一种门冬氨酸洛美沙星中洛美沙星异构体的分离测定方法与流程
本发明涉及药物分析领域,具体涉及一种门冬氨酸洛美沙星中洛美沙星异构体的分离测定方法。
背景技术:
门冬氨酸洛美沙星结构式存在2个手性碳,分别来自门冬氨酸和洛美沙星。对门冬氨酸洛美沙星原料进行比旋度测试,结果发现门冬氨酸洛美沙星原料比旋度介于﹣9.9°到﹢7.8°之间,为弄清比旋度差异的原因,本发明人进行了门冬氨酸异构体测定研究,发现所有原料和制剂中门冬氨酸均为l-门冬氨酸,由此说明门冬氨酸洛美沙星原料比旋度的差异是由洛美沙星左右旋异构体比例之间差异所致。由于左右旋洛美沙星在抑菌性能和细胞毒性特性上均存在较大差异,建立一种门冬氨酸洛美沙星中洛美沙星异构体的分离测定方法来控制和监测门冬氨酸洛美沙星中左右旋洛美沙星异构体的量非常必要。
查询相关文献,企业标准和药典中均未有对门冬氨酸洛美沙星中洛美沙星异构体测定方法的报道。
技术实现要素:
本发明的目的在于提供一种门冬氨酸洛美沙星中洛美沙星异构体的分离测定方法,该方法能够实现门冬氨酸洛美沙星中左右旋洛美沙星峰的完全分离,且该方法具有很好的专属性、稳定性和精密度等,此外,该方法具有很好的灵敏度,其检出限和定量限均达到纳克级。可有效地应用于门冬氨酸洛美沙星的质量控制。
本发明的上述目的是采用以下技术方案来实现:
本发明提供一种门冬氨酸洛美沙星中洛美沙星异构体的分离测定方法,所述方法采用高效液相色谱法进行,所述高效液相色谱法采用改性β-环糊精为填料的手性色谱柱,并以高氯酸盐溶液和甲醇组成流动相体系。
在本发明的技术方案中,优选地,所述改性β-环糊精为填料的手性色谱柱型号为chiralcd-ph。
在本发明的技术方案中,优选地,所述高氯酸盐溶液的浓度为0.1~0.5mol/l;更优选地,所述高氯酸盐溶液的浓度为0.5mol/l。
在本发明的技术方案中,优选地,所述高氯酸盐为高氯酸钠或高氯酸钾;更优选地,所述高氯酸盐为高氯酸钠。
在本发明的技术方案中,优选地,所述高氯酸盐溶液的ph为3.0~4.0,所述ph的调节剂是高氯酸;更优选地,所述高氯酸盐溶液的ph为3.5。
在本发明的技术方案中,优选地,所述门冬氨酸洛美沙星中门冬氨酸为l-型门冬氨酸。
在本发明的技术方案中,优选地,所述高氯酸盐溶液与所述甲醇的体积比为(30~50):(50~70);更优选地,所述高氯酸盐溶液与所述甲醇的体积比为40:60。
作为一种具体的实施方式,所述方法包括以下步骤:
(1)用50%甲醇溶解门冬氨酸洛美沙星原料药,配制成每1ml含有洛美沙星0.5~2.0mg的样品溶液;
(2)设置流动相流速为0.5~1.5ml/min,检测波长为280~290nm,色谱柱柱温为40~50℃;
(3)取步骤(1)中的样品溶液5~20μl注入色谱仪,记录色谱图。
作为一种具体的实施方式,所述方法还包括提供包含盐酸洛美沙星对照品的系统适用性溶液,并采用步骤(2)中的方法对所述系统适用性溶液进行检测,记录色谱图。
在本发明的技术方案中,优选地,所述系统适用性溶液每1ml含有洛美沙星1.0mg。
本发明提供的一种门冬氨酸洛美沙星中洛美沙星异构体的测定方法的有益效果:本发明提供的方法采用高效液相色谱法进行门冬氨酸洛美沙星中洛美沙星异构体的测定,采用改性β-环糊精为填料的手性色谱柱,并以高氯酸盐溶液和甲醇组成流动相体系。该方法能够实现门冬氨酸洛美沙星中左右旋洛美沙星峰的完全分离,其分离度为2.0,且该方法具有很好的专属性、稳定性和精密度等,此外,该方法具有很好的灵敏度,其检出限和定量限均达到纳克级,可有效地应用于门冬氨酸洛美沙星的质量控制。
附图说明
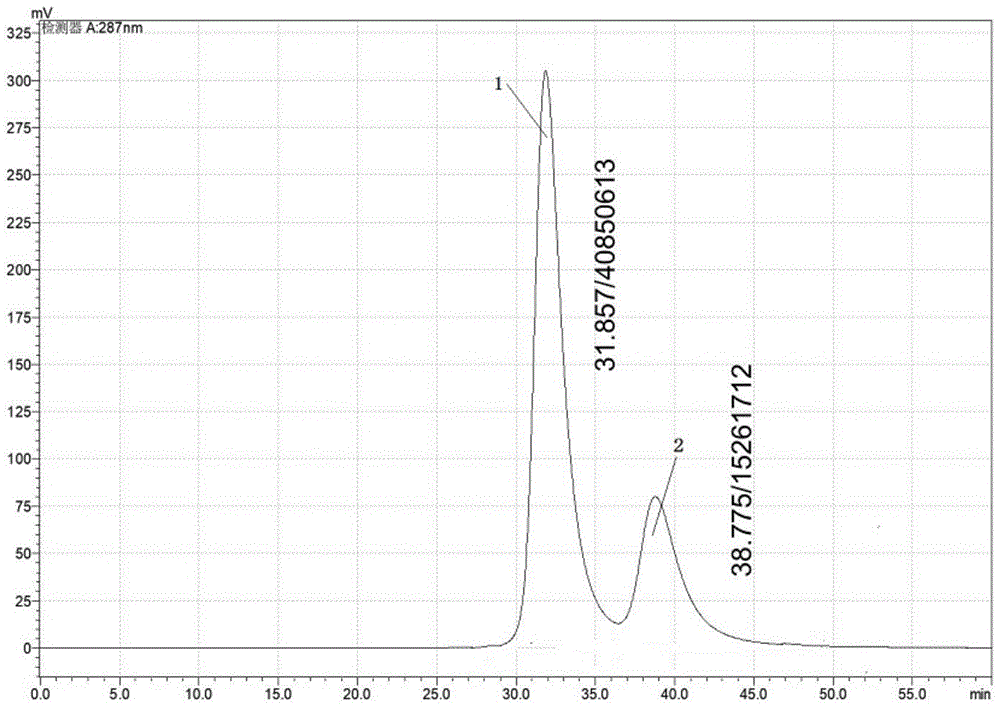
图1为实施例2所得系统适用性溶液的高效液相色谱图;
图2为实施例2所得a厂1号原料的高效液相色谱图;
图3为实施例2所得c厂1号原料的高效液相色谱图;
图4为实施例3所得50%甲醇溶液的高效液相色谱图;
图5为实施例6所得洛美沙星线性关系图;
图6为实施例7所得定量限的高效液相色谱图;
图7为实施例7所得检出限的高效液相色谱图;
图中,峰1代表右旋洛美沙星峰;峰2代表左旋洛美沙星峰。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
在下面的实施例中使用的对照品(盐酸)洛美沙星来源于中国食品药品检定研究院(批号:130452-201603,含量:90.4%,其中的有效成分含量均以洛美沙星计算);化学试剂高氯酸钠和高氯酸为分析纯,甲醇为色谱纯,水为纯化水。高效液相色谱仪型号为lc-20ad,购自岛津公司;电子天平型号为ms205du,购自mettler公司。
以下结合实施例对本发明的特征和性能作进一步的详细描述。
实施例1:色谱条件建立
(1)系统适用性溶液的制备
称取(盐酸)洛美沙星对照品33.18mg,置10ml容量瓶,加50%甲醇溶解并稀释至刻度,摇匀,制成含洛美沙星约为3mg/ml的溶液,作为对照品贮备溶液。精密量取该对照品贮备溶液3.3ml,加50%甲醇稀释至10ml,摇匀,制成含洛美沙星约为1mg/ml的溶液,作为系统适用性溶液。
(2)供试品溶液的制备
取门冬氨酸洛美沙星原料适量,加50%甲醇溶液溶解并稀释制成每1ml中含洛美沙星1mg的溶液,摇匀,作为供试品溶液。
(3)色谱条件
流动相:以0.5mol/l高氯酸钠(用高氯酸调ph至3.5)与甲醇按照体积比为40:60配成流动相体系;
检测波长:287nm;
色谱柱:chiralcd-ph;4.6mmi.d.×250mm(β-环糊精键合硅胶为填充剂);
流速:1.0ml/min;
柱温:45℃;
进样量10μl。
实施例2:系统适用性试验
取实施例1提供的系统适用性溶液10μl注入液相色谱仪,按照实施例1中的色谱条件进行高效液相色谱分析,记录色谱图,检测结果见图1,图中,峰1代表右旋洛美沙星;峰2代表左旋洛美沙星。
由图1可知,右旋洛美沙星峰与左旋洛美沙星峰之间分离度为2.0(大于1.5),说明本测试方式对右旋洛美沙星与左旋洛美沙星的分离度很好。
其中,左右旋洛美沙星的出峰顺序通过以下方法确定:
取四个厂家的天冬氨酸洛美沙星原料,先进行比旋度测定,结果如表1所示,然后将表1所示各原料分别按照实施例1的方法配制供试品溶液,取各供试品溶液10μl,分别注入液相色谱仪,按照实施例1的色谱条件进行高效液相色谱分析,记录色谱图,a厂1号原料的检测结果见图2,c厂1号原料的检测结果见图3;
表1门冬氨酸洛美沙星原料比旋度测定结果表
备注:表1中原料名称1号、2号、3号分别表示同批次原料的不同包装袋中的原料;表1中a,b,c厂的门冬氨酸洛美沙星原料均由d厂供应。
从表1可知,a厂1号原料旋光呈左旋,c厂1号原料旋光呈右旋。从图2可知,a厂1号原料峰1和峰2的面积比为41:59,由图3可知,c厂1号原料峰1和峰2的面积比为73:27。将图2~3结合表1提供的原料旋光性可知,峰1为右旋洛美沙星峰;峰2为左旋洛美沙星峰。
实施例3:专属性试验
取50%甲醇溶液10μl,分别注入液相色谱仪,记录色谱图见图4,结果显示50%的甲醇在左、右旋洛美沙星保留时间处均无色谱峰,不存在干扰。表明该方法专属性良好。
实施例4:稳定性试验
取天冬氨酸洛美沙星原料(d厂3号原料,批号20151201)25mg,置25ml量瓶中,加50%甲醇稀释至刻度,摇匀,按照实施例1的色谱条件进行高效液相色谱分析,结果见表2:
表2门冬氨酸洛美沙星原料中洛美沙星稳定性试验数据
由表2可知,右旋洛美沙星峰面积的rsd为0.2%,左旋洛美沙星峰面积的rsd为0.3%,说明左右旋洛美沙星在24h内稳定性很好。
实施例5:精密度试验
取系统适用性溶液连续进样6针,按照实施例1的条件进行高效液相色谱分析,记录色谱图,结果见表3:
表3洛美沙星精密度试验结果
由表3可知,右旋洛美沙星峰面积和左旋洛美沙星峰面积rsd分别为0.1%和0.2%,说明本方法精密度很好。
实施例6:线性关系
精密量取对照品贮备溶液3.3ml和2.5ml,分别置5ml量瓶中,加50%甲醇稀释至刻度,摇匀,制成含洛美沙星浓度约为2mg/ml和1.5mg/ml的溶液;精密量取2mg/ml的洛美沙星溶液2.5ml、1.25ml和625μl,分别置5ml量瓶中,加50%甲醇稀释至刻度,摇匀,制成浓度约为1mg/ml、500μg/ml和250μg/ml的洛美沙星溶液;精密量取1mg/ml的洛美沙星溶液1ml,置10ml量瓶中,加50%甲醇稀释至刻度,摇匀,制成100μg/ml的洛美沙星溶液。取上述溶液进行色谱分析,以峰面积(左、右旋洛美沙星峰面积和)为纵坐标,对照品浓度为横坐标,绘制标准曲线,回归方程为y=63282664x+11764,r2=1,结果见图5。
由图5可知,洛美沙星在浓度为99.98μg/ml~2mg/ml之间呈良好线性。
实施例7:检出限、定量限
精密量取浓度约为100μg/ml的洛美沙星对照品溶液(按照实施例6提供的方法配制)100μl和300μl,置50ml量瓶中,加50%甲醇稀释至刻度,摇匀,制成浓度约为200ng/ml和600ng/ml的洛美沙星溶液。按照实施例1中的色谱条件分别进行测定,记录色谱图,其中定量限的色谱图如图6所示,检出限的色谱图如图7所示,结果发现该方法洛美沙星的检出限和定量限分别为2ng和6ng,左旋洛美沙星的检出限和定量限分别为1ng和3ng,右旋洛美沙星的检出限和定量限分别为1ng和3ng,说明本发明的方法灵敏度高。
实施例8:重复性试验
取d厂3号原料(批号20151201)6份各25mg,置25ml量瓶中,加50%甲醇稀释至刻度,摇匀,按照实施例1的色谱条件进行高效液相色谱分析,结果见表4:
表4原料中洛美沙星重复性试验数据
由上表可知,样品中右旋洛美沙星rsd为0.6%,左旋洛美沙星rsd为0.6%,表明该方法重复性较好。
实施例9:样品测定
按照实施例1中方法配制供试品溶液,取各供试品溶液10μl,按照实施例1的色谱条件进行高效液相色谱分析进行测定,记录色谱图,根据右旋洛美沙星和左旋洛美沙星的峰面积比例计算得出各原料右旋洛美沙星和左旋洛美沙星的一一相对含量百分比,结果见表5:
表5对照品及原料洛美沙星手性异构体检测数据
备注:表5中原料名称1号、2号、3号分别表示同批次原料的不同包装袋中原料;表5中a,b,c厂的门冬氨酸洛美沙星原料均由d厂供应。
由表5可知,本发明提供的测定方法能够对门冬氨酸洛美沙星中左右旋洛美沙星进行分离并分析原料中左右旋洛美沙星的一一相对含量百分比,可有效地应用于门冬氨酸洛美沙星的质量控制。
以上所描述的实施例是本发明一部分实施例,而不是全部的实施例。本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!