一种维生素K2含量的检测方法与流程
本发明涉及利用吸附作用,例如色谱法将材料分离成各个组分来测试材料,尤其是在保健食品、药品、预混料中测量维生素k2含量的检测方法。
背景技术:
维生素k2是一种脂溶性维生素,具有叶绿醌生物活性的萘醌基团的衍生物,也被称为甲萘醌,其结构是由二甲基-1,4-萘醌母核及c-3位上数目不等的异戊二烯侧链组成,根据其单元个数差异,维生素k2共分为14种,以维生素k2(mk-5),k2(mk-7)最为重要。维生素k2具有治疗和预防骨质疏松症、增加骨密度,预防肝癌,促进凝血,利尿,降血压等药理活性。
维生素k2易溶于有机溶剂,正己烷、异丙醇等。其遇碱和见光不稳定,而耐热、耐酸。维生素k2技术上常采用包衣技术增加稳定性。目前,已公开的维生素k2的检测方法比较单一,且保留时间较长,色谱条件较复杂。因此,为控制维生素k2相关产品质量,需要建立更高效便捷的检测方法。
技术实现要素:
为了克服现有维生素k2检测方法存在应用单一、保留时间较长、色谱条件较复杂的不足,本发明的目的在于提供一种操作便捷、检测效率高、应用范围广的维生素k2含量的检测方法。
本发明解决其技术问题所采用的技术方案是:一种维生素k2含量的检测方法,其特征在于:其经过以下步骤:
(1)称取待测样品并且置于棕色容量瓶中,加入10mln,n—二甲基甲酰胺,涡旋震荡分散均匀后,加入25ml异丙醇,震荡至均匀后,超声15-30min,冷却至室温,用异丙醇定容至刻度,摇匀,得待测样品溶液;
(2)将步骤1的待测样品溶液进行高效液相色谱分析,其条件为:色谱柱为c18的反相柱,直径为4-5mm,长度为25mm,填料粒径为5微米;流动相包含60-80重量份的乙醇和40-20重量份的甲醇;流动相流速为0.8-1.2ml/min;柱温为25-35摄氏度;进样量为5-20微升;使用紫外可见248nm下进行检测;
(3)配制标准样品溶液:称取维生素k2标准样品,制成1.0ug/ml、2.0ug/ml、5ug/ml、10ug/ml、20.0ug/ml的标准样品溶液;
(4)建立标准曲线:将步骤3得到的标准样品溶液在步骤2的条件下进行高效液相分析,并且统计其色谱峰的面积,并且以峰面积为纵坐标,已知浓度为横坐标,建立一元一次标准曲线;
(5)将步骤2中相应位置的峰面积与标准曲线进行对比,得到其浓度。
在本发明优选的方面,所述的方法经过以下步骤:
(1)称取待测样品并且置于棕色容量瓶中,加入10mln,n-二甲基甲酰胺,涡旋震荡分散均匀后,加入25ml异丙醇,震荡至均匀后,超声22min,冷却至室温,用异丙醇定容至刻度,摇匀,得待测样品溶液;
(2)将步骤1的待测样品溶液进行高效液相色谱分析,其条件为:色谱柱为4.6×250mm的c18的反相柱,填料粒径为5微米;流动相包含70重量份的乙醇和30重量份的甲醇;流动相流速为1.0ml/min,柱温为30摄氏度;进样量为10微升;使用紫外可见248nm下进行检测;
(3)配制标准样品溶液:维生素k2标准样品,制成1.0ug/ml、2.0ug/ml、5ug/ml、10ug/ml、20.0ug/ml的标准样品溶液;
(4)建立标准曲线:将步骤3得到的标准样品溶液在步骤2的条件下进行高效液相色谱分析,并且统计其色谱峰的面积,并且以峰面积为纵坐标,已知浓度为横坐标,建立一元一次标准曲线;
(5)将步骤2中相应位置的峰面积与标准曲线进行对比,得到待测样品溶液中维生素k2的浓度,并且计算待测样品中维生素k2浓度。
在本发明优选的方面,所述的步骤1中,超声的频率是45khz,超声功率为1kw。
在本发明优选的方面,所述的步骤1中,异丙醇和n,n-二甲基甲酰胺使用分子筛进行除水,并且在使用前均使用氮气鼓泡30-50秒。
在本发明优选的方面,所述的步骤2中,所述的流动相包含70重量份的乙醇、30重量份的甲醇和0.15重量份的三氟乙酸。
在本发明优选的方面,在步骤5中,所述的维生素k2含量按照下式来计算:
式中,c:由液相色谱仪计算得到的待测样品浓度,ug/ml;
50:待测样品稀释体积,ml;
m:待测样品称样量,g;
1000:浓度转化系数。
本发明采用n,n-二甲基甲酰胺及异丙醇做提取剂可以方便地提取待测样品中的维生素k2,并采用甲醇与无水乙醇的混合溶剂做流动相能够快速有效地分离待测样品溶液中的维生素k2。本发明的含量测定方法提取便捷,检测效率高,准确度高,应用范围广泛,各试剂易得且毒性低。本发明适用于保健食品、药品、预混料、特殊膳食食品中维生素k2的测定。
附图说明
下面结合附图和实施例对本发明做进一步说明。
图1是本发明实施例1的标准样品溶液的高效液相色谱图;
图2是本发明实施例1的待测样品溶液的高效液相色谱图;
图3是本发明实施例2的标准样品溶液的高效液相色谱图;
图4是本发明实施例2的待测样品溶液的高效液相色谱图;
图5是本发明实施例3的标准样品溶液的高效液相色谱图;
图6是本发明实施例3的待测样品溶液的高效液相色谱图。
具体实施方式
除非另外地说明,本发明所测定的维生素k2是mk-7,其cas号为2124-57-4。
除非另外地说明,在各个实施例的步骤1中,超声的频率是45khz,超声功率为1kw。
实施例1
一种维生素k2含量的检测方法,其经过以下步骤:
(1)从待测样品中提取维生素k2;
在本实施例中,待测样品的产品名称:维生素k2粉,批号:vimk1500-r2,生产厂家:上海技源科技有限公司。
准确称取0.2014g粉剂的待测样品于50ml棕色容量瓶中,加入10mln,n-二甲基甲酰胺,涡旋震荡分散均匀后,加入25ml异丙醇,震荡至均匀后,超声25min,冷却至室温,用异丙醇定容至刻度,摇匀,经0.45um微孔滤膜过滤,得到待测样品溶液;
(2)待测样品溶液的高效液相色谱分析;将步骤1的样品溶液进行高效液相色谱分析,其条件为:色谱柱:agilenteclipsexdb-c18(4.6×250mm,5μm)或等效色谱柱;柱温:30℃;流动相:无水乙醇:甲醇=70:30;流速:1.0ml/min;进样量:10μl;紫外线检测波长:248nm;
(3)标准样品溶液的制备;精密称取9.88mg维生素k2(mk-7)的标准样品于50ml容量瓶中,加入30ml异丙醇,超声使粉末溶解,用异丙醇定容至刻度,作为储备液。将储备液用异丙醇稀释制成0.988ug/ml、1.976ug/ml、4.940ug/ml、9.88ug/ml、19.76ug/ml的标准样品溶液。阴凉避光保存;
(4)标准曲线建立;将步骤3得到的标准样品溶液在步骤2的条件下进行高效液相分析,其条件为:色谱柱:agilenteclipsexdb-c18(4.6×250mm,5μm)或等效色谱柱;流动相:无水乙醇:甲醇=70:30;柱温:30℃;流速:1.0ml/min;进样量:10μl;紫外线检测波长:248nm;并且统计其色谱峰的面积,并且以峰面积为纵坐标,已知浓度为横坐标,建立一元一次标准曲线;
(5)浓度获取。将步骤2中相应位置的峰面积与标准曲线进行对比,得到待测样品溶液的浓度。其中,所述的维生素k2含量按照下式来计算:
式中c:由液相色谱仪计算得到的待测样品溶液的浓度,ug/ml;
50:待测样品稀释体积,ml;
m:待测样品称样量,g;
1000:浓度转化系数。
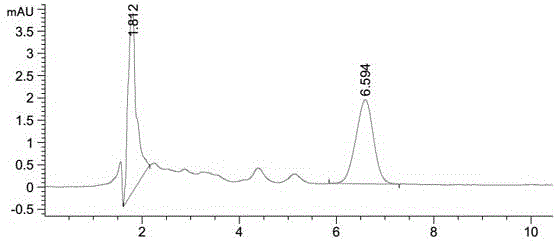
本实施例中,对标准样品溶液进行高效液相色谱测定,以保留时间定性(色谱图如图1所示),以得到维生素k2(mk-7)色谱峰的峰面积;并且结合待测样品的称样量,获得了待测样品溶液内的浓度定量(色谱图如图2所示)。经计算得到维生素k2(mk-7)的含量为2.040mg/g。
实施例2
一种维生素k2含量的检测方法,其经过以下步骤:
(1)从待测样品中提取维生素k2;
在本实施例中,待测样品的产品名称:产品名称:维生素k2粉,批号:yk20151012a01,生产厂家:广东双骏生物科技有限公司。
称取0.2g粉剂的待测样品于50ml棕色容量瓶中,加入10mln,n-二甲基甲酰胺,涡旋震荡分散均匀后,加入25ml异丙醇,震荡至均匀后,超声20min,冷却至室温,用异丙醇定容至刻度,摇匀。经0.45um微孔滤膜过滤,得到待测样品溶液;
(2)待测样品溶液的高效液相色谱分析;将步骤1的待测样品溶液进行高效液相色谱分析,其条件为:色谱柱:agilenteclipsexdb-c18(4.6×250mm,5μm)或等效色谱柱;流动相:无水乙醇:甲醇=60:40;柱温:25℃;流速:0.8ml/min;进样量:10μl;紫外线检测波长:248nm;
(3)标准样品溶液制备;精密称取9.88mg维生素k2(mk-7)标准样品于50ml容量瓶中,加入30ml异丙醇,超声使粉末溶解,用异丙醇定容至刻度,作为储备液。将储备液用异丙醇稀释制成0.988ug/ml、1.976ug/ml、4.940ug/ml、9.88ug/ml、19.76ug/ml的标准样品溶液。阴凉避光保存;
(4)标准曲线建立;将步骤3得到的标准样品溶液在步骤2的条件下进行高效液相色谱分析,其条件为:色谱柱:agilenteclipsexdb-c18(4.6×250mm,5μm)或等效色谱柱;流动相:无水乙醇:甲醇=60:40;柱温:25℃;流速:0.8ml/min;进样量:10μl;紫外线检测波长:248nm;并且统计其色谱峰的面积,并且以峰面积为纵坐标,已知浓度为横坐标,建立一元一次标准曲线;
(5)浓度获取。将步骤2中相应位置的峰面积与标准曲线进行对比,得到待测样品溶液的浓度。其中,所述的维生素k2含量按照下式来计算:
式中c:由液相色谱仪计算得到的待测样品浓度,ug/ml;
50:待测样品稀释体积,ml;
m:待测样品称样量,g;
1000:浓度转化系数。
对标准样品溶液进行高效液相色谱测定,以保留时间定性(色谱图如图3所示),以得到维生素k2(mk-7)色谱峰的峰面积;并且结合待测样品称样量,获得了待测样品溶液内的浓度定量(色谱图如图4所示)。经计算得到维生素k2(mk-7)的含量为2.091mg/g。
实施例3
一种维生素k2含量的检测方法,其经过以下步骤:
(1)待测样品提取;
在本实施例中,待测样品的产品名称:鱼胶原牡蛎k2片,批号:试压制小样,生产厂家:威海百合生物技术股份有限公司
取20粒片剂待测样品,粉碎,混合均匀,精密称取2.0g于50ml棕色容量瓶中,加入10mln,n-二甲基甲酰胺,涡旋震荡分散均匀后,加入25ml异丙醇,震荡至均匀后,超声30min,冷却至室温,用异丙醇定容至刻度,摇匀。经0.45um微孔滤膜过滤,得待测样品溶液;
(2)待测样品溶液的高效液相色谱分析;将步骤1的待测样品溶液进行高效液相色谱分析,其条件为:色谱柱:agilenteclipsexdb-c18(4.6×250mm,5μm)或等效色谱柱;流动相:无水乙醇:甲醇=80:20;柱温:35℃;流速:1.2ml/min;进样量:10μl;紫外线检测波长:248nm;
(3)标准样品溶液的制备;精密称取9.88mg维生素k2(mk-7)标准样品于50ml容量瓶中,加入30ml异丙醇,超声使粉末溶解,用异丙醇定容至刻度,作为储备液。将储备液用异丙醇稀释制成0.988ug/ml、1.976ug/ml、4.940ug/ml、9.88ug/ml、19.76ug/ml的标准样品溶液。阴凉避光保存;
(4)标准曲线建立;将步骤3得到的标准样品溶液在步骤2的条件下进行高效液相色谱分析,其条件为:色谱柱:agilenteclipsexdb-c18(4.6×250mm,5μm)或等效色谱柱;流动相:无水乙醇:甲醇=80:20;柱温:35℃;流速:1.2ml/min;进样量:10μl;紫外线检测波长:248nm;并且统计其色谱峰的面积,并且以峰面积为纵坐标,已知浓度为横坐标,建立一元一次标准曲线;
(5)浓度获取。将步骤2中相应位置的峰面积与标准曲线进行对比,得到待测样品溶液的浓度。其中,所述的维生素k2含量按照下式来计算:
式中c:由液相色谱仪计算得到的待测样品溶液的浓度,ug/ml;
50:待测样品稀释体积,ml;
m:待测样品称样量,g;
1000:浓度转化系数。
对标准样品溶液进行高效液相色谱测定,以保留时间定性(色谱图如图5所示),以得到维生素k2(mk-7)色谱峰的峰面积;并且结合待测样品的称样量,获得了待测样品溶液内的浓度定量(色谱图如图6所示)。经计算得到维生素k2(mk-7)的含量为1.372mg/100g。
实施例4
本实施例中的样品、条件和实施例2基本相同,其区别在于:异丙醇和n,n-二甲基甲酰胺使用分子筛进行除水,并且在使用前均使用氮气鼓泡45秒。在本实施例中,加入待测样品量为240.3mg,测量含量为2.051mg/g。
实施例5
本实施例中的样品、条件和实施例2基本相同,其区别在于,所述的流动相包含70重量份的乙醇、30重量份的甲醇和0.15重量份的三氟乙酸。在本实施例中,加入待测样品量为211.8mg,测量含量为2.044mg/g。
实施例6
本实施例中的样品、条件和实施例2基本相同,其区别在于:异丙醇和n,n-二甲基甲酰胺使用分子筛进行除水,并且在使用前均使用氮气鼓泡45秒;所述的流动相包含70重量份的乙醇、30重量份的甲醇和0.15重量份的三氟乙酸。在本实施例中,加入待测样品量为223.5mg,测量含量为2.133mg/g。
实施例7
本实施例中的样品、条件和实施例5基本相同,其区别在于,在超声提取完毕之后,定容之前,向容量瓶内加入实施例1的储备液500微升,即加入了0.09880毫克的维生素k。在本实施例中,加入待测样品量为214.7mg,测量含量为2.486mg/g。
实施例8
本实施例中的样品、条件和实施例6基本相同,其区别在于,在超声提取完毕之后,定容之前,向容量瓶内加入实施例1的储备液500微升,即加入了0.09880毫克的维生素k。在本实施例中,加入待测样品量为225.0mg,测量含量为2.458mg/g。
实施例9
本实施例中的样品、条件和实施例7基本相同,其区别在于,在超声提取完毕之后,定容之前,向容量瓶内加入实施例1的储备液500微升,即加入了0.09880毫克的维生素k。在本实施例中,加入待测样品量为190.9mg,测量含量为2.642mg/g。
实验分析:对以上的实施例1、4-9的实验数据分析如下:
实施例7-9中,在样品中额外加入0.09880毫克的维生素k,理论上应当使得其含量提升水平计算并列入表1中。同时还计算得到了实际上其含量提升的数量。两者相比,计算了其含量提升的实际水平和理论水平的偏差,从而评估了内标法下,各组试验方法的样品回收率的高低。偏差越小,说明其试验方法更为准确。
其中,理论含量按照如下方法计算:
理论含量=(测量含量*待测样品重量+额外加入维生素k2的重量)/待测样品重量
理论差值=额外加入维生素k2的重量/待测样品重量
实际差值=测量浓度-对应方法无内标试验组的测量浓度
在表1中,额外加入维生素k2的重量简称为“加料量”;
“待测样品重量”简称为“样品量”;
“待测样品测量的浓度”简称为“测量含量”;
表1:实施例7-9中内标法下的测量值相对于理论的偏差
从表1中可以看到,实施例7-8中均具有一定量的偏差。而在实施例9的条件(也就是实施例6的条件)下其偏差最小,内标的加入的还原程度最高。
- 还没有人留言评论。精彩留言会获得点赞!