银杏洋参胶囊中大孔树脂残留溶剂的检查方法与流程
本发明涉及中医药检测技术领域,具体是一种银杏洋参胶囊中大孔树脂残留溶剂的检查方法。
背景技术:
银杏洋参胶囊是一种处方药,由银杏叶和西洋参2味中药制备而成,用于气虚血瘀症、冠心病(胸痹)的辅助治疗,对胸闷、憋气、心悸等症状有改善作用。大孔树脂(macroporousresin)又称全多孔树脂、聚合物吸附剂,它是一类以吸附为特点,对有机物具有浓缩、分离作用的高分子聚合物。大孔吸附树脂主要以苯乙烯、二乙烯苯等为原料,在0.5%的明胶溶液中,加入一定比例的致孔剂聚合而成。其中,苯乙烯为聚合单体,二乙烯苯为交联剂,甲苯、二甲苯等作为致孔剂,它们互相交联聚合形成了大孔吸附树脂的多孔骨架结构。由于大孔吸附树脂新技术的引进,使中草药有效单体成分或复方中某一单体成分的指标得到提高;它具有快速、高效、方便、灵敏、选择性好等优点,因而发展速度很快,应用面很广。银杏洋参胶囊的制备过程中,用到大孔吸附树脂来提高成分的指标,因大孔树脂在生产、存储过程中易存在正己烷、苯、甲苯、二甲苯、苯乙烯及二乙基苯等毒性溶剂的残留。为保障药品的安全、有效,国家食品药品监督管理局对于使用大孔树脂提取的产品均要求进行残留溶剂的检查。而对于银杏洋参胶囊中大孔树脂残留溶剂的检查方法暂未见有相关的报道。
技术实现要素:
本发明的目的是提供一种银杏洋参胶囊中大孔树脂残留溶剂的检查方法,该方法是采用顶空-气相色谱法同时测定银杏洋参胶囊中正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯的含量,检查大孔树脂残留溶剂是否符合要求;为全面、准确地控制银杏洋参胶囊中的大孔树脂残留溶剂,保障药品安全、有效,提供了技术支撑。
一种银杏洋参胶囊中大孔树脂残留溶剂的检查方法,具体包括以下内容:
(1)色谱条件:色谱柱为db-wax宽口径毛细管柱(直径0.53mm×30m),柱温:40℃;气化室温度为200℃;检测器温度为250℃;载气:氮气,柱头压100pa;流速:约2ml/min;
顶空条件:平衡温度85℃,定量环110℃,传输管120℃,平衡时间30min,加压时间0.20min,定量环填充时间0.20min,定量环平衡时间0.05min,进样时间1min,进样量1ml。
(2)供试品溶液的制备:取银杏洋参胶囊装量差异项下的内容物,精密称取0.5g,置顶空瓶中,精密加水5ml,振摇使样品分散溶解,加盖密封,即得。
(3)对照品溶液的制备:
1)精密称取正己烷110.0mg、苯107.0mg、甲苯123.5mg、间二甲苯112.2mg、苯乙烯85.4mg、1,4—二乙基苯109.8mg、1,2—二乙基苯140.3mg分别置50ml量瓶中,加n,n—二甲基甲酰胺溶解并稀释至刻度,作为对照品储备液;
2)分别精密量取上述7种对照品储备液各适量,置同一250ml量瓶中,加水稀释至刻度,配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各0.25μg的溶液,得到混合对照品溶液①;配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各0.5μg的溶液,得到混合对照品溶液②;配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各1.0μg的溶液,得到混合对照品溶液③;配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各1.5μg的溶液,得到混合对照品溶液④;配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各2.0μg的溶液,得到混合对照品溶液⑤;
3)精密量取以上5个浓度梯度的混合对照品溶液各5ml,分别置不同顶空瓶中,加盖密封,即得。
(4)测定:取对照品溶液和供试品溶液的顶空气各1ml,分别注入气相色谱仪,记录色谱图,按外标峰面积法,以标准曲线计算含量。
所述方法还包括取浓度为0.5μg/ml的混合对照品溶液②用n,n—二甲基甲酰胺逐步稀释,以信噪比s/n=3计算检测限;在此试验条件下,苯、甲苯、二甲苯、苯乙烯的检测限为0.1μg/ml,正己烷、1,2—二乙基苯、1,4—二乙基苯的检测限为0.2μg/ml。
本发明的有益效果是:
银杏洋参胶囊是本申请人自主研发的独家产品,为了更好的控制产品质量及稳定性,本申请人开展了银杏洋参胶囊质量标准的研究。本发明通过顶空-气相色谱法同时测定银杏洋参胶囊中大孔树脂残留溶剂正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯的含量,可以有效地将大孔树脂残留溶剂控制在《中国药典》所要求的范围内,再结合银杏洋参胶囊中萜类内酯的含量测定方法,建立了银杏洋参胶囊的产品质量标准,为提高银杏洋参胶囊的稳定性提供了科学依据。通过建立银杏洋参胶囊的产品质量标准,可以有效地控制银杏洋参胶囊的产品质量,保证银杏洋参胶囊的药效,也可以避免伪劣药品进入市场扰乱市场秩序。
本发明的方法采用气体进样,可专一性收集样品中的易挥发性成分,与液-液萃取和固相萃取相比既可避免在除去溶剂时引起挥发物的损失,又可降低共提物引起的噪音,具有更高的灵敏度和分析速度,对分析人员和环境危害小,操作简便,是一种符合“绿色分析化学”要求的分析手段。顶空技术的使用,可以免除冗长烦琐的样品前处理过程,避免有机溶剂带入的杂质对分析造成干扰,减少对色谱柱及进样口的污染。而且本发明的方法经过了实验条件的考察以及方法学验证,色谱条件和顶空条件均选择最优参数,其专属性及系统适应性、线性、精密度、稳定性、检测限均符合《中国药典》中药质量标准分析方法验证指导原则的要求。本发明的方法为全面、准确地控制银杏洋参胶囊中的大孔树脂残留溶剂,保障药品安全、有效,提供了技术支撑。
附图说明
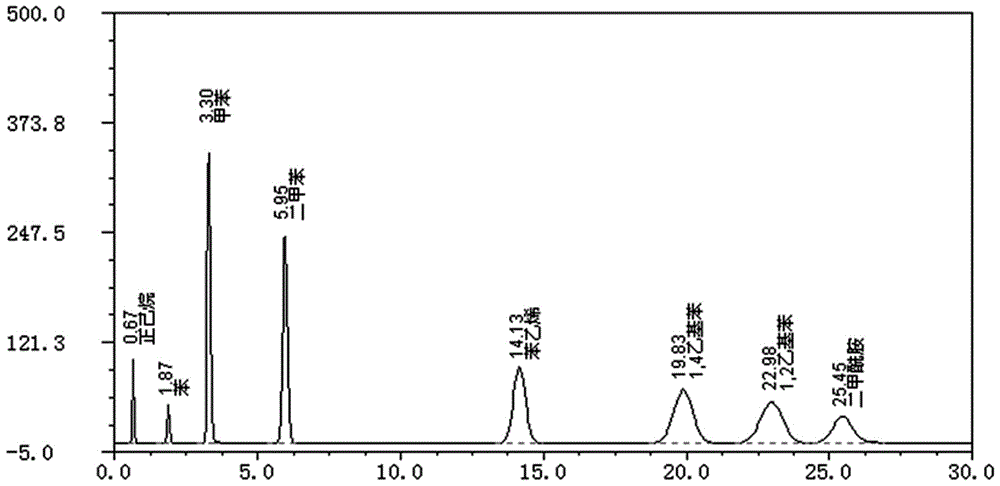
图1是样品中大孔树脂残留溶剂测定时对照品溶液的色谱图;
图2是样品(批号20031105)中大孔树脂残留溶剂测定的色谱图;
图3是样品(批号20031107)中大孔树脂残留溶剂测定的色谱图;
图4是样品(批号20031109)中大孔树脂残留溶剂测定的色谱图;
图5是样品(批号20031109)中大孔树脂残留溶剂测定色谱图的局部放大图,即图4的局部放大图;
图6是n,n-二甲基甲酰胺溶剂的色谱图;
图7是混合对照品溶液③的色谱图(混合对照品溶液的浓度为1.0μg/ml);
图8是混合对照品溶液③中正己烷的色谱图;
图9是混合对照品溶液③中苯的色谱图;
图10是混合对照品溶液③中甲苯的色谱图;
图11是混合对照品溶液③中间二甲苯的色谱图;
图12是混合对照品溶液③中苯乙烯的色谱图;
图13是混合对照品溶液③中1,4-二乙基苯的色谱图;
图14是混合对照品溶液③中1,2-二乙基苯的色谱图;
图15是供试品溶液的色谱图;
图16是树脂提取溶液的色谱图;
图17是阴性空白溶液的色谱图;
图18是标准曲线图;图中从各曲线最高那一点看从下至上分别是正己烷、苯、苯乙烯、甲苯、间二甲苯、1,2—二乙基苯、1,4—二乙基苯;
图19是正己烷、、1,2—二乙基苯、1,4—二乙基苯最低检测限的色谱图(各溶剂检测限浓度为0.2μg/ml);
图20是苯、甲苯、二甲苯、苯乙烯最低检测限的色谱图(各溶剂检测限浓度为0.1μg/ml)。
具体实施方式
为了更加详细的介绍本发明,下面结合实施例,对本发明做进一步说明。
实施例1
一种银杏洋参胶囊中大孔树脂残留溶剂的检查方法,采用顶空-气相色谱法同时测定银杏洋参胶囊中正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯的含量,检查大孔树脂残留溶剂是否符合要求,具体包括以下内容:
(1)色谱条件:色谱柱为db-wax宽口径毛细管柱(直径0.53mm×30m),柱温:40℃;气化室温度为200℃;检测器温度为250℃;载气:氮气,柱头压100pa;流速:约2ml/min;
顶空条件:平衡温度85℃,定量环110℃,传输管120℃,平衡时间30min,加压时间0.20min,定量环填充时间0.20min,定量环平衡时间0.05min,进样时间1min,进样量1ml。
(2)供试品溶液的制备:取银杏洋参胶囊装量差异项下的内容物(即测了装量后,银杏洋参胶囊内的药物),精密称取0.5g,置顶空瓶中,精密加水5ml,振摇使样品分散溶解,加盖密封,即得。
(3)对照品溶液的制备:
1)精密称取正己烷110.0mg、苯107.0mg、甲苯123.5mg、间二甲苯112.2mg、苯乙烯85.4mg、1,4—二乙基苯109.8mg、1,2—二乙基苯140.3mg分别置50ml量瓶中,加n,n—二甲基甲酰胺溶解并稀释至刻度,作为对照品储备液;
2)分别精密量取上述7种对照品储备液各适量,置同一250ml量瓶中,加水稀释至刻度,配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各0.25μg,得到混合对照品溶液①;配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各0.5μg,得到混合对照品溶液②;配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各1.0μg,得到混合对照品溶液③;配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各1.5μg,得到混合对照品溶液④;配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各2.0μg,得到混合对照品溶液⑤;
3)精密量取以上5个浓度梯度的混合对照品溶液各5ml,分别置不同顶空瓶中,加盖密封,即得。
(4)测定:取5个浓度梯度的混合对照品溶液和供试品溶液的顶空气各1ml,分别注入气相色谱仪,记录色谱图,按外标峰面积法,以标准曲线计算含量。
在此试验条件下,苯、甲苯、二甲苯、苯乙烯的检测限为0.1μg/ml,正己烷、1,2—二乙基苯、1,4—二乙基苯的检测限为0.2μg/ml。
测试结果表明,样品含苯量约为《中国药典》二部2005版附录规定限度的1/2,正己烷、甲苯等则大大低于药典的限度值,1,2—二乙基苯、1,4—二乙基苯在样品中未检出,结果见表1-5、图1-5。
表2对照品溶液测定的数据
表2中,组分苯的浓度为0.2μg/ml,正己烷、甲苯、二甲苯、苯乙烯、1,2—二乙基苯、1,4—二乙基苯的浓度均为2μg/ml(当苯的浓度为2μg/ml时,苯的峰值太高会影响后面的峰,因此,把苯的浓度设定为0.2μg/ml使苯的色谱峰能与其他成分的色谱峰出现在同一色谱图中)。
表3样品(批号20031105)中大孔树脂残留溶剂测定的数据
表4样品(批号20031107)中大孔树脂残留溶剂测定的数据
表5样品(批号20031109)中大孔树脂残留溶剂测定的数据
所述方法还包括实验条件的考察和方法学验证,具体内容如下:
1.实验条件的选择:
(1)提取溶剂的选择:
供试品按通常方法以水为溶剂,但在对照品溶液的配制中,因被测成分均为脂溶性,宜先将其溶解后再用水稀释。经考察不同的有机溶剂(乙醇、正丙醇、正丁醇、二甲亚砜等),其中n,n-二甲基甲酰胺对各成分溶解性能较好,并且出峰时间在各成分之后,不干扰测定,见图6,故选用n,n-二甲基甲酰胺作为对照品的提取溶剂。
(2)色谱柱的选择:
毛细管柱由于其具有很高的分离效能,是有机溶剂残留检测之首选。在极性柱和非极性柱中选择,经比较db-wax和db-1毛细管柱,在拟测的成分中,db-1分离效果不及db-wax。同时考虑到宽口径毛细管柱使用的方便性,故选定色谱柱为db-wax宽口径毛细管柱(直径0.53mm×30m)。
(3)色谱条件的选择:
1)柱温:程序升温方法对后出峰的组分(如二乙基苯)峰形会有所改善,但保留时间重现性不好,并且由于供试品中未检出后出峰的组分,故仍采用等温顶空进样方法。经逐步降温比较80~40℃柱温范围内,考察各组分的分离情况,结果在40℃时,不仅各组分分离完全,而且供试品中其它未知组分也不干扰主成分的测定。
2)顶空进样器参数设置:根据测定所用溶剂的性质,通常顶空瓶加热温度应为70~80℃,加热时间为30~60min。经试验,最终参数设置为:平衡温度85℃,定量环110℃,传输管120℃,平衡时间30min,加压时间0.20min,定量环填充时间0.20min,定量环平衡时间0.05min,进样时间1min,进样量1ml。
2.方法学验证:
(1)专属性试验
1)对照品溶液的制备:精密称取正己烷110.0mg、苯107.0mg、甲苯123.5mg、间二甲苯112.2mg、苯乙烯85.4mg、1,4—二乙基苯109.8mg、1,2—二乙基苯140.3mg分别置50ml量瓶中,加n,n—二甲基甲酰胺溶解并稀释至刻度,即得各对照品溶液。
2)混合对照品溶液的制备:分别精密量取上述7种对照品溶液各适量,置同一250ml量瓶中,加水稀释至刻度,配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各1.0μg的混合溶液。
3)银杏洋参胶囊供试品溶液的制备:称取银杏洋参胶囊内容物0.5g,精密称定,置顶空瓶中,精密加水5ml,振摇使样品分散溶解,加盖密封,作为供试品溶液。
4)树脂提取物溶液的制备:称取d101大孔树脂0.5g,精密称定,置顶空瓶中,精密加水5ml,振摇使样品分散溶解,加盖密封,作为树脂提取溶液。
5)阴性空白溶液的制备:按处方比例配制不含银杏叶的阴性样品,精密称取适量(相当于0.5g银杏洋参胶囊内容物),置顶空瓶中,精密加水5ml,振摇使样品分散溶解,加盖密封,作为阴性空白溶液。
6)测定:分别取各对照品溶液、混合对照品溶液、供试品溶液、树脂提取溶液及阴性空白溶液各1ml,注入气相色谱仪,记录色谱图,按外标峰面积法,以标准曲线计算含量。结果被测各组分均完全分离,理论板数按苯峰计,不低于2500,各组分之间的分离度均大于2。见图7-17。
(2)线性关系考察
1)对照品溶液的制备:精密称取正己烷110.0mg、苯107.0mg、甲苯123.5mg、间二甲苯112.2mg、苯乙烯85.4mg、1,4—二乙基苯109.8mg、1,2—二乙基苯140.3mg分别置50ml量瓶中,加n,n—二甲基甲酰胺溶解并稀释至刻度,即得各对照品储备液。
2)分别精密量取上述7种对照品储备液各适量,置同一250ml量瓶中,加水稀释至刻度,配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各0.25μg的溶液,得到混合对照品溶液①(浓度为0.25μg/ml);配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各0.5μg的溶液,得到混合对照品溶液②(浓度为0.5μg/ml);配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各1.0μg的溶液,得到混合对照品溶液③(浓度为1.0μg/ml);配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各1.5μg的溶液,得到混合对照品溶液④(浓度为1.5μg/ml);配成每1ml含正己烷、苯、甲苯、间二甲苯、苯乙烯、1,4—二乙基苯、1,2—二乙基苯各2.0μg的溶液,得到混合对照品溶液⑤(浓度为2.0μg/ml);
3)从以上5个浓度梯度的混合对照品溶液中各精密量取5ml,分别置顶空瓶中,加盖密封,按上述色谱、顶空条件测定。分别以对照品峰面积为纵坐标,对照品浓度为横坐标进行线性回归。结果表明,当进样顶空气为1ml时,各组分在0.25~2.0µg/ml浓度范围内,线性关系良好,见表6、图18。
(3)精密度试验
精密量取专属性试验项下混合对照品溶液5ml5份,分别置5个顶空瓶中,加盖密封,按上述色谱、顶空条件操作,连续进样5次,结果见表7。试验表明,本法的精密度良好。
(4)稳定性试验
精密量取专属性试验项下混合对照品溶液5ml3份,分别置3个顶空瓶中,加盖密封,按上述色谱、顶空条件操作,分别在0小时、24小时、36小时进样测定,结果表明,在36小时内混合对照品溶液稳定,各成分峰面积基本不变,见表8。
(5)检测限
取线性关系考察项下浓度为0.5μg/ml的混合对照品溶液②进行逐级稀释,按上述色谱条件操作,以该成分峰高/基线噪音(s/n)≈3时的浓度为检测限。试验结果,当浓度在0.2μg/ml时,正己烷、1,4—二乙基苯、1,2—二乙基苯已达到s/n≈3,见图19;浓度在0.1μg/ml时,苯、甲苯、二甲苯、苯乙烯也已达到s/n≈3,见图20。因此,苯、甲苯、二甲苯、苯乙烯的检测限为0.1μg/ml(0.5μg),正己烷、1,4—二乙基苯、1,2—二乙基苯的检测限为0.2μg/ml(1.0μg)。
- 还没有人留言评论。精彩留言会获得点赞!