一种氯乙酰肼的检测方法和一种检测磷酸西格列汀中氯乙酰肼的方法与流程
本发明属于分析化学领域,具体涉及一种氯乙酰肼的检测方法和一种检测磷酸西格列汀中氯乙酰肼的方法。
背景技术:
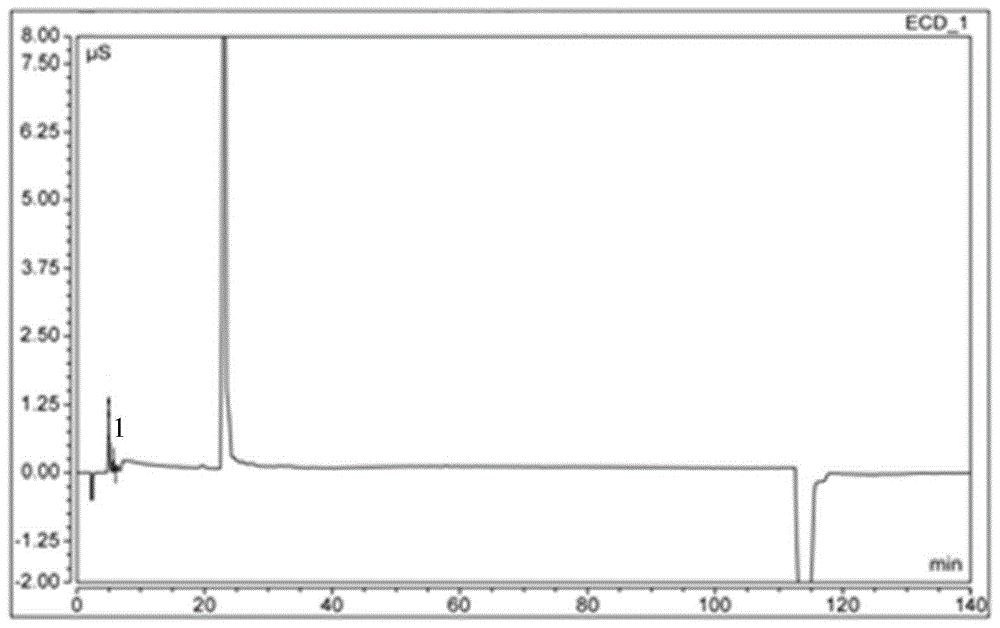
:糖尿病是一种以糖代谢紊乱为主的慢性综合性疾病,包括胰岛素依赖型糖尿病(i型糖尿病)、非胰岛素依赖型糖尿病(ii型糖尿病,占比90%)及妊娠期糖尿病。国际糖尿病联盟发布的全球糖尿病地图(第8版)显示,2017年全球糖尿病患病率(20~79岁)大约为8.8%,约有4.25亿成人糖尿病患者,到2045年,这一数字可能达到6.29亿。中国是糖尿病患者人数最多的国家,约占世界总数的四分之一。糖尿病作为一种高发慢性病,具有病情不可逆、后期并发症多等特点。血糖若控制不佳,糖尿病慢性化可引发糖尿病肾病、糖尿病视网膜病变等并发症,造成肾衰、失明等严重后果,给社会和家庭造成巨大负担。同时,糖尿病也是引起众多心血管疾病的危险因素,如心肌梗死、脑卒中等。糖尿病的临床治疗需求一直未得到充分满足。相关降糖机制研究表明,人体内血糖升高时可促进活性肠促胰岛激素胰高血糖素样肽-1(glp-1)和葡萄糖依赖性促胰岛素分泌肽(gip)的分泌,进而激发胰岛素的合成分泌过程,最终达到控制血糖的效果。体内的glp-1和gip容易被二肽基肽酶iv(dpp-iv)降解失活;因此,二肽基肽酶iv成为糖尿病治疗的靶点。磷酸西格列汀是首个获得美国食品药品监督管理局(fda)批准用于治疗ii型糖尿病的二肽基肽酶-4抑制剂(dpp-4抑制剂),可有效抑制胰高血糖素样肽-1(glp-1)和葡萄糖依赖性促胰岛素分泌多肽(gip)的灭活,提高内源性glp-1和gip的水平,促进胰岛β细胞释放胰岛素,同时抑制胰岛α细胞分泌胰高血糖素,从而提高胰岛素水平,降低血糖,且不易诱发低血糖和增加体重。磷酸西格列汀,化学名称:7-[(3r)-3-氨基-1-氧-4-(2,4,5-三氟苯基)丁基]-5,6,7,8-四氢-3-(三氟甲基)-1,2,4-三唑酮[4,3-a]吡嗪磷酸盐(1∶1)一水合物,化学分子式c16h15f6n5o·h3po4·h2o,化学结构如式(i)所示。3-(三氟甲基)-5,6,7,8-四氢-[1,2,4]三唑并[4,3-a]吡嗪盐酸盐(以下简称“xg-sm2”),结构式如式(ii)所示,是合成磷酸西格列汀过程中的一个重要中间体。氯乙酰氯是xg-sm2合成的起始物料之一,其非常易与肼(合成西格列汀原料药时所用到的另一种起始物料)反应生成氯乙酰肼。而氯乙酰酰肼不再参与其他反应,可能会在终产物中残留。酰肼具有基因毒警示结构;在药品安全性日益得到重视的情况下,药物中残留的极微量杂质的检测也逐步纳入药品质量标准或企业内控标准。因此,有必要建立氯乙酰肼的检测方法,并将其应用于磷酸西格列汀中可能存在的微量氯乙酰肼的检测方法,以确保临床患者用药的安全。现有技术中,酰肼类的检测多采用反相高效液相。如公告号为cn101876649b(公告日2012年12月5日)的中国发明专利,公开了以硅胶键合c18为固定相,以0.1m的醋酸水溶液(用醋酸铵调节ph=4.8)为流动相检测烟草或烟草制品中马来酰肼的残留量。又如公告号cn103592381b(公告日2015年1月14日)的中国发明专利,公开了以sinochromods-bp为固定相,体积比10∶90的已腈和离子对缓冲液(含有1.7g/l的十二烷基磺酸钠和1.0ml/l的磷酸)为流动相检测磺酸苯肼硫酸盐。但是迄今尚未见到基于高效液相色谱法检测氯乙酰肼的报道。发明人研究发现氯乙酰肼分子极性较大,在普通的反相硅胶色谱柱中保留极差,因此不能采用普通的反相高效液相色谱法来检测氯乙酰肼。离子色谱是以离子交换树脂或化学键合离子交换剂为固定相,利用被分离组分离子交换能力的差别或选择性系数的差别而实现分离的色谱方法,是一类比较特殊的高效液相色谱法。现有技术中已经出现多篇利用离子色谱检测极性肼类化合物的报道。如公告号cn103293262b(公告日2015年2月25日)的中国发明专利公开了利用离子色谱法检测叔丁基肼盐酸盐的方法,采用diobexionpaccs12a色谱柱,以10-20mmol/l甲基磺酸为流动相。但是发明人经过试验发现,离子色谱法对氯乙酰肼的响应较差,不能将氯乙酰肼与杂质有效分离。技术实现要素:为了克服现有技术的不足,本发明提供一种利用亲水作用色谱hilic(hydrophilicinteractionliquidchromatography)检测氯乙酰肼的方法,并将该方法应用于对磷酸西格列汀中可能存在的微量氯乙酰肼进行定性和/或定量检测。根据本发明的方法,氯乙酰肼的出峰时间在4min左右,最低线性检出浓度为0.17μg/ml,最低检出浓度为0.05μg/ml;且通过加样回收实验表明,磷酸西格列汀中其他成分对氯乙酰肼的检测无干扰。本发明可实现在磷酸西格列汀中快速、有效定性/定量检测氯乙酰肼含量的目的,填补现有技术空白。为了实现上述发明目的。本发明采用了如下的技术方案:一种氯乙酰肼的检测方法,所述方法基于高效液相法,色谱条件为:色谱柱:hilic色谱柱;检测器:紫外检测器;检测波长:204nm~210nm;流动相:乙腈为a相,0.1%磷酸水溶液为b相,等度洗脱,两相的体积比为:a∶b=78~82%∶18~22%;流速:0.7ml/min~1.4ml/min;柱温:35℃~45℃;进样量:5μl。优选地,所述色谱柱为watershilic色谱柱,规格250mm*4.6mm,5μm。优选地,所述检测波长为207nm。优选地,所述流动相中,所述两相的体积比为a∶b=80%∶20%优选地,所述流速为1.0~1.1ml/min。还优选地,所述柱温为40℃~42℃。本发明提供的检测方法中,对照品溶液用超纯水配制。优选地,对照品溶液的制备方法为:精密量取氯乙酰肼盐酸盐适量,用超纯水溶解并稀释成每1ml含0.1~10μg氯乙酰肼的溶液,即得。优选地,本发明提供的检测方法还包括定性检测和/或定量测定,具体步骤包括:定性检测:在所述色谱条件下,精密量取对照品溶液5μl,注入液相色谱仪,调节检测器灵敏度,使主成分的色谱峰高为满量程的10%~20%;再精密量取供试品溶液和对照品溶液各5μl,分别进样,注入液相色谱仪,记录色谱图;与对照品溶液的色谱图比较,观察供试品溶液的色谱图中是否在相应的保留时间出现相应的色谱峰;和/或定量测定:在所述色谱条件下,精密量取对照品溶液5μl,注入离子色谱仪,调节检测器灵敏度,使主成分的色谱峰高为满量程的10%~20%;再精密量取供试品溶液和对照品溶液各5μl,分别进样,注入离子色谱仪,按外标法计算供试品溶液中氯乙酰肼的含量。优选地,本发明提供的检测方法,最低线性检出浓度为0.17μg/ml,按照信噪比3∶1计算最低检出浓度为0.05μg/ml。本发明还有一个目的在于提供上述的检测方法在磷酸西格列汀杂质定性和/或定量检测中的应用,所述杂质为氯乙酰肼。作为一个优选的实施方式,本发明提供一种磷酸西格列汀中的氯乙酰肼的检测方法,包括色谱条件建立、对照品溶液的制备、供试品溶液的制备、定性检测和/或定量测定,其中,色谱条件的建立:色谱柱:watershilic色谱柱,规格250mm*4.6mm,5μm;检测器:紫外检测器;检测波长:204nm~210nm,优选207nm流动相:乙腈为a相,0.1%磷酸水溶液为b相,等度洗脱,两相的体积比为:a∶b=78~82%∶18~22%,优选a∶b=80%∶20%;流速:0.7ml/min~1.4ml/min,优选1.0~1.1ml/min;柱温:35℃~45℃,优选40℃~42℃;进样量:5μl;对照品溶液的制备:精密量取氯乙酰肼盐酸盐适量,用超纯水溶解并稀释成每1ml约含0.9±0.09μg氯乙酰肼的溶液,即得;供试品溶液的制备:取磷酸西格列汀适量,用超纯水溶解并稀释制成每1ml约含60±6mg磷酸西格列汀的溶液,即得;定性检测:在所述色谱条件下,精密量取所述对照品溶液5μl,注入离子色谱仪,调节检测器灵敏度,使主成分的色谱峰高为满量程的10%~20%;再精密量取所述供试品溶液和所述对照品溶液各5μl,分别进样,注入液相色谱仪,记录色谱图;与对照品溶液的色谱图比较,观察供试品溶液的色谱图中是否在相应的保留时间出现相应的色谱峰;和/或定量测定:在所述色谱条件下,精密量取所述对照品溶液5μl,注入离子色谱仪,调节检测器灵敏度,使主成分的色谱峰高为满量程的10%~20%;再精密量取所述供试品溶液和所述对照品溶液各5μl,分别进样,注入液相色谱仪,按外标法计算供试品溶液中氯乙酰肼的含量。本发明通过优选建立的测定氯乙酰肼及检测磷酸西格列汀中氯乙酰肼的方法,基于亲水作用液相色谱hilic,对氯乙酰肼的保留作用较强,氯乙酰肼在该色谱柱上具有适中的保留时间。一般情况下,不主张hilic色谱柱的供试品和对照品溶液用水配制,因为水会降低柱效。但是发明人经过试验发现,氯乙酰肼和磷酸西格列汀在水中稳定性均较好;经过耐用性实验考察,色谱柱的理论塔板数以及氯乙酰肼等的吸收峰的峰型均没有明显变化,说明在本发明中采用水作为供试品和对照品的稀释剂是可行的。本领域技术人员应该理解,除了超纯水,去离子水也满足离子色谱对水的要求,因此去离子水也可以用来配制本发明所述的流动相b。本发明的方法专属性强,所述磷酸西格列汀中其他成分对氯乙酰肼检测无干扰。本发明的方法灵敏度高,最低线性检出浓度为0.17μg/ml,最低检出浓度为0.05μg/ml。而且本发明提供的方法完成一个样品检测的时间短、可操作性强。因此,本发明提供的氯乙酰肼的检测方法可以用于有效、快速地检测磷酸西格列汀中的氯乙酰肼,从而更好地控制产品的质量,为药品的安全使用提供保障。附图说明下面结合附图,对本发明做详细的说明。图1示出的是实施例1中离子色谱法i下得到的氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图2示出的是实施例1中离子色谱法ii下氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图3示出的是实施例1中反相高效液相法得到的氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图4示出的是实施例1中亲水作用色谱hilic法下氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图5示出的是实施例2中检测波长为204nm的氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图6示出的是实施例2中检测波长为210nm的氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图7示出的是实施例2中流动相为a(乙腈)∶b(0.1%磷酸水溶液)=78%∶22%,流速为1.0ml/min下氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图8示出的是实施例2中流动相为a(乙腈)∶b(0.1%磷酸水溶液)=82%∶18%,流速为1.0ml/min时的氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图9示出的是实施例2中流动相为a(乙腈)∶b(0.1%磷酸水溶液)=80%∶20%,流速为0.7ml/min时的氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图10示出的是实施例2中流动相为a(乙腈)∶b(0.1%磷酸水溶液)=80%∶20%,流速为1.4ml/min时的氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图11示出的是实施例2中流动相为a(乙腈)∶b(0.1%磷酸水溶液)=80%∶20%,柱温为35℃时的氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图12示出的是实施例2中流动相为a(乙腈)∶b(0.1%磷酸水溶液)=80%∶20%,柱温为40℃时的氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图13示出的是实施例2中流动相为a(乙腈)∶b(0.1%磷酸水溶液)=80%∶20%,柱温为42℃时的氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图14示出的是实施例2中流动相为a(乙腈)∶b(0.1%磷酸水溶液)=80%∶20%,柱温为45℃时的氯乙酰肼对照品的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图15示出的是实施例3中专属性试验中,氯乙酰肼对照品溶液的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图16示出的是实施例3中专属性试验中,磷酸西格列汀供试品溶液的色谱图。图17示出的是实施例3中线性范围试验中对照品溶液的回归曲线,其中横坐标(x)为氯乙酰肼浓度,纵坐标(y)为色谱峰面积。图18出的是实施例3中检出限试验中对照品溶液(氯乙酰肼浓度为0.05μg/m1)的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图19示出的是实施例3中加样回收试验中,批号171101的供试品溶液的色谱图;图中,标号为1的色谱峰为氯乙酰肼的色谱峰。图20示出的是实施例4中批号为171101的磷酸西格列汀的色谱图。具体实施方式以下参照具体的实施例来说明本发明。本领域技术人员能够理解,这些实施例仅用于说明本发明,其不以任何方式限制本发明的范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的原料、试剂材料等,如无特殊说明,均为市售购买产品。其中,部分试剂和仪器设备购买情况如下:液相色谱仪:waterse2695,沃特世科技有限公司;电子天平:cpa225d,赛多利斯科学仪器(北京)有限公司;色谱柱:1)watersatlantichilic(4.6mm*250mm,5um),沃特世科技有限公司;2)agilentzorbaxsbaq(4mm*150mm,5um),安捷伦科技有限公司;离子色谱仪:thermodionexaq离子色谱仪,赛默飞世尔科技有限公司;色谱柱:thermodionexionpaccs12a阳离子柱(4mm*250mm),赛默飞世尔科技有限公司;保护柱:thermodionexionpaccg12保护柱(4mm*50mm),赛默飞世尔科技有限公司;试剂以及试药:氯乙酰肼盐酸盐:苏州克瑞斯特医药科技有限公司超纯水:杭州娃哈哈集团乙腈:色谱纯,astoon美国阿斯顿化学技术有限公司甲醇:色谱纯,astoon美国阿斯顿化学技术有限公司磷酸:色谱纯,国药集团化学试剂有限公司磷酸西格列汀:通化东宝药业股份有限公司,批号171101、171102、171103、160501、160502、160503。实施例1:检测方法的选择本实施例的目的在于考察适于氯乙酰肼的检测方法。本实施例均采用氯乙酰肼对照品溶液进行测定,氯乙酰肼对照品溶液的制备方法为:精密称取氯乙酰肼盐酸盐适量,用超纯水溶解并稀释成每1ml约含0.93μg氯乙酰肼的溶液,即得。测定方法:精密量取对照品溶液5μl,注入液相色谱仪,记录色谱图。1.1离子色谱法i色谱条件如下离子色谱仪:thermodionexaquion型离子色谱仪;色谱柱:thermodionexionpaccs12a阳离子柱(4mm*250mm);保护柱:thermodionexionpaccg12保护柱(4mm*50mm);流动相:15mmol/l→50mmol/l甲磺酸;梯度洗脱程序见下表:时间(min)甲磺酸(mmol/l)0.01520.01520.150.0110.050.0110.115.0140.015.0流速:1.0ml/min;柱温:28-35℃;检测器:电导检测仪进样量:25μl。色谱图见图1。从图1中可以看出,在上述色谱条件下氯乙酰肼保留时间5.81min,但是峰型不佳,响应较差,且出现明显的梯度峰。因此,上述色谱方法不适于氯乙酰肼的检测。1.2离子色谱法ii将流动相调整为10mmol/l→40mmol/l甲磺酸;梯度洗脱程序见下表:时间(min)甲磺酸(mmol/l)0.01020.01020.14040.04040.11060.010其它的条件均与“1.1”项下的离子色谱法i相同,色谱图见图2。从图2可以看出,与离子色谱法i的分离结果类似,氯乙酰肼峰型也不佳,与其他杂质仍未有效分离,响应较差,也出现明显的梯度峰。1.3反相高效液相法色谱仪:waterse2695型高效液相色谱仪;色谱柱:agilentzorbaxsbaq(4mm*150mm,5um);流动相:0.1%磷酸水溶液-乙腈,梯度洗脱,梯度洗脱见下表:时间(min)0.1%磷酸水溶液乙腈0.095.05.02.095.05.010.010.09010.195.05.016.095.05.0流速:1.0ml/min;柱温:40℃;检测器:紫外检测器;检测波长:207nm;色谱图见图3。从图3可以看出,采用上述反相高效液相色谱法,出现较大的基线波动,同时与氯乙酰肼色谱峰有明显干扰。因此,反相高效液相色谱法也适于氯乙酰肼的分离和检测。1.4亲水作用色谱hilic法色谱仪:waterse2695型高效液相色谱仪;色谱柱:watersatlantichilic(4.6mm*250mm,5um)流动相:乙腈(a)-0.1%磷酸水溶液(b),体积比a∶b=80%∶20%,等度洗脱;流速:1.0ml/min;柱温:40℃;检测器:紫外检测器;波长:207nm。色谱图见图4。从图4可以看出,采用上述色谱方法可以实现氯乙酰肼的基线分离,保留时间适当。因此,基于本实施例的结果,初步确定采用hilic色谱柱对氯乙酰肼进行分离和检测。实施例2hilic色谱条件的优化按照实施例1所述的方法制备氯乙酰肼对照品溶液,基本色谱条件为:色谱仪:waterse2695型高效液相色谱仪;色谱柱:watersatlantichilic(4.6mm*250mm,5um)流动相:乙腈(a)-0.1%磷酸水溶液(b),等度洗脱;检测器:紫外检测器。2.1检测波长的考察流动相:体积比a∶b=80%∶20%,等度洗脱;流速:1.0ml/min;柱温:40℃;检测器:紫外检测器;波长:204nm、210nm。两种检测波长下氯乙酰肼对照品溶液的色谱图分别见图5和图6。将图5、图6和图4比较,2.2流动相的优选在流速1.0ml/min,柱温40℃下,分别以a(乙腈)∶b(0.1%磷酸水溶液)=78%∶22%和82%∶18%为流动相,进行洗脱和测定,色谱图见图7和图8。将图7、图和图4比较,2.3流动相流速的考察(1)流速0.7ml/min柱温40℃下,以a(乙腈)∶b(0.1%磷酸水溶液)=80%∶20%为流动相,分别考察流速0.7ml/min、1.1ml/min、1.4ml/min下氯乙酰肼的进行洗脱情况,记录色谱图。将得到的色谱图与图4比较,发现流速0.7ml/min条件下,氯乙酰肼出峰时间较晚,洗脱耗时长,色谱峰相应较低,参见图9;流速1.4ml/min条件下,氯乙酰肼的色谱峰响应虽然较好,但出峰时间较早,参见图10;流速1.1ml/min时的色谱图(略)与图4非常类似。上面的试验结果显示,流速在0.7ml/min~1.4ml/min之间氯乙酰肼色谱峰的响应均较好,尤其是流速为1.0ml/min、1.1ml/min时,氯乙酰肼的保留时间最适宜,色谱峰峰型好,没有拖尾,基线平滑。因此,特别优选的流动相的流速为1.0~1.1ml/min。2.4柱温的考察(1)柱温35℃以乙腈∶0.1%磷酸水溶液=80∶20为流动相,流速1.0ml/min,柱温35℃下进行洗脱和测定,色谱图见图11。从图中可以看出,柱温35℃下,色谱峰响应较低,色谱峰拖尾。(2)柱温40℃以乙腈∶0.1%磷酸水溶液=80∶20为流动相,流速1.0ml/min,柱温40℃下进行洗脱和测定,色谱图见图12。从图中可以看出,柱温40℃下,色谱峰响应好,保留时间适当。(3)柱温42℃乙腈∶0.1%磷酸水溶液=80∶20为流动相,流速1.0ml/min,柱温42℃下进行洗脱和测定,色谱图见图13。从图中可以看出,柱温42℃下,色谱峰响应较好,保留时间适当。(4)柱温45℃乙腈∶0.1%磷酸水溶液=80∶20为流动相,流速1.0ml/min,柱温35℃下进行洗脱和测定,色谱图见图14。从图中可以看出,柱温45℃下,色谱峰响应较低,出峰过早。上面的试验结果显示,柱温在40℃左右(40~42℃)时氯乙酰肼色谱峰的响应均较好,尤其是柱温在40℃时,分离效果最好。因此,本发明方法柱温可以在35~45℃之间,最优选为40~42℃。实施例3本发明的氯乙酰肼检测方法的方法学研究在实施例2的基础上,建立了氯乙酰肼的检测方法,具体的:i.色谱条件色谱柱:hilic色谱柱;检测器:紫外检测器;检测波长:204nm-210nm,优选为207nm;流动相:乙腈为a相,0.1%磷酸水溶液为b相,等度洗脱,两相的体积比为:a:b=78~82%:18~22%;流速:0.7ml/min~1.4ml/min,优选为1.0~1.1ml/min;柱温:35℃~45℃,优选为40℃~42℃;进样量:5μl。ii.对照品溶液的制备:精密量取氯乙酰肼盐酸盐适量,用超纯水稀释成每1ml约含0.9±0.09μg氯乙酰肼的溶液,即得;供试品溶液的制备:取磷酸西格列汀适量,加超纯水溶解并稀释制成每1ml约含60±6mg磷酸西格列汀的溶液,即得。iii.定性检测:在所述色谱条件下,精密量取所述对照品溶液5μl,注入离子色谱仪,调节检测器灵敏度,使主成分的色谱峰高为满量程的10%~20%;再精密量取所述供试品溶液和所述对照品溶液各5μl,分别进样,注入液相色谱仪,记录色谱图;与对照品溶液的色谱图比较,观察供试品溶液的色谱图中是否在相应的保留时间出现相应的色谱峰。iv.定量测定:在所述色谱条件下,精密量取所述对照品溶液5μl,注入离子色谱仪,调节检测器灵敏度,使主成分的色谱峰高为满量程的10%~20%;再精密量取所述供试品溶液和所述对照品溶液各5μl,分别进样,注入液相色谱仪,按外标法计算供试品溶液中氯乙酰肼的含量。本实施例对建立的氯乙酰肼的检测方法从专属性、稳定性、精密度、线性检测范围、最低检出限、耐用性、准确性(加样回收法)等方面进行方法学的验证。3.1专属性精密量取供试品溶液和对照品溶液各5μl,分别注入高效液相色谱仪,记录色谱图。结果见表1及图15和图16。.表1专属性试验结果样品称样量(mg)浓度(ug/ml)保留时间(min)峰面积理论塔板数含量(%)氯乙酰肼盐酸盐12.000.874.267698112693/磷酸西格列汀600.0160001.00/000试验结果表明,本发明的方法能够有效检测出氯乙酰肼,供试品溶液的色谱图在氯乙酰肼保留时间附近没有色谱峰,说明供试品溶液中其它成分对氯乙酰肼没有干扰,方法专属性强。3.2稳定性3.1项下配制好的对照品溶液和供试品溶液制备好后,在室温(18-23℃)中放置,分别在0h、2h、10h、15h测定,结果见表2。表2稳定性试验结果对照溶液保留时间(min)峰面积供试品溶液保留时间(min)峰面积0h4.26661990h/02h4.26461852h/010h4.271601510h/015h4.267558515h/0平均值4.2675996平均值/0rsd%0.07%4.78%rsd%//表2的数据示出,供试品溶液以及对照品溶液在室温(18~23℃)放置至少15小时内稳定,方法实用性强。3.3线性范围取氯乙酰肼盐酸盐对照品适量,用超纯水分别配制成浓度为0.17μg/ml、0.44μg/ml、0.87μg/ml、1.31μg/ml和2.62μg/ml的一系列的溶液,各精密吸取5ul注入高效液相色谱仪,记录色谱图,以氯乙酰肼浓度为横坐标(x),以色谱峰面积(y)为纵坐标,进行线性回归,结果见图17,线性回归方程为:y=6,306.7890x-220.5457(r2=0.9980)结果表明,当氯乙酰肼浓度在0.17μg/ml-2.62μg/ml的范围内,其浓度与峰面积具有良好的线性关系(r=0.9990)。本法对氯乙酰肼的最低线性检出浓度为0.17μg/ml,检出线性范围为20%~300%。3.4检测限氯乙酰肼对照品溶液用超纯水多次稀释后分别进样,按照信噪比3∶1计算氯乙酰肼的最低检出浓度为0.05μg/ml。氯乙酰肼浓度为0.05μg/ml时的色谱图见图18。3.5精密度重复测定氯乙酰肼对照品溶液6次,结果见表3,rsd为1.09%。说明本发明的方法精密度符合要求。表3精密度试验结果3.6耐用性精密称取12.02mg氯乙酰肼盐酸盐,用超纯水配制成浓度为0.87μg/ml的溶液,即得对照品溶液。精密称取599.76mg磷酸西格列汀,用超纯水配制成浓度为59976.0μg/ml的溶液,即得供试品溶液。按照本发明提供氯乙酰肼的检测方法,用对照品溶液和供试品溶液,使用同一根色谱柱,分别微调流速和柱温,结果供试样品中其他成分对氯乙酰肼检测无干扰,表明所建方法耐用性好。结果见表4。表4耐用性试验结果3.7准确度(加样回收实验)精密称取氯乙酰肼盐酸盐12.02mg,用超纯水配制成浓度为8.72μg/ml的贮备液,然后取贮备液适量,用超纯水稀释成浓度为0.872μg/ml的对照品溶液。精密称取9份磷酸西格列汀(批号171101),每份约600mg,用超纯水配制成浓度约为60mg/ml的供试品溶液。分别精密吸取5μl,注入高效液相色谱仪,记录色谱图,在与对照品溶液色谱图相应的位置没有出现吸收峰。然后按照表所示分别加入相应体积的贮备液(其中,50%加入量为加入0.5ml贮备液,100%加入量为加入1ml贮备液,150%加入量为加入1.5ml贮备液),摇匀后,分别精密吸取5μl,注入高效液相色谱仪,进行测定,记录色谱图,计算氯乙酰肼的含量,计算回收率,结果见表5,批号171101的加样回收测试样品溶液的色谱图见图19。表5加样回收率试验结果表5的数据表明本发明的方法准确度较高,图19也表明磷酸西格列汀对氯乙酰肼检出无影响。总之,通过上述方法学研究证明,本发明提供的利用高效液相色谱检测磷酸西格列汀中氯乙酰肼的方法,专属性、稳定性和耐用性好,精密度和准确性高,以信噪比3∶1确定的最低检出浓度为0.05μg/ml,最低线性检出浓度为0.17μg/ml。实施例4使用本发明方法对6批磷酸西格列汀进行测定具体测定结果如下:分别按照表所示称取磷酸西格列汀适量,加超纯水溶解并稀释制成每1ml约含60mg的溶液,作为供试品溶液;按照表所示精密量取氯乙酰肼盐酸盐适量,用超纯水稀释成每1ml约含0.9μg的溶液,作为对照溶液。在实施例3所述的色谱条件下,精密量取对照品溶液5μl,注入高效液相色谱仪,调节仪器灵敏度,使主成分的色谱峰高为满量程的10%~20%。再精密量取供试品溶液和对照品溶液各5μl,分别进样,注入高效液相色谱仪,记录色谱图,按外标法计算磷酸西格列汀中氯乙酰肼含量。结果见表6,其中批号171101的磷酸西格列汀的色谱图见图20。表6六批磷酸西格列汀测定结果名称批号称样量(mg)浓度(μg/ml)保留时间(min)峰面积含量(%)氯乙酰肼盐酸盐12.080.884.2675008/磷酸西格列汀171101599.9259992.00/00磷酸西格列汀171102600.0160001.00/00磷酸西格列汀171103599.7359973.00/00磷酸西格列汀160501599.9459994.00/00磷酸西格列汀160502599.9159991.00/00磷酸西格列汀160503599.9059990.00/00表的数据示出,在检测的六批磷酸西格列汀中,按照本发明提供的方法均未检测出氯乙酰肼;说明受检产品在该项检测项目中均合格。总之,本发明提供了一种基于亲水作用高效液相色谱法检测氯乙酰肼的方法,进而提供了一种基于所述亲水作用高效液相色谱法定性/定量检测磷酸西格列汀中氯乙酰肼的方法。本发明的方法专属性强、灵敏度高,最低线性检出浓度为0.17μg/ml,以信噪比3:1确定的最低检出浓度为0.05μg/ml。因此,本发明提供的氯乙酰肼的检测方法可以用于有效、快速地检测磷酸西格列汀中可能残存的氯乙酰肼,从而更好地控制产品的质量,保障临床用药的安全。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种检测醛酮类化合物的方法与制造工艺
- 中氮茚罗丹明酰肼类铜离子比率荧光探针及其应用的制造方法与工艺
- 用作选择性抗菌剂的含2‑吡啶酮部分的芳基酰肼的制造方法与工艺
- 硫代杯[4]芳烃酰腙类希夫碱衍生物及其合成方法和应用与制造工艺
- 一种识别肼的小分子荧光探针及其应用的制造方法与工艺
- 一种2-甲基-1,3,4-噁二唑-5(4H)-酮生产工艺的制造方法与工艺
- 吩嗪‑1‑羧酸双酰肼类化合物及含其化合物的杀菌组合物的制造方法与工艺
- 吲哚啉‑2‑酮的肼基二硫代甲酸甲酯衍生物的铂配合物及其制备方法和用途与制造工艺
- 3‑(2‑氨基‑2‑硫代乙基)苯甲酸甲酯的合成方法与制造工艺
- 3‑(2‑氨基‑2‑硫代乙基)苯甲酸甲酯的制备方法与制造工艺