用于生物胺的衍生化的方法和试剂盒与流程
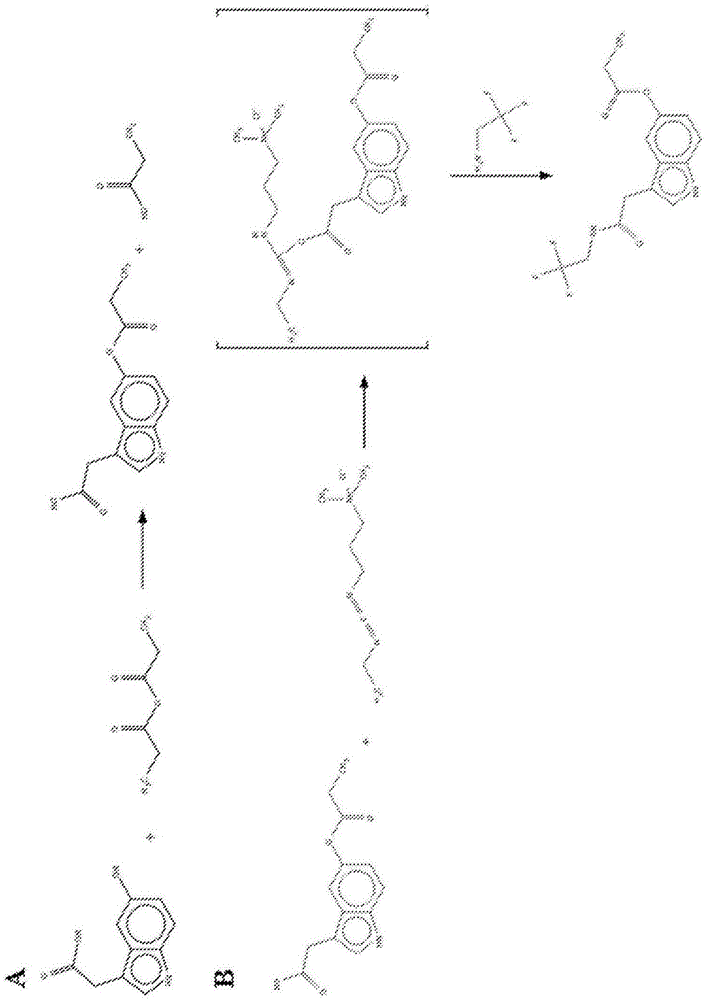
生物胺是具有一个或多个胺基的生物物质。它们是主要通过氨基酸的脱羧或通过醛和酮的氨基化和转氨基化形成的碱性含氮化合物。生物胺是具有低分子量的有机碱,并且由微生物、植物和动物代谢合成。生物胺的一些突出的实例包括经典的单胺,如组胺、血清素和儿茶酚胺类神经递质。组胺是衍生自氨基酸组氨酸的物质,其充当介导唤醒和注意力的神经递质,以及响应于变态反应或组织损伤而从肥大细胞释放的促炎信号。组胺也是胃通过组胺h2受体分泌hcl的重要刺激物。血清素是衍生自氨基酸色氨酸的中枢神经系统神经递质,并参与调节情绪、睡眠、食欲和性欲。儿茶酚胺去甲肾上腺素(降肾上腺素)是参与睡眠和觉醒、注意力和摄食行为的神经递质,以及由调节交感神经系统的肾上腺释放的应激激素。肾上腺素(epinephrine/adrenaline)是肾上腺应激激素,以及以较低水平存在于脑中的神经递质。多巴胺是参与身体运动的动机、奖励、成瘾、行为增强和协调的神经递质。因为诸如儿茶酚胺的生物胺在有机体的体内平衡中起神经递质的重要作用,所以它们在体内的动力学分析已经成为主要的研究挑战。然而,生物流体中的生物胺及其代谢物以低内源性浓度存在。最近尝试在液相色谱-串联质谱法或lc-ms-ms中分析药理学相关的生物单胺和氨基酸。这是将高选择性与高灵敏度相结合的分析技术。通常,在lc-ms-ms中,首先使用液相色谱法(lc)在柱上分离分析物的混合物。收集在某一时间从lc洗脱的一种或多种(衍生化的)分析物并将其转移至第一ms。这些过程通常在线发生。在本文中,发生一种或多种洗脱的分析物的离子化,通常使用大气压化学离子化(apci)或电喷雾离子化(esi)技术实施该离子化。在第一ms中选择由此形成的相应的一个或多个分子离子。这些分子离子(其全部具有一个m/z值,但其可能对应于一个或多个不同的化学身份)然后被转移至碰撞室中。在该室中,分子离子分裂为特征片段。在第二ms中检测具有一个特定的m/z值的一个片段,其为目的分析物的特征。可以采用lc-ms-ms以定量测定样品中的分析物。允许定量的一种方式是通过比较分析精确已知浓度的分析物。ms-ms的大选择性涉及选择两种特征离子(一种用于分子离子,其包含分析物的分子离子(母离子)),一种用于离子片段,其为分析物的独特特征(产物离子)),ms-ms的大选择性不仅有助于将分析物与样品中的其它化合物区分开,它还极大地有助于定量分析的准确性和再现性。几种生物胺衍生化策略已经公布或获得专利。参见例如zhao等人,2015;zhang等人,2012;vandemerbel等人,2011;tan等人,2014或ford等人,2007。然而,每种已知的方案需要冗长的提取,包括在衍生化之前的液-液提取或离线固相提取或蒸发,或者它们仅选择性地靶向胺基或羧酸基团。例如,使用所谓的symdaq试剂所建立的衍生化方法具有以下缺点:其仅靶向伯胺基团,所形成的衍生物不稳定,因此必须被立即分析。此外,通过lc-ms/ms的代谢物分析通常受到低灵敏度(不特定的水损失和/或氨损失)的限制。其原因是(i)在较低质量范围内的较高背景噪声;(ii)非特异性产物离子和/或(iv)没有丰富的产物离子。因此,本发明人旨在改进生物胺和其他低分子量分子的hplc质谱检测,所述低分子量分子具有<200amu的质量并且含有伯胺基、仲胺基、羟基和/或羧酸。此外,期望开发简单的衍生化策略,其可直接(即原位)适用于含水样品,特别是适用于复杂的生物流体样品(血浆、唾液、尿液、csf、细胞裂解物、细胞培养上清液等)中,而不需要先前提取目的化合物。已经发现,上述目标可以通过提供涉及胺基团、羟基基团和/或羧酸基团的衍生化的双重衍生化方法来实现,使得例如儿茶酚胺的完整的代谢途径可以在一个运行中以高灵敏度和高选择性进行分析。在衍生化后,可以将小瓶放置在分析系统中,并且可以运行(在线)-spe-lc-ms/ms程序。所形成的衍生物显示了密集的前体离子和非常特定的产物离子图(productionionpattern)(高强度产物),几乎没有灵敏度损失。即使当分析非常小的样品体积(例如仅50μl)的样品时,测试化合物的灵敏度也在皮摩尔浓度的范围内。所形成的衍生物是稳定的,甚至可以在形成后数周进行分析,而不损失灵敏度。因此,本发明提供了用于将含水样品中的至少一种生物胺、其代谢物和/或其前体进行原位衍生化的方法,所述衍生化包括如下步骤:(i)在具有ph7.0至9.0、优选ph8.0至8.5的磷酸盐缓冲液的存在下,使所述样品与丙酸酐/乙腈溶液接触,并且使得所述生物胺或其代谢物的胺部分和/或羟基部分转化以形成所述生物胺的丙酰基衍生物;随后(ii)向在步骤(i)中获得的反应混合物添加碳二亚胺试剂和亲电的含胺化合物,并且使得生物胺、其代谢物和/或其前体的羧酸部分进行碳二亚胺介导的衍生化。本发明的方法在本领域中是未知的或未建议的。bourgogne等人(anal.andbioanal.chem.(2011),第402卷,第1期,第449-459页)公开了用于生物胺的衍生化的方法,其包括在(hilic)-ms/ms分析之前,使用丙酸酐将脑透析液中的组胺和1-甲基组胺进行柱前衍生化。使衍生化的样品经历提取步骤,然后将其注入lc-ms/ms设备。然而,bourgogne等人未公开在本发明中公开的2步衍生化步骤,本发明允许将衍生化的样品直接注入lc-ms/ms设备,并且无需进一步净化。生物胺是具有一个或多个胺基的生物物质。它们是主要通过氨基酸的脱羧或通过醛和酮的氨基化和转氨基化形成的碱性含氮化合物。根据本发明,生物胺、其代谢物和/或其前体至少含有一个或多个伯胺基、仲胺基、羟基和/或羧酸,其对衍生化反应敏感。例如,代谢物或前体可通过存在于酪氨酸、色氨酸、组氨酸或精氨酸的代谢途径中的酶或赖氨酸降解途径(kegg途径)中的酶形成。在一个实施方案中,生物胺是单胺或多胺,其参与器官的生长、修复和代谢和/或用于肠(内脏)的正常的运行和免疫系统的高代谢活性。在另一个实施方案中,生物胺可充当自由基清除剂或具有抗氧化剂活性。在另一个实施方案中,生物胺具有毒理学风险,并且在严重的情况下诱发副作用,例如头痛、低血压或高血压、恶心、心悸和甚至死亡。当有利于微生物或生物化学活性的条件持续时,生物胺可能形成于含有蛋白质或游离氨基酸的食物和饮料中。此类食物和饮料的实例包括但不限于鱼、鱼制品、肉制品(香肠)、蛋、奶酪、发酵的乳制品、巧克力、坚果、发酵的水果和新鲜水果、蔬菜如酸泡菜和大豆制品,包括酱油、啤酒和葡萄酒。因此,食物中生物胺的存在和浓度被认为与腐败和发酵有关,特别是通过微生物。已知啤酒和葡萄酒含有各种量和组成的许多不同的生物胺。组胺、酪胺、腐胺、异戊胺和β-苯乙胺是在葡萄酒中发现的一些常见的生物胺。由于生物胺也可以用作由细菌引起的食物腐败的指示剂(即,间接检测细菌的存在),本发明有利地不仅用于检测低水平的药理学相关的胺,而且还用于衍生化和检测可以用作健康并发症的指示剂的化合物,所述健康并发症包括癌症、细菌感染和食物中毒等,仅举几例而言。食物腐败(例如肉和鱼腐败)随着在屠宰时间后不久细菌开始生长而发生。在食物腐败的初始阶段期间,游离的氨基酸被侵入的腐败微生物释放的酶脱羧。脱羧的产物包括生物胺,即腐胺和尸胺。这两种胺在气味方面特别独特,并且与表面细菌计数良好地相关。另一种产物组胺是令人感兴趣的,这是由于其宣称的引起组胺中毒的能力,一种与腐败鱼的消耗相关的食物中毒的形式。本文所公开的方法能够在相同或相似类型的分析物中区分分析物。如下文所证实的,本发明的方法有利地应用于生物样品中的生物胺神经递质的原位衍生化。优选地,所述生物胺选自肾上腺素、去甲肾上腺素、多巴胺、血清素、5-羟基吲哚乙酸(5-hiaa)、变肾上腺素、去甲变肾上腺素和3-甲氧基酪胺。根据本发明的待衍生化的其它生物胺包括组胺、酪胺、β-苯基乙胺、色胺、腐胺、尸胺、精胺、亚精胺、丁胺、二甲胺、乙醇胺、乙胺、己胺、吲哚、异丙胺、异戊胺、甲胺、2-甲基丁胺、吗啉、戊胺、哌啶、丙胺、吡咯烷、亚精胺和腐胺。生物胺的示例性前体包括氨基酸,例如组氨酸(组胺前体)、酪氨酸(酪胺前体)、羟基色氨酸(血清素前体)、色氨酸(血清素/色胺前体)、赖氨酸(尸胺前体)、鸟氨酸(腐胺前体)和精氨酸(胍基丁胺、精胺、亚精胺的前体)。在一个实施方案中,含水样品是分离的生物样品,优选地选自尿液、血浆、唾液、csf、细胞裂解物和细胞培养上清液。样品通常与哺乳动物对象(优选为人对象)分离。例如,样品是来自患者的尿液样品,患者被诊断患有或跟踪与生物胺的异常水平相关的疾病,如儿茶酚胺,例如多巴胺、去甲肾上腺素或肾上腺素。示例性的应用包括(a)血浆游离或尿游离的变肾上腺素用于嗜铬细胞瘤诊断。(b)色氨酸、5-羟基色氨酸、血清素和5-hiaa在富含血小板的血浆中的分布用于诊断神经内分泌肿瘤。(c)csf和/或尿液中的儿茶酚胺和酸性代谢物、血清素和5-hiaa(高香草酸、香草扁桃酸(vanillylmandelicacid)、3-甲氧基-4-羟基苯乙二醇)的检测用于检测例如酶缺陷(酪氨酸羟化酶、芳族氨基酸脱羧酶、多巴胺β-羟化酶)神经母细胞瘤。在另一个实施方案中,样品是食物样品,例如疑似含有一种或多种可引起健康问题的生物胺的食物样品。本发明方法的步骤(i)包括在具有ph7.0至9.0的磷酸盐缓冲液的存在下使含水样品与丙酸酐/乙腈溶液接触。该步骤允许生物胺或其代谢物的胺部分和/或羟基部分转化以形成丙酰基衍生物。参见用于示意性反应的图1a和图2a。ph为7.0至9.0、优选为7.5至8.8、更优选为8.0至8.5的碱性磷酸盐缓冲液确保发生向丙酰基衍生物的转化,同时其碱性不太大以免引起生物胺的降解。通常,在室温下5-60分钟的孵育时间是足够的。在一个实施方案中,丙酸酐/乙腈溶液是含有10%v/v-50%v/v丙酸酐的乙腈。用含有20%v/v-30%v/v丙酸酐的乙腈获得了非常好的结果。在一个具体的方面,磷酸盐缓冲液的ph为8.0至8.5。例如,合适地使用0.5m磷酸氢二钾缓冲液ph8.5,任选包含1-10mmedta。优选地,将一种或多种内标物添加至(生物)样品中。分析化学中的内标物是在化学分析中以恒定量添加至样品、空白和/或校准标准物中的化学物质。然后,作为标准物的分析物浓度的函数,通过绘制分析物信号与内标物信号的比率,该物质可用于进行校准。这样做是为了校正样品制备期间或样品入口的分析物的损失。内标物通常是非常类似的但不与样品中的目的化学物质相同的化合物,因为在理想情况下,样品制备的作用(相对于各物质的量)对于来自内标物的信号应该与对于来自目的物质的信号相同。本领域技术人员将能够根据衍生化方法的目的和环境选择合适的内标物。氘化的内标物是特别有用的。例如,该标准物选自l-dopa-d3、多巴胺-d4-hcl、去甲肾上腺素-d6-hcl、血清素-d4肌酸酐硫酸盐、5-羟基吲哚乙酸(hiaa)-d2、肾上腺素-d3、3-甲氧基酪胺-d4-hcl、dl-变肾上腺素-d3-hcl和dl-去甲变肾上腺素-d3-hcl。在一个实施方案中,本发明方法的步骤(i)包括混合1体积(生物)样品、1体积内标物和5体积磷酸盐缓冲液,并使混合物与1体积含丙酸酐的乙腈接触。将生物胺转化成丙酰基衍生物后,直接进行第二衍生化步骤,通过向在步骤(i)中获得的反应混合物中添加碳二亚胺试剂和含伯胺的化合物,优选亲电性含胺化合物,并且允许生物胺、其代谢物和/或其前体的羧酸部分进行碳二亚胺介导的衍生化。参见图1b和图2b,用于步骤(ii)中的反应的示意图描述。在一个实施方案中,步骤(ii)包括向1体积的步骤(i)中获得的反应混合物中添加2体积碳二亚胺试剂和2体积含胺化合物。优选地,使用约等摩尔量的碳二亚胺试剂和含伯胺的化合物。根据样品类型,可以添加一定体积的水以稀释步骤(ii)的反应混合物,使得衍生化的生物胺的最终浓度在合适的检测范围内。例如,在尿液样品的情况下,生物胺的浓度相对高,并且优选加入若干体积(例如4-5体积)的水,以防止ms检测信号的饱和。在室温下,第二衍生化步骤的孵育时间通常为约10分钟至60分钟,优选为20分钟-40分钟。令人惊讶地观察到,在第一衍生化步骤中形成的羧基提供了第二衍生化步骤所需的酸性ph,因此不需要样品纯化、改变缓冲液等的中间步骤。水溶性碳二亚胺已在本领域中用作用于将羧酸与胺偶联以产生酰胺衍生物的脱水剂。碳二亚胺介导的衍生化的描述性机理也是已知的(参见例如ford等人,2007)。给定的羧酸盐(酯)和碳二亚胺反应以形成中间体复合物,所述中间体复合物受到诸如胺的亲电体攻击而被活化。用于本发明的合适的水溶性碳二亚胺试剂包括n-乙基-n'-(3-二甲基氨基丙基)碳二亚胺(edc)和1-环己基-3-(2-吗啉代乙基)碳二亚胺(cmc)。含伯胺的化合物置换碳二亚胺以产生相应的酰胺。技术人员将理解,可使用任何的亲电性含胺化合物,其可引入允许质量增加和改善的信噪比的部分。例如,通过引入卤素原子或不是天然存在于生物胺中的其它部分。合适的含胺化合物包括o-苄基羟基胺(o-bha)和2,2,2-三氟乙胺(tfea)。在一个具体的方面,步骤(ii)包括添加2,2,2-三氟乙胺(tfea)和n-乙基-n'-(3-二甲基氨基丙基)碳二亚胺(edc)。优选为0.4mtfea和0.4medc。当含(伯)胺的化合物在碳二亚胺试剂之前添加,优选地在其间有涡旋时,获得非常好的结果。例如,在一个实施方案中,步骤(ii)包括在edc之前添加tfea。在完成步骤(ii)后,该方法可涉及离心步骤。例如,在血浆样品衍生化的情况下,将最终的反应混合物有利地进行离心并且将所得的上清液用于随后的分析。根据本发明的两步衍生化方法优选地随后检测至少一种衍生化的生物胺,例如通过使衍生化的样品经历(在线或离线)spe-lc-ms/ms或任何其它合适的分析技术。鉴于使用本文所述的方法形成的衍生物的高稳定性,无需在衍生化之后立即进行检测。相反,出于实用和经济的原因,可能优选地在不同的日子制备和收集多个(一系列)衍生化的样品,并且在稍后的时间点使它们经历检测步骤。例如,在一个实施方案中,在衍生化之后至少一周进行检测(例如使用spe-lc-ms/ms)。在一个实施方案中,检测步骤包括通过电喷雾离子化在质谱仪中使生物胺或其代谢物离子化,以产生生物胺或其代谢物的质子化的分子离子;使质子化的分子离子碎裂以产生产物离子,并检测质子化的分子离子或产物离子中的至少一种的存在或其量,其中所检测的离子的存在或其量与生物样品中的生物胺或其代谢物的存在或其量有关。本发明的另一个方面涉及用于实施根据本发明的原位衍生化方法的试剂盒部件。所述试剂盒至少包含(i)第一容器,其包含10%v/v-50%v/v丙酸酐的乙腈溶液;(ii)第二容器,其包含碳二亚胺试剂,优选n-乙基-n'-(3-二甲基氨基丙基)碳二亚胺(edc)或1-环己基-3-(2-吗啉代乙基)碳二亚胺(cmc);以及(iii)第三容器,其包含含伯胺的化合物,优选2,2,2-三氟乙胺(tfea)。所述试剂盒可还包含ph为7.0至9.0的磷酸盐缓冲液和/或至少一种生物胺内标物。优选地,内标物是氘化的标准物。在一个实施方案中,所述试剂盒包含一种或多种内标物,所述内标物选自l-dopa-d3、多巴胺-d4-hcl、去甲肾上腺素-d6-hcl、血清素-d4肌酸酐硫酸盐、5-hiaa-d3、肾上腺素-d3、3-甲氧基酪胺-d4-hcl、dl-变肾上腺素-d3-hcl和dl-去甲变肾上腺素-d3-hcl。附图的图例图1.步骤(i)中生物胺(在这种情况下,5-hiaa)与丙酸酐的示意性衍生化反应(图a),随后在步骤(ii)中采用碳二亚胺试剂(此处:edc)和亲电性含胺化合物(此处:tfea)的生物胺的羧酸部分的衍生化(图b)。图2.步骤(i)中生物胺(在这种情况下,l-dopa)与丙酸酐的示意性衍生化反应(图a),随后在步骤(ii)中采用碳二亚胺试剂(此处:edc)和亲电性含胺化合物(此处:tfea)的生物胺的羧酸部分的衍生化(图b)。图3.根据本发明的双重衍生化方案衍生化的并进行在线spe和lc-ms/ms分析的尿液样品的代表性色谱图。图4.得自根据本发明的双重衍生化方案衍生化的并进行在线spe和lc-ms/ms分析的尿液样品的代表性色谱图。详情参见实施例2。图5.得自根据本发明的双重衍生化方案衍生化的并进行在线spe和lc-ms/ms分析的csf样品的典型色谱图。详情参见实施例3。实验部分实施例1:尿液中的双重衍生化方法和衍生物的分析。材料和方法试剂lc-ms级乙腈、异丙醇、甲醇、甲酸和乙酸铵购自biosolvebv(valkenswaard,thenetherlands)。抗坏血酸、磷酸氢二钾和盐酸(32%)来自merckmillipore(darmstadt,germany)。氢氧化铵溶液(28%-30%)、2,2,2-三氟乙胺盐酸盐(tfea)、n-(3-二甲基氨基丙基)-n’-乙基碳二亚胺盐酸盐(edc)、丙酸酐、k2edta二水合物、l-dopa、多巴胺-hcl、去甲肾上腺素、肾上腺素、3-甲氧基酪胺、dl-变肾上腺素-hcl、dl-去甲变肾上腺素-hcl、血清素、5-羟基吲哚乙酸(5-hiaa)和l-dopa-d3(所有都是分析纯度)都购自sigmaaldrich(missouri,usa)。多巴胺-d4-hcl、去甲肾上腺素-d6-hcl、血清素-d4肌酸酐硫酸盐、5-hiaa-d2和肾上腺素-d3的稳定的氘化同位素来自cdnisotopes(pointe-claire,加拿大),并且3-甲氧基酪胺d4-hcl和dl-变肾上腺素-d3-hcl来自cambridgeisotopes(massachusetts,usa)以及dl-去甲变肾上腺素-d3-hcl来自medicalisotopes(newhampshire,usa)。使用内部净化系统(merckmillipore,massachusetts,usa)产生超纯水。衍生化在分析样品之前,将等分的尿液(50μl)与50μl内标物工作溶液、200μl水、250μl的0.5m磷酸氢二钾、4mmk2edta,ph8.5在2.0ml96-深孔板(greinerbio-one,kremsmünster,austria)中混合。随后,添加50μl的含20%(v/v)丙酸酐的乙腈,将该板涡旋三十分钟。此后,将100μl的0.4mtfea、100μl的0.4medc和水添加至所有孔中,以填满至1.0ml的体积。将所述板涡旋三十分钟,并在1,500xg下离心十五分钟。将50μl的每个校准物和样品注入在线spelc-ms/ms系统中,如下所述。lc-ms/ms使用全自动sparkhollandsymbiosistm系统在提取液相色谱法(xlc)模式中进行在线固相提取(spe),如先前所描述的(dejong,w.h.a.等人,plasmafreemetanephrinemeasurementusingautomatedonlinesolid-phaseextractionhplctandemmassspectrometry.clin.chem.53,1684–93(2007))。如下药盒用于在线spe:oasishlb10×1mm,30μm(waters)。每个药盒最初在左侧夹具位置用500μl乙腈、500μl的含有0.2%甲酸的甲醇/异丙醇/乙腈/水(1∶1∶1∶1)混合物调节,然后以5000μl/min的流速用500μl水平衡。吸取样品(100μl)并以2000μl/min的流速加载到具有500μl水的药盒中。用三种不同的溶剂组合物进行三次洗涤步骤:1)500μl20%甲醇、4mm乙酸铵和0.4%氢氧化铵,2500μl/min的流速,2)500μl20%甲醇、4mm乙酸铵和0.4%甲酸,2500μl/min的流速和3)250μl20%乙腈、4mm乙酸铵和0.4%甲酸,2500μl/min的流速。在洗涤后,将药盒转移至右侧夹具,并通过使用梯度洗脱来洗脱皮质醇和褪黑激素:用流动相洗脱药盒,起始梯度持续3:00min。在进行洗脱后,用500μl40%乙腈、0.1%甲酸以5000μl/min的流速,500μl的混合物甲醇/异丙醇/乙腈/水(4∶1∶1∶1)和0.2%甲酸以5000μl/min的流速,500μl乙腈以5000μl/min的流速和最后500μl水以5000μl/min的流速冲洗右侧夹具。将新的药盒放置在左侧夹具中,使得下一个样品经历spe,同时对先前的样品进行色谱分析。用700μl10%乙腈、750μl40%乙腈、0.1%甲酸,然后用750μl的甲醇/异丙醇/乙腈/水(4:2:2:2(v/v))混合物和0.2%甲酸,然后再用700μl10%乙腈洗涤自动进样器。用二元梯度系统在lunaphenyl-hexyl2.0×150mm3μm柱上进行液相色谱法,所述二元梯度系统由含有0.1%甲酸的10mm乙酸铵(洗脱液a)和含0.1%甲酸的乙腈(洗脱液b)组成。初始条件为80:20(v/v),洗脱液a:洗脱液b,流速为0.3ml/min,随后洗脱液b线性增加至55%,历时8.5分钟,然后快速线性增加至100%b,此时保持恒定持续1.0分钟。此后,将泵的流速和比例恢复至起始条件并保持恒定再持续两分钟。总运行时间为12分钟。以正离子化模式在quattropremier上分析所有分析物。通过在选择性反应监测模式(srm)中进行调节来优化质谱仪设置。在整个研究中应用以下设置:毛细管电压0.5kv、去溶剂化温度450℃、去溶剂化气体流速1000l/h、锥气体流速50l/h和碰撞气体流速0.20ml/min。针对所有分析物和各自的转变优化锥电压和碰撞能量,并列在表1中。通过使用分析物和相应的内标物的定量仪转换(quantifiertransition)的峰面积响应比来进行定量。用targetlynxtm软件(waters,milford,usa)进行计算。表1.列出了每种化合物的定量仪和定性仪的质谱仪设置。实施例2:尿液中的双重衍生化方法和衍生物的分析。材料和方法试剂lc-ms级乙腈、异丙醇、甲醇、甲酸和乙酸铵购自biosolvebv(valkenswaard,thenetherlands)。抗坏血酸、磷酸氢二钾和盐酸(32%)来自merckmillipore(darmstadt,germany)。氢氧化铵溶液(28%-30%)、2,2,2-三氟乙胺盐酸盐(tfea)、n-(3-二甲基氨基丙基)-n’-乙基碳二亚胺盐酸盐(edc)、丙酸酐、k2edta二水合物、对羟基苯乙酸(pohpaa)、高香草酸(hva)、4-羟基-3-甲氧基苯乙醇(mopet)、4-羟基-3-甲氧基-甲氧基苯乙二醇(mopeg)、香草扁桃酸(vma)、香草乳酸(vla)和3,4-二羟基苯乙酸(dopac)(所有都是分析纯度)都购自sigmaaldrich(missouri,usa)。购买或内部合成用于vma、dopac、pohpaa、mopet和vla的稳定的标记的同位素。使用内部净化系统(merckmillipore,massachusetts,usa)产生超纯水。衍生化在分析样品之前,将等分的尿液(50μl)与50μl内标物工作溶液、400μl水、250μl的0.5m磷酸氢二钾、4mmk2edta,ph8.5在2.0ml96-深孔板(greinerbio-one,kremsmünster,austria)中混合。随后,添加50μl含20%(v/v)丙酸酐的乙腈,并且将该板涡旋三十分钟。此后,将100μl的0.4mtfea、100μl的0.4medc和水添加至所有孔中,以填满至1.0ml的体积。将所述板涡旋三十分钟。将50μl的每个校准物和样品注入在线spelc-ms/ms系统中,如下所述。lc-ms/ms使用全自动sparkhollandsymbiosistm系统在提取液相色谱法(xlc)模式中进行在线固相提取(spe),如先前所描述的(dejong,w.h.a.等人,plasmafreemetanephrinemeasurementusingautomatedonlinesolid-phaseextractionhplctandemmassspectrometry.clin.chem.53,1684–93(2007))。如下药盒用于在线spe:oasishlb10×1mm,30μm(waters)。每个药盒最初在左侧夹具位置用500μl乙腈、500μl的含有0.2%甲酸的甲醇/异丙醇/乙腈/水(1∶1∶1∶1)混合物调节,然后以4000μl/min的流速用1000μl0.1%甲酸(fa)水溶液平衡。吸取样品(50μl)并以2000μl/min的流速加载到具有600μl0.1%fa水溶液的药盒中。用三种不同的溶剂组合物进行三次洗涤步骤:1)500μl20%甲醇、4mm乙酸铵和0.4%氢氧化铵,2500μl/min的流速,2)500μl20%甲醇、4mm乙酸铵和0.4%甲酸,2500μl/min的流速和3)250μl20%乙腈、4mm乙酸铵和0.4%甲酸,2500μl/min的流速。在洗涤后,将药盒转移至右侧夹具,并通过使用梯度洗脱来洗脱衍生物:用流动相洗脱药盒,起始梯度持续3:00min。在进行洗脱后,用750μl40%乙腈、0.1%甲酸以5000μl/min的流速,750μl甲醇/异丙醇/乙腈/水(1:1:1:1)混合物和0.2%甲酸以5000μl/min的流速,750μl乙腈以5000μl/min的流速和最后750μl水以5000μl/min的流速冲洗右侧夹具。将新的药盒放置在左侧夹具中,使得下一个样品经历spe,同时对先前的样品进行色谱分析。用700μl10%乙腈、750μl40%乙腈、0.1%甲酸,然后用750μl甲醇/异丙醇/乙腈/水(4:2:2:2(v/v))混合物和0.2%甲酸,然后再用700μl10%乙腈洗涤自动进样器。用二元梯度系统在lunaphenyl-hexyl2.0×150mm3μm柱上进行液相色谱法,所述二元梯度系统由含有0.1%甲酸的10mm乙酸铵(洗脱液a)和含0.1%甲酸的乙腈(洗脱液b)组成。初始条件为75:25(v/v),洗脱液a:洗脱液b,流速为0.3ml/min,随后洗脱液b线性增加至45%,历时2分钟,然后线性增加至65%b,历时4分钟,然后快速线性增加至90%b,此时保持恒定持续1.0分钟。此后,将泵的流速和比例恢复至起始条件并保持恒定再持续两分钟。总运行时间为9.5分钟。以正离子化模式在quattropremier上分析所有分析物。通过在选择性反应监测模式(srm)中进行调节来优化质谱仪设置。在整个研究中应用以下设置:毛细管电压1.0kv、去溶剂化温度450℃、去溶剂化气体流速1100l/h、锥气体流速100l/h和碰撞气体流速0.15ml/min。针对所有分析物和各自的转变优化锥电压和碰撞能量,并列在表2中。通过使用分析物和相应的内标物的定量仪转换的峰面积响应比来进行定量。用targetlynxtm软件(waters,milford,usa)进行计算。表2.列出了每种化合物的定量仪和定性仪的质谱仪设置。化合物名称前体(m/z)产物(m/z)锥(v)碰撞(v)1pohpaa-quan291.00233.9530.0014.002pohpaa-qual291.00106.9530.0026.003pohpaa-d4-quan295.00237.9530.0014.004pohpaa-d4-qual295.00110.9530.0026.005mopet-quan298.00150.9516.0018.006mopet-qual298.0090.9516.0045.007mopet-d3-quan301.00153.9516.0018.008mopet-d3-qual301.0093.9516.0045.009hva-quan337.00136.9516.0028.0010hva-qual337.00264.0016.0015.0011hva-13c6-quan345.00144.9516.0028.0012hva-13c6-qual345.00272.0016.0015.0013mopeg-quan258.00166.9514.0013.0014mopeg-qual258.00106.9514.0027.0015mopeg-13c6-quan264.00172.9514.0013.0016mopeg-13c6-qual264.00112.9514.0027.0017vma-quan353.00229.9516.0032.0018vma-qual353.00109.9516.0045.0019vma-d3-quan356.00229.9516.0032.0020vma-d3-qual356.00109.9516.0045.0021dopac-quan306.00122.9535.0022.0022dopac-qual306.00178.9535.0011.0023dopac-d3-quan309.00125.9535.0022.0024dopac-d3-qual309.00181.9535.0011.0025vla-quan350.00176.9525.0018.0026vla-qual350.00148.9525.0030.0027vla-d3-quan353.00178.9525.0018.0028vla-d3-qual353.00150.9525.0030.00实施例3:脑脊液(csf)中的双重衍生化方法和衍生物的分析。试剂lc-ms级乙腈、异丙醇、甲醇、甲酸和乙酸铵购自biosolvebv(valkenswaard,thenetherlands)。抗坏血酸、磷酸氢二钾和盐酸(32%)来自merckmillipore(darmstadt,germany)。氢氧化铵溶液(28%-30%)、2,2,2-三氟乙胺盐酸盐(tfea)、n-(3-二甲基氨基丙基)-n’-乙基碳二亚胺盐酸盐(edc)、丙酸酐、k2edta二水合物、高香草酸(hva)、4-羟基-3-甲氧基-甲氧基苯乙二醇(mopeg)、多巴胺、5-hiaa和3,4-二羟基苯乙酸(dopac)(所有都是分析纯度)都购自sigmaaldrich(missouri,usa)。购买或内部合成稳定的标记的同位素。使用内部净化系统(merckmillipore,massachusetts,usa)产生超纯水。衍生化除了使用100μlcsf样品和350μl水之外,与实施例1和实施例2相同。lc-ms/ms与实施例2相同,但是使用表3中提及的ms设置。表3.列出了每种化合物的定量仪和定性仪的质谱仪设置。参考文献1.zhaox-e,zhus,yangh,youj,songf,liuz等人,simultaneousdeterminationofaminoacidandmonoamineneurotransmittersinpc12cellsandratsmodelsofparkinson’sdiseaseusingasensitizingderivatizationreagentbyuhplc-ms/ms.jchromatogrbanalyttechnolbiomedlifesci.2015;995-996c:15–23。2.zhangg,zhangy,jic,mcdonaldt,waltonj,groeberea等人,ultrasensitivemeasurementofendogenousepinephrineandnorepinephrineinhumanplasmabysemi-automatedspe-lc-ms/ms.jchromatogrbanalyttechnolbiomedlifesci.2012;895-896:186–90。3.vandemerbelnc,hendriksg,imbosr,tuunainenj,rouruj,nikkanenh.quantitativedeterminationoffreeandtotaldopamineinhumanplasmabylc-ms/ms:theimportanceofsamplepreparation.bioanalysis.2011;3:1949–61。4.tanb,luz,dongs,zhaog,kuom-s.derivatizationofthetricarboxylicacidintermediateswitho-benzylhydroxylamineforliquidchromatography-tandemmassspectrometrydetection.analbiochem.2014;465:134–47。5.fordql,burnsjm,ferryjl.aqueousinsituderivatizationofcarboxylicacidsbyanioniccarbodiimideand2,2,2-trifluoroethylamineforelectron-capturedetection.jchromatogra.2007;1145:241–5。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1