一种食品中化学残留物质的高通量分析方法与流程
本发明涉及一种食品中化学残留物质的高通量分析方法。
背景技术:
随社会工业化的发展,环境致病因素,如大气、水和食品污染,对健康的影响日益显著。食品的化学污染一直是受到广泛关注的环境致病因素。目前已知的外源性化学物质达500万种以上,其中至少6万种已经进入人们的生产和生活,如各种工业原料及产品、医药、农药、化妆品和生物毒素等。这些化学物质可通过食品的原材料生产、加工、烹调、包装、贮存和运输等环节进入食品成为食品污染物。食品污染物包括食品生产中使用的物质和在食品上市的其他环节引入的环境化学或生物污染物,如农药、兽药、霉菌毒素、工业污染物等。
兽药在防治动物疾病、提高生产率、改善畜产品质量方面起着十分重要的作用。然而,由于生产人员对科学知识的缺乏及一味地追求经济效益,致使滥用兽药现象在当前畜牧业中普遍存在。随着人们对动物源性食品由需求型向质量型的转变,动物源性食品中的兽药残留已逐渐成为全世界关注的一个焦点。兽药残留是指用药后蓄积或存留于畜禽机体或产品(如鸡蛋、奶品、肉品等)中原型药物或其代谢产物,包括与兽药有关的杂质残留。滥用兽药极易造成动物源性食品中有害物质的残留,这对人体健康造成直接危害。对动物源性食品中的兽药残留进行监控的一个重要手段就是建立快速准确的残留分析方法。
申请公布日为2017年7月28日、申请公布号为cn106990195a的中国专利公开了一种检测动物源性食品中磺胺类、喹诺酮类兽药残留的方法,其包括(1)标准溶液的配制;(2)样品前处理;(3)液质分离检测;其通过1%甲酸乙腈提取、正己烷去脂,结合高分辨质谱检测兽药残留。
研究发现,磺胺类兽药的水溶性低,喹诺酮类兽药在水中有一定溶解度但其在有机溶剂中也有一定的溶解度,因此在采用现有技术的样品前处理方法时,在1%甲酸乙腈提取时,磺胺类兽药提取率有一定下降,而在正己烷去脂时磺胺类兽药和喹诺酮兽药均有一定的损失。现有技术中对磺胺类和喹诺酮兽药的提取仍有一定的改善空间,提高提取率可提高含量测定的准确率,降低分析样品的检测量,具有较为重要的价值。
技术实现要素:
本发明的目的是提供一种食品中化学残留物质的高通量分析方法,具有提高兽药提取率的优点。
本发明的上述技术目的是通过以下技术方案得以实现的:
一种食品中化学残留物质的高通量分析方法,包括依次设置的样品预处理、标准工作溶液配制和uplc-ms/ms测定,样品预处理包括以下步骤:
①粉碎干燥
取动物源性食品为待测样品,若待测样品为液体则将其在40~50℃下浓缩至含水量低于2%,粉碎过50~100目筛;若待测样品为固体则将其搅碎并在40~50℃下干燥至含水量低于2%,粉碎过50~100目筛;
②提取
取粉碎干燥后的待测样品,加水、氯化钾和烟酰胺,在55~60℃下搅拌反应5~10min,过滤取滤液,待测样品、水、氯化钾和烟酰胺的质量比为1:10~15:0.2~0.3:0.05~0.08;将滤液和壳聚糖按质量比1:0.3~0.4混合,在45~50℃下搅拌反应5~10min,过滤取滤液,将滤液在40~50℃下浓缩至含水量低于1%,得到待测固体,将待测固体用初始流动相溶解,用于uplc-ms/ms测定。
优选为,②提取步包括以下步骤:
取粉碎干燥后的待测样品,加水、氯化钾和烟酰胺,在55~60℃下搅拌反应5~10min,加入金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000,搅拌分散5~10min,过滤取滤液,待测样品、水、氯化钾、烟酰胺、金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000的质量比为1:10~15:0.2~0.3:0.05~0.08:0.01~0.02:0.01~0.02:0.01~0.02;将滤液和壳聚糖按质量比1:0.3~0.4混合,在45~50℃下搅拌反应5~10min,过滤取滤液,将滤液在40~50℃下浓缩至含水量低于1%,得到待测固体,将待测固体用初始流动相溶解,用于uplc-ms/ms测定。
优选为,标准工作溶液配制包括以下步骤:准确称取所有目标兽药的标准品,用初始流动相配制成1000mg/l的标准储存液,用初始流动相将标准储存液稀释至5个浓度点,将该5个浓度点作为标准工作溶液;所述目标兽药选自磺胺类和/或喹诺酮类;
uplc-ms/ms测定的条件如下:
色谱条件:watersacquityuplcbehc18色谱柱,流动相a为甲醇,流动相b为1wt%的甲酸水溶液,洗脱方式为梯度洗脱;以体积比计,梯度洗脱程序为:0~2.0min10%的流动相a+90%的1wt%的甲酸水溶液,2.1~8.0min30%的流动相a+70%的1wt%的甲酸水溶液,8.1~14.0min90%的流动相a+10%的1wt%的甲酸水溶液,14.1~16.0min10%的流动相a+90%的1wt%的甲酸水溶液;
质谱条件:电喷雾离子源(esi+),温源为110℃,脱溶剂温度为350℃,脱溶剂气流速度为600l·h-1,锥孔反吹气流温度为50l·h-1,质谱扫描方法为多时间段的多反应检测扫描方法。
优选为,包括以下步骤:
(1)样品预处理
①粉碎干燥
取动物源性食品为待测样品,若待测样品为液体则将其在40~50℃下浓缩至含水量低于2%,粉碎过50~100目筛;若待测样品为固体则将其搅碎并在40~50℃下干燥至含水量低于2%,粉碎过50~100目筛;
②提取
取粉碎干燥后的待测样品,加水、氯化钾和烟酰胺,在55~60℃下搅拌反应5~10min,过滤取滤液,待测样品、水、氯化钾和烟酰胺的质量比为1:10~15:0.2~0.3:0.05~0.08;将滤液和壳聚糖按质量比1:0.3~0.4混合,在45~50℃下搅拌反应5~10min,过滤取滤液,将滤液在40~50℃下浓缩至含水量低于1%,得到待测固体,将待测固体用初始流动相溶解,用于uplc-ms/ms测定;
(2)标准工作溶液配制
准确称取所有目标兽药的标准品,用初始流动相配制成1000mg/l的标准储存液,用初始流动相将标准储存液稀释至5个浓度点,将该5个浓度点作为标准工作溶液;所述目标兽药选自磺胺类和/或喹诺酮类;
(3)uplc-ms/ms测定
按如下方法进行测定:
色谱条件:watersacquityuplcbehc18色谱柱,流动相a为甲醇,流动相b为1wt%的甲酸水溶液,洗脱方式为梯度洗脱;以体积比计,梯度洗脱程序为:0~2.0min10%的流动相a+90%的1wt%的甲酸水溶液,2.1~8.0min30%的流动相a+70%的1wt%的甲酸水溶液,8.1~14.0min90%的流动相a+10%的1wt%的甲酸水溶液,14.1~16.0min10%的流动相a+90%的1wt%的甲酸水溶液;
质谱条件:电喷雾离子源(esi+),温源为110℃,脱溶剂温度为350℃,脱溶剂气流速度为600l·h-1,锥孔反吹气流温度为50l·h-1,质谱扫描方法为多时间段的多反应检测扫描方法。
优选为,包括以下步骤:
(1)样品预处理
①粉碎干燥
取动物源性食品为待测样品,若待测样品为液体则将其在40~50℃下浓缩至含水量低于2%,粉碎过50~100目筛;若待测样品为固体则将其搅碎并在40~50℃下干燥至含水量低于2%,粉碎过50~100目筛;
②提取
取粉碎干燥后的待测样品,加水、氯化钾和烟酰胺,在55~60℃下搅拌反应5~10min,加入金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000,搅拌分散5~10min,过滤取滤液,待测样品、水、氯化钾、烟酰胺、金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000的质量比为1:10~15:0.2~0.3:0.05~0.08:0.01~0.02:0.01~0.02:0.01~0.02;将滤液和壳聚糖按质量比1:0.3~0.4混合,在45~50℃下搅拌反应5~10min,过滤取滤液,将滤液在40~50℃下浓缩至含水量低于1%,得到待测固体,将待测固体用初始流动相溶解,用于uplc-ms/ms测定;
(2)标准工作溶液配制
准确称取所有目标兽药的标准品,用初始流动相配制成1000mg/l的标准储存液,用初始流动相将标准储存液稀释至5个浓度点,将该5个浓度点作为标准工作溶液;所述目标兽药选自磺胺类和/或喹诺酮类;
(3)uplc-ms/ms测定
按如下方法进行测定:
色谱条件:watersacquityuplcbehc18色谱柱,流动相a为甲醇,流动相b为1wt%的甲酸水溶液,洗脱方式为梯度洗脱;以体积比计,梯度洗脱程序为:0~2.0min10%的流动相a+90%的1wt%的甲酸水溶液,2.1~8.0min30%的流动相a+70%的1wt%的甲酸水溶液,8.1~14.0min90%的流动相a+10%的1wt%的甲酸水溶液,14.1~16.0min10%的流动相a+90%的1wt%的甲酸水溶液;
质谱条件:电喷雾离子源(esi+),温源为110℃,脱溶剂温度为350℃,脱溶剂气流速度为600l·h-1,锥孔反吹气流温度为50l·h-1,质谱扫描方法为多时间段的多反应检测扫描方法。
本发明技术效果主要体现在以下方面:
在55~60℃的水环境中,粉碎后的动物源性食品中的喹诺酮类兽药溶解在水中,磺胺类兽药在该温度下和水中的烟酰胺发生反应得到磺胺类兽药的烟酰胺共晶,在反应产生共晶时以氯化钾作为离子浓度调节剂,该类共晶在水中溶解度较大,改变了磺胺类兽药在水中难溶的性质,又因为动物源性食品中的油脂等难溶于水,因此在水环境中提取后得到的滤液浓缩物i为水溶解度较佳的磺胺类兽药烟酰胺共晶、喹诺酮类兽药和极少量油脂等杂质,且由于其水溶性和无其他溶剂参与,所以磺胺类兽药和喹诺酮类兽药的提取率得到明显增加,提取过程环保安全;之后将滤液浓缩物i和壳聚糖作用,磺胺类兽药烟酰胺共晶中的磺胺类兽药被壳聚糖置换,游离出的磺胺类兽药在壳聚糖和壳聚糖烟酰胺共晶水溶液中仍能保持较佳的溶解度,将游离后的产品进行测定即可获得较佳的检测效果;另外在②提取步中加入金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000,可选择性吸附其中的油脂等杂质并将该类杂质包裹形成固体,减少乳液状或油状物在过滤步骤中的难度以及在处理过程中对反应过程的表观判断,在添加该些物质后还可减少油脂等杂质对兽药的包裹,进一步提高兽药的提取率;适用于低含量检测,对高通量分析有较为重要的意义。
附图说明
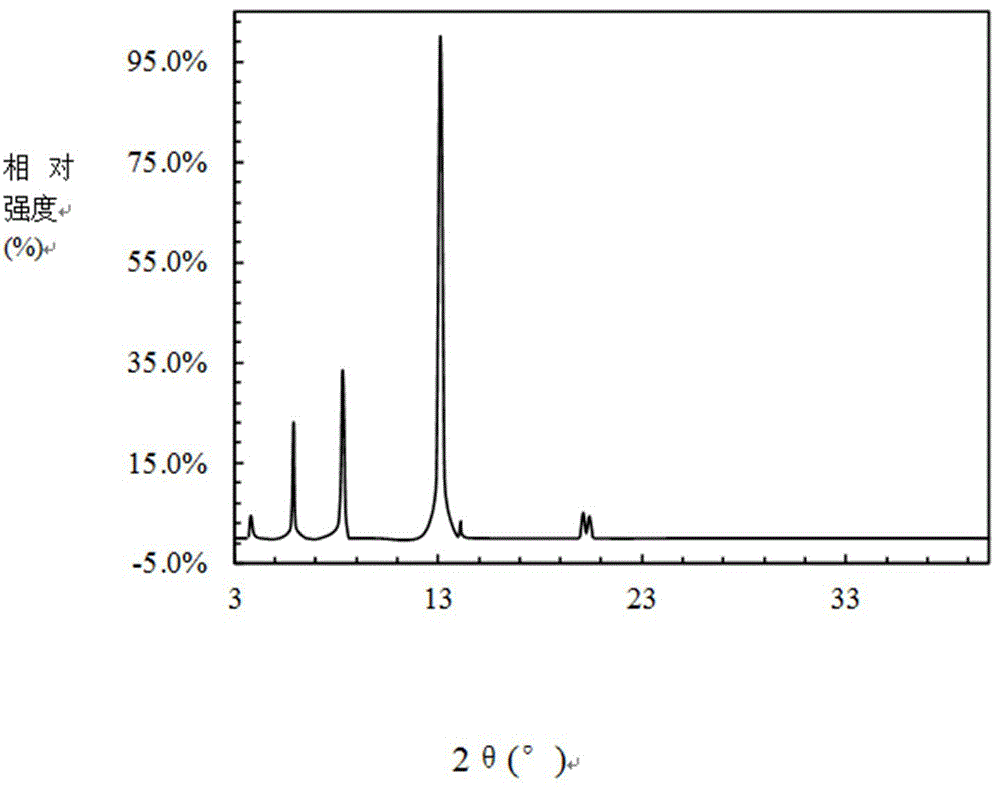
图1为磺胺甲基嘧啶烟酰胺共晶的xrd图(摩尔比为1:1);
图2为磺胺噻唑烟酰胺共晶的xrd图(摩尔比为1:1);
图3为磺胺喹噁啉烟酰胺共晶的xrd图(摩尔比为1:1)。
具体实施方式
以下结合附图,对本发明的具体实施方式作进一步详述,以使本发明技术方案更易于理解和掌握。
实施例1:一种食品中化学残留物质的高通量分析方法,包括以下步骤:
(1)样品预处理
①粉碎干燥
取动物源性食品为待测样品,若待测样品为液体则将其在45℃下浓缩至含水量低于2%,粉碎过100目筛;若待测样品为固体则将其搅碎并在45℃下干燥至含水量低于2%,粉碎过100目筛;
②提取
取粉碎干燥后的待测样品,加水、氯化钾和烟酰胺,在58℃下搅拌反应8min,加入金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000,搅拌分散8min,过滤取滤液,待测样品、水、氯化钾、烟酰胺、金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000的质量比为1:12:0.24:0.06:0.014:0.015:0.015;将滤液和壳聚糖按质量比1:0.33混合,在48℃下搅拌反应8min,过滤取滤液,将滤液在45℃下浓缩至含水量低于1%,得到待测固体,将待测固体用初始流动相溶解,用于uplc-ms/ms测定;
(2)标准工作溶液配制
准确称取所有目标兽药的标准品,用初始流动相配制成1000mg/l的标准储存液,用初始流动相将标准储存液稀释至5个浓度点,将该5个浓度点作为标准工作溶液;所述目标兽药选自磺胺类和/或喹诺酮类;
(3)uplc-ms/ms测定
按如下方法进行测定:
色谱条件:watersacquityuplcbehc18色谱柱,流动相a为甲醇,流动相b为1wt%的甲酸水溶液,洗脱方式为梯度洗脱;以体积比计,梯度洗脱程序为:0~2.0min10%的流动相a+90%的1wt%的甲酸水溶液,2.1~8.0min30%的流动相a+70%的1wt%的甲酸水溶液,8.1~14.0min90%的流动相a+10%的1wt%的甲酸水溶液,14.1~16.0min10%的流动相a+90%的1wt%的甲酸水溶液;
质谱条件:电喷雾离子源(esi+),温源为110℃,脱溶剂温度为350℃,脱溶剂气流速度为600l·h-1,锥孔反吹气流温度为50l·h-1,质谱扫描方法为多时间段的多反应检测扫描方法。
实施例2:一种食品中化学残留物质的高通量分析方法,与实施例1的区别在于,(1)样品预处理用下述方法替代:
①粉碎干燥
取动物源性食品为待测样品,若待测样品为液体则将其在40℃下浓缩至含水量低于2%,粉碎过50目筛;若待测样品为固体则将其搅碎并在40℃下干燥至含水量低于2%,粉碎过50目筛;
②提取
取粉碎干燥后的待测样品,加水、氯化钾和烟酰胺,在55℃下搅拌反应10min,加入金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000,搅拌分散10min,过滤取滤液,待测样品、水、氯化钾、烟酰胺、金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000的质量比为1:10:0.2:0.05:0.01:0.01:0.01;将滤液和壳聚糖按质量比1:0.3混合,在45℃下搅拌反应10min,过滤取滤液,将滤液在40℃下浓缩至含水量低于1%,得到待测固体,将待测固体用初始流动相溶解,用于uplc-ms/ms测定。
实施例3:一种食品中化学残留物质的高通量分析方法,与实施例1的区别在于,(1)样品预处理用下述方法替代:
①粉碎干燥
取动物源性食品为待测样品,若待测样品为液体则将其在50℃下浓缩至含水量低于2%,粉碎过100目筛;若待测样品为固体则将其搅碎并在50℃下干燥至含水量低于2%,粉碎过100目筛;
②提取
取粉碎干燥后的待测样品,加水、氯化钾和烟酰胺,在60℃下搅拌反应5min,加入金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000,搅拌分散5min,过滤取滤液,待测样品、水、氯化钾、烟酰胺、金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000的质量比为1:15:0.3:0.08:0.02:0.02:0.02;将滤液和壳聚糖按质量比1:0.4混合,在50℃下搅拌反应5min,过滤取滤液,将滤液在50℃下浓缩至含水量低于1%,得到待测固体,将待测固体用初始流动相溶解,用于uplc-ms/ms测定。
实施例4:一种食品中化学残留物质的高通量分析方法,与实施例1的区别在于,(1)样品预处理的②提取步骤用下述方法替代:
取粉碎干燥后的待测样品,加水、氯化钾和烟酰胺,在58℃下搅拌反应8min,过滤取滤液,待测样品、水、氯化钾和烟酰胺的质量比为1:12:0.24:0.06;将滤液和壳聚糖按质量比1:0.33混合,在48℃下搅拌反应8min,过滤取滤液,将滤液在45℃下浓缩至含水量低于1%,得到待测固体,将待测固体用初始流动相溶解,用于uplc-ms/ms测定。
实施例5:一种食品中化学残留物质的高通量分析方法,与实施例1的区别在于,(3)uplc-ms/ms测定参照cn101358953b的实施例1的方法测定。
对比实施例1:一种食品中化学残留物质的高通量分析方法,与实施例1的区别在于,(1)样品预处理用下述方法替代:
参照cn103760269b的实施例1,即将动物性肌肉组织陷用料理机充分粉碎,冷冻保存,准确称取解冻后的动物源性食品5.0g,置于50ml离心管内,加10wt%三氯乙酸:乙腈提取液(v:v=20:80)10.0ml,充分匀浆后,超声提取10min,10000r·min-1离心10min,上清液转移到另一50ml离心管中,残渣再用提取液10.0ml重复提取一次,合并提取液,将提取液于30℃下氮气吹干。残余物用初始流动相2.0ml溶解,涡旋混匀,以10000r·min-1的速率高速离心10min。取适量上清液,用0.45μm微孔滤膜过滤,供uplv-ms/ms测定。
对比实施例2:一种食品中化学残留物质的高通量分析方法,与实施例1的区别在于,(1)样品预处理用下述方法替代:
取粉碎干燥后的待测样品,加水和烟酰胺,在58℃下搅拌反应8min,过滤取滤液,待测样品、水和烟酰胺的质量比为1:12:0.06;将滤液和壳聚糖按质量比1:0.33混合,在48℃下搅拌反应8min,过滤取滤液,将滤液在45℃下浓缩至含水量低于1%,得到待测固体,将待测固体用初始流动相溶解,用于uplc-ms/ms测定。
对比实施例3:一种食品中化学残留物质的高通量分析方法,与实施例1的区别在于,(1)样品预处理用下述方法替代:
取粉碎干燥后的待测样品,加水和氯化钾,在58℃下搅拌反应8min,过滤取滤液,待测样品、水和氯化钾的质量比为1:12:0.24;将滤液和壳聚糖按质量比1:0.33混合,在48℃下搅拌反应8min,过滤取滤液,将滤液在45℃下浓缩至含水量低于1%,得到待测固体,将待测固体用初始流动相溶解,用于uplc-ms/ms测定。
对比实施例4:一种食品中化学残留物质的高通量分析方法,与实施例1的区别在于,(1)样品预处理的②提取步骤用下述方法替代:
取粉碎干燥后的待测样品,加水、氯化钾和烟酰胺,在58℃下搅拌反应8min,过滤取滤液,待测样品、水、氯化钾和烟酰胺的质量比为1:12:0.24:0.06;将滤液在45℃下浓缩至含水量低于1%,得到待测固体,将待测固体用初始流动相溶解,用于uplc-ms/ms测定。
性能测试
1、分析方法效果比较
选取经检测且检测结果为无磺胺类和喹诺酮兽药残留的猪肺,向其注入一定含量的磺胺类和喹诺酮兽药,单一兽药在动物源性食品中的质量比控制在0.1、1.0和7.0μg/kg,注入的兽药包括磺胺甲基嘧啶、磺胺噻唑、磺胺喹噁啉、洛美沙星、环丙沙星和诺氟沙星。分别按实施例和对比实施例的方法对样品进行处理,按实施例1的条件进行uplc-ms/ms测定,测试结果如表1-3所示,平行试验5组,取平均值。
表1-3显示,相比对比实施例1,实施例1-4的提取率有较大提高,测试含量更接近配制含量,测试准确率更高,且实施例1-4在低含量测试中也具有较为明显的优势,适用于痕量检测,明显优于现有技术;相比对比实施例2-4,实施例1-4的提取率有较大提高,说明实施例1-4的技术方案联合具有意想不到的结果。
表1分析方法效果比较(0.1μg/kg)
表2分析方法效果比较(1.0μg/kg)
表3分析方法效果比较(7.0μg/kg)
2、检测过程中样品表征
选取经检测且检测结果为无磺胺类和喹诺酮兽药残留的猪肺,向其注入一定含量的磺胺类兽药,单一兽药在动物源性食品中的质量比控制在7.0μg/kg,注入的兽药分别为磺胺甲基嘧啶、磺胺噻唑和磺胺喹噁啉。分别按以下方法对样品进行处理:①粉碎干燥:将其搅碎并在45℃下干燥至含水量低于2%,粉碎过100目筛;②提取:取粉碎干燥后的待测样品,加水、氯化钾和烟酰胺,在58℃下搅拌反应8min,加入金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000,搅拌分散8min,过滤取滤液,待测样品、水、氯化钾、烟酰胺、金属有机框架材料uio-66、纳米二氧化钛和聚乙二醇2000的质量比为1:12:0.24:0.06:0.014:0.015:0.015;将滤液和壳聚糖按质量比1:0.33混合,在48℃下搅拌反应8min,过滤取滤液,将滤液在45℃下浓缩至含水量低于1%,得到待测固体,进行xrd测试。测试结果如图1-图3和表4-表6所示。
表4磺胺甲基嘧啶烟酰胺共晶的xrd数据(与图1相对,摩尔比为1:1)
表5磺胺噻唑烟酰胺共晶的xrd数据(与图2相对,摩尔比为1:1)
表6磺胺喹噁啉烟酰胺共晶的xrd数据(与图3相对,摩尔比为1:1)
当然,以上只是本发明的典型实例,除此之外,本发明还可以有其它多种具体实施方式,凡采用等同替换或等效变换形成的技术方案,均落在本发明要求保护的范围之内。
- 还没有人留言评论。精彩留言会获得点赞!