米催化剂。
作为技术方案的进一步改进,步骤1)中,所述钴源、镍源和磷源按co:ni:p的原子比为1:1:1。
优选地,步骤1)中,所述电解液中co、ni、p的浓度为0.08-0.12moll-1。
优选地,步骤1)中,所述超声的时间为12-18min。
优选地,步骤1)中,所述钴源、镍源和磷源分别是六水合硝酸钴、六水合硝酸镍和次亚磷酸钠。
优选地,步骤1)中,所述电解池容量为100ml,添加的电解液量为50ml。
作为技术方案的进一步改进,步骤2)中,所述超声的时间为4-6min。
优选地,所述泡沫镍裁切成片的尺寸宽*长为1*2cm。
优选地,步骤2)中,所述清洗的次数为2-4次。
优选地,步骤3)中,所述循环伏安法的参数设定为:扫描次数20-60次,扫描速率100mv/s,扫描区域为0~-1.2v至0~-2.0vvssce。
作为技术方案的进一步改进,步骤4)中,所述次亚磷酸钠的添加量为500~1000mg。
为解决上述第二个技术问题,本发明采用上述制得的异质结丰富的conip-p纳米催化剂在电解水制氢中应用。
本发明所记载的任何范围包括端值以及端值之间的任何数值以及端值或者端值之间的任意数值所构成的任意子范围。
如无特殊说明,本发明中的各原料均可通过市售购买获得,本发明中所用的设备可采用所属领域中的常规设备或参照所属领域的现有技术进行。
与现有技术相比较,本发明具有如下有益效果:
本发明得到的异质结丰富的conip-p催化剂通过双重磷掺杂形成了丰富晶态nip和非晶态co-o-p交界的异质结构,成为促进水解催化反应的高活性位点,本系列催化剂在电解水析氢和析氧反应催化中展现出优异的活性和稳定性,是理想的非贵金属电解水双功能催化剂。
附图说明
下面结合附图对本发明的具体实施方式作进一步详细的说明
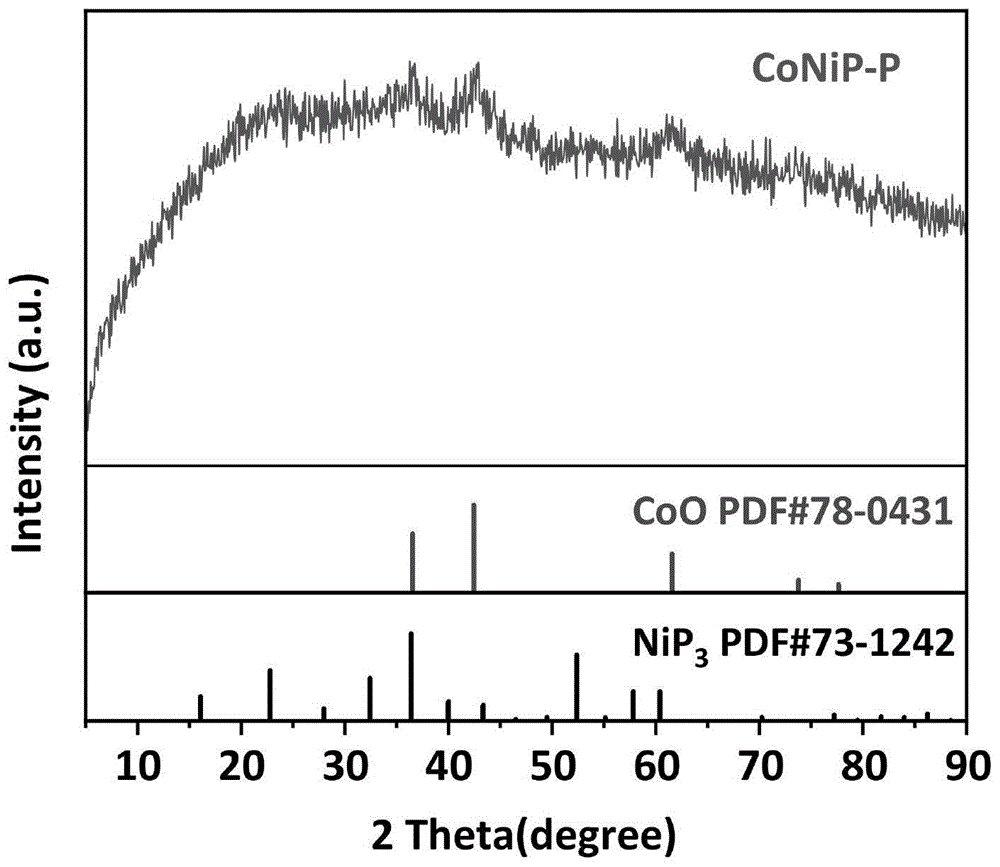
图1为本发明实施例1异质结丰富的conip-p纳米催化剂的xrd图谱;
图2为本发明实施例1异质结丰富的conip-p纳米催化剂的扫描电镜图;
图3为本发明实施例1异质结丰富的conip-p纳米催化剂的透射电镜图;
图4为本发明对比例1conip-p纳米催化剂的扫描电镜图;
图5为本发明对比例2nip-p纳米催化剂的扫描电镜图;
图6为本发明对比例3cop-p纳米催化剂的扫描电镜图;
图7为本发明对比例5conip-p纳米催化剂的扫描电镜图;
图8为本发明对比例6conip-p纳米催化剂的扫描电镜图;
图9为本发明对比例8conip-p纳米催化剂的扫描电镜图;
图10为本发明对比例9conip-p纳米催化剂的扫描电镜图。
具体实施方式
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
作为本发明的一个方面,本发明一种异质结丰富的conip-p纳米催化剂的制备方法,包括如下步骤:
1)将钴源、镍源和磷源添加至去离子水中,配制成电解液,随后在室温下超声,将电解液倒入电解池中;
2)将泡沫镍裁切成片浸泡在稀盐酸溶液中并超声,随后分别用去离子水和无水乙醇清洗2-4次,烘干;
3)采用三电极体系,其中以饱和甘汞电极作为参比电极,碳棒为辅助电极,泡沫镍作为工作电极,泡沫镍完全浸入电解液面,采用循环伏安法进行电沉积制备,参数设定为:扫描次数20-60次,扫描速率100mv/s,扫描区域为0~-1.2v至0~-2.0vvssce;电沉积完成后取出泡沫镍,分别用去离子水和乙醇冲洗,随后置于干燥箱中室温真空干燥,得到生长有conip前驱体的泡沫镍;
4)将生长有conip前驱体的泡沫镍和次亚磷酸钠分开置于两个瓷舟内,将两个瓷舟顺序装入石英管中,其中装有次亚磷酸钠的瓷舟置于上游,装有泡沫镍的瓷舟置于下游,随后将石英管置于管式炉中;在常温下以90-110mlmin-1的气速通入氮气吹扫1h,随后升温至300℃,并保持2h;升温后将气速降低为15-25mlmin-1,自然降温,得到产物conip-p纳米催化剂。本步骤中,升温后温度过低会导致磷化不完全或无法磷化,温度过高会破坏催化剂活性。
根据本发明的某些实施例,步骤1)中,所述电解液中co、ni、p的浓度为0.08-0.12moll-1。添加量过少或过多均无法形成理想的异质结丰富的纳米结构,导致催化性能降低。
根据本发明的某些实施例,步骤1)中,所述超声的时间为12-18min。
根据本发明的某些实施例,步骤1)中,所述钴源、镍源和磷源分别是六水合硝酸钴、六水合硝酸镍和次亚磷酸钠;如果采用其他金属源如氯化钴,氯化镍,将无法在泡沫镍上沉积得到目标conip前驱体。
根据本发明的某些实施例,步骤1)中,所述电解池容量为100ml,添加的电解液量为50ml,电解池容量加大,相应所需电解液体积增加,工作电极和辅助电极之间的间隔增大,会导致溶液阻抗增加,相同电位下的电流密度降低,从而造成催化剂沉积量降低;同理,电解池容量变小会导致过量沉积。
根据本发明的某些实施例,步骤2)中,所述超声的时间为4-6min;时间过短可能无法达到清洁表面的目的,时间过长会使泡沫镍表面被过度腐蚀。
根据本发明的某些实施例,步骤3)中,所述循环伏安法的参数设定为:扫描次数20-60次,扫描速率100mv/s,扫描区域为0~-1.2v至0~-2.0vvssce。扫描次数过少,会导致泡沫镍表面conip前驱体沉积量过低,无法完全覆盖,扫描次数过多,对造成过度沉积乃至脱落;扫描区域过窄,会导致conip前驱体沉积不完全,无法形成纳米形貌,扫描区域过宽,会使conip前驱体沉积过度,造成堆叠。
根据本发明的某些实施例,步骤4)中,所述次亚磷酸钠的添加量为500~1000mg。添加量过少导致催化剂磷化不完全,添加量过高不利于生产安全,同时导致不必要的试剂浪费和环境污染。
作为本发明的另一个方面,本发明采用上述制得的异质结丰富的conip-p纳米催化剂在电解水制氢中应用。本发明异质结丰富的conip-p纳米催化剂在电解水的析氢和析氧反应中均表现出优良的催化活性和稳定性,是具有商业应用潜力的非贵金属电解水催化剂。
实施例1
一种异质结丰富的conip-p纳米催化剂的制备方法,包括如下步骤:
1)制备电解质溶液,将钴源,镍源和磷源按co:ni:p=1:1:1的原子比添加至去离子水中,配制成0.1moll-1的溶液作为电解液,随后在室温下超声15min,将制备得到电解液倒入电解池中。
2)集流体的前处理,制备体积浓度为5%的稀盐酸溶液,将裁切成1*2cm的泡沫镍浸泡在稀盐酸溶液中并超声5min,随后分别用去离子水和无水乙醇清洗三次,烘干。
3)电沉积制备,采用三电极体系,其中以饱和甘汞电极作为参比电极,碳棒为辅助电极,泡沫镍作为工作电极,泡沫镍完全浸入液面,采用循环伏安法进行制备,设定参数为:扫描次数40次,扫描速率100mv/s,扫描区域为0~-1.5vvssce,电沉积完成后取出泡沫镍,用去离子水和乙醇冲洗,随后置于干燥箱中室温真空干燥12h。
4)将生长有conip前驱体的泡沫镍置于瓷舟,并在上游用瓷舟装取500mg次亚磷酸钠,随后置于管式炉中,首先在常温下以100mlmin-1的气速通入氮气吹扫1h,随后以3℃min-1的升温速率升温至300℃,并保持2h,在开始升温后将气速降低为20mlmin-1,最后使其自然降温,得到目标样品conip-p纳米催化剂。
经检测:
图1为本发明实施例1异质结丰富的conip-p纳米催化剂的xrd图谱;
图2为本发明实施例1异质结丰富的conip-p纳米催化剂的扫描电镜图;
图3为本发明实施例1异质结丰富的conip-p纳米催化剂的透射电镜图。
实施例2
重复实施例1,其不同之处在于:步骤1)中电解质溶液中六水合硝酸镍的添加浓度为0.08moll-1。
经检测:实施例2能得到与实施例1相当的技术效果。与实施例1相比,ni相对于co的含量有所降低,但整体维持丰富的异质界面,析氢和析氧催化性能与实施例1相当。
实施例3
重复实施例1,其不同之处仅在于:步骤3)中电沉积设定参数的扫描范围为0~
-2.0vvssce。
实施例4
重复实施例1,其不同之处仅在于:步骤3)中电沉积设定参数的扫描次数为20次。
经检测:实施例3与实施例4均能得到与实施例1相当的技术效果。与实施例1相比,实施例3泡沫镍表面催化剂负载量有所提升,但维持丰富的异质界面,对催化性能影响不明显;与实施例1相比,实施例4中泡沫镍表面催化剂负载量有所降低,但均能完全覆盖泡沫镍表面,对催化性能影响不明显。
实施例5
重复实施例1,其不同之处仅在于:步骤4)中次亚磷酸钠的添加量为1000mg。
经检测:实施例5能得到与实施例1相当的技术效果,表明对于催化剂材料而言,过量磷源不会影响催化剂的形貌,结构以及性能。
对比例1
重复实施例1,其不同之处仅在于:步骤1)中,用cocl2代替co(no3)26h2o作为钴源,用nicl2代替ni(no3)2·6h2o作为镍源制备电解质溶液。
经检测:如图4所示,采用cocl2和nicl2制备的电解液无法通过电沉积法生成目标conip前驱体,表明硝酸钴和硝酸镍的不可替代性。
对比例2
重复实施例1,其不同之处仅在于:步骤1)中,不添加钴源,只添加镍源和磷源。
经检测:如图5所示,不添加钴源,最终生成的nip-p为块状结构,oer和her活性相对conip-p均有一定程度下降,表明金属元素co的添加会产生协同作用。
对比例3
重复实施例1,其不同之处仅在于:步骤1)中,不添加镍源,只添加钴源和磷源。
经检测:如图6所示,不添加镍源,最终生成的cop-p为纳米片阵列结构,而其oer和her活性相对conip-p亦均有一定程度下降,结合对比例2可以发现,过渡金属元素co和ni会产生协同作用,此外,元素ni的添加会导致纳米片阵列结构崩塌,形成表面粗糙不平的块状纳米结构,这也许和晶态/非晶态异质界面的形成有关。
对比例4
重复实施例1,其不同之处仅在于:步骤1)中,步骤1)中电解质溶液中六水合硝酸镍的添加浓度为0.05moll-1。
经检测:显著降低镍源的添加量后,催化剂整体催化性能下降,异质界面显著减少。
对比例5
重复实施例1,其不同之处仅在于:步骤3)中,电沉积设定参数中扫描次数为10次。
经检测:如图7所示,得到的conip-p催化剂具有类似的形貌,但其电解水析氢和析氧反应性能与实施例1相比下降明显。
对比例6
重复实施例1,其不同之处仅在于:步骤3)中,电沉积设定参数中扫描区域为0~-1.0vvssce。
经检测:如图8所示,得到的conip-p催化剂在泡沫镍上生成量较少,无法完全覆盖其表面,其电解水析氢和析氧反应性能与实施例1相比下降明显。
对比例7
重复实施例1,其不同之处仅在于:步骤4)中,次亚磷酸钠的添加量为200mg。
经检测:得到的conip-p双功能催化性能下降较为明显,且异质界面显著减少,表明低温磷化不完全。
对比例8
重复实施例1,其不同之处仅在于:步骤4)中,磷化温度控制为200℃。
经检测:如图9所示,得到的泡沫镍材料表面未完全变黑,部分保持conip前驱体的纳米片装阵列结构,且双功能催化性能下降较为明显,表明磷化不完全。
对比例9
重复实施例1,其不同之处仅在于:步骤4)中,磷化温度控制为500℃。
经检测:如图10所示,经测试,得到的conip-p双功能催化性能下降较为明显,表明过高的掺杂温度会降低催化剂活性。
综上所述,本发明的异质结丰富的conip-p纳米催化剂的制备方法中,钴源和镍源的选择,溶液浓度,电沉积参数设定范围,低温磷化参数设定等相互协调、相互配合形成一个完整的技术方案,才可以制得本发明要求的异质结丰富的conip-p纳米催化剂。
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无法对所有的实施方式予以穷举。凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
- 还没有人留言评论。精彩留言会获得点赞!