白术药材UPLC特征图谱的构建方法和检测方法与流程
本发明涉及医药
技术领域:
,特别是涉及一种白术药材uplc特征图谱的构建方法和检测方法。
背景技术:
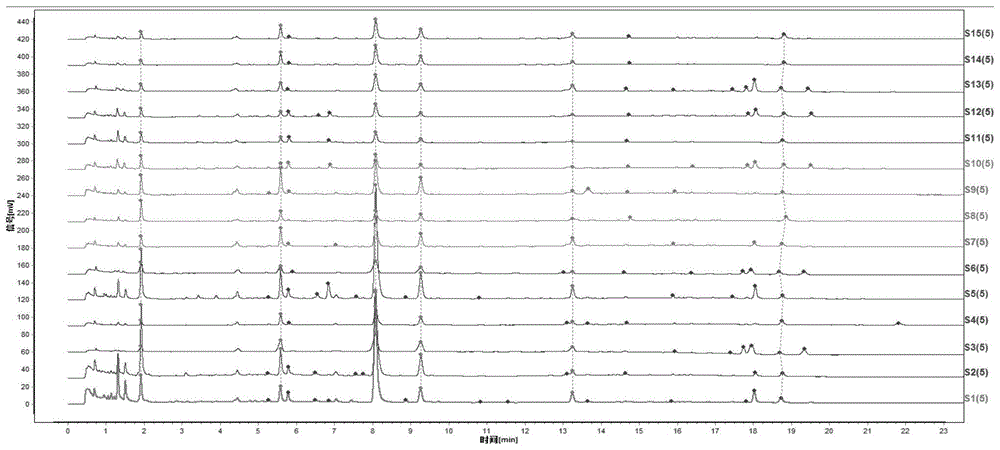
:白术为菊科植物白术atractylodesmacrocephalakoidz.的干燥根茎,为临床常用中药。其具有健脾益气,燥湿利水,止汗,安胎的作用,临床上多用于治疗脾虛食少,腹胀泄泻,痰饮眩悸,水肿,自汗,胎动不安等,具有较强的临床应用价值,市场需求广阔,被开发成相关复方制剂如玉屏风颗粒,以及中药配方颗粒广泛用于临床。中药的临床使用多以传统汤剂为主。中药汤剂的物质基础,是中医理论指导下防治疾病的基础。现行的法定标准没有定量控制指标,不能全面反映中药成分的整体作用。在现阶段,中药绝大多数有效成分未明确的情况下,中药指纹图谱/特征图谱的建立能大大提高中药质量控制的技术水平及科技含量。《中国药典》2015年版白术药材没有收载含量测定项,并不能全面地评价白术质量。目前文献研究报道的关于白术指纹图谱或特征图谱多采用的是常规的hplc法,且均只针对原药材的物质基础,指标成分多以脂溶性成分为研究对象;主要用于中药材的定性鉴别,不能全部反映中药汤剂的物质基础特征。技术实现要素:基于此,本发明提供一种白术药材uplc特征图谱的构建方法。该方法重现性良好,准确可靠,通过所建立的uplc特征图谱可以实现对白术药材质量的有效控制。具体技术方案为:一种白术药材uplc特征图谱的构建方法,包括以下步骤:参照物溶液的制备:以绿原酸为对照品,加溶剂溶解,制备对照品参照物溶液;取白术对照药材,加入提取溶剂提取,滤过,取续滤液作为对照药材参照物溶液;供试品溶液的制备:取白术药材,加入提取溶剂提取,滤过,取续滤液作为白术药材供试品溶液;取白术饮片,加入提取溶剂提取,滤过,取续滤液作为白术饮片供试品溶液;取麸炒白术饮片,加入提取溶剂提取,滤过,取续滤液作为麸炒白术饮片供试品溶液;取白术标准汤剂,加入提取溶剂提取,滤过,取续滤液作为白术标准汤剂供试品溶液;取麸炒白术标准汤剂,加入提取溶剂提取,滤过,取续滤液作为麸炒白术标准汤剂供试品溶液;测定:将所述对照品参照物溶液、对照药材参照物溶液、白术药材供试品溶液、白术饮片供试品溶液、麸炒白术饮片供试品溶液、白术标准汤剂供试品溶液和麸炒白术标准汤剂供试品溶液注入超高效液相色谱仪中测定,标定水溶性共有峰,即得所述白术药材uplc特征图谱。本发明还提供一种白术药材的检测方法,包括以下步骤:参照物溶液的制备:以绿原酸为对照品,加溶剂溶解,制备对照品参照物溶液;取白术对照药材,加入提取溶剂提取,滤过,取续滤液作为对照药材参照物溶液;待测溶液的制备:取待测样品,加入提取溶剂提取,滤过,取续滤液作为待测样品溶液;测定:将所述对照品参照物溶液、对照药材参照物溶液、和待测样品溶液注入超高效液相色谱仪中测定,即得所述待测样品的uplc图谱。与现有技术相比,本发明具有以下有益效果:本发明采用超高效液相色谱(uplc)法构建白术药材的特征图谱,引入白术饮片、麸炒白术饮片、白术标准汤剂和麸炒白术标准汤剂的特征图谱进行研究,针对饮片、汤剂与白术药材共有的水溶性特征成分进行研究,并作为特征图谱峰确定的依据。采用uplc-ms和相关对照品确认了3个水溶性特征成分,分别为新绿原酸、绿原酸、隐绿原酸成分,并作为白术药材特征图谱特征峰确定的依据;同时,采用了对照品、对照药材双重对照,可以有效克服液相条件指纹图谱固有存在的耐用性偏差问题,比较更加全面。本发明以未知峰1作为参照峰,规定每个特征峰的相对峰面积限量范围,实现多个特征成分的定量化,符合中药的整体作用理念。本发明采用了uplc法,与常规hplc法相比,更加高效、快速、环保;采用本发明构建的特征图谱及方法,重现性好,准确可靠,可以快速、全面实现对白术药材多个特征成分的质量监控,既提升了白术药材的质量控制水平,又提升和稳定白术药材的内在质量;保持了白术药材、白术饮片与临床汤剂质量的一致性,为临床提供符合白术标准汤剂要求的原料,为白术相关的制剂工艺生产过程提供重要的多指标参数依据。附图说明图1为15批次白术标准汤剂uplc特征图谱的叠加图(峰2:新绿原酸、峰3:绿原酸、峰4:隐绿原酸);图2为15批次白术饮片uplc特征图谱的叠加图;图3为15批次白术药材uplc特征图谱的叠加图;图4为15批次麸炒白术标准汤剂uplc特征图谱的叠加图;图5为15批次麸炒白术饮片uplc特征图谱的叠加图;图6为白术标准汤剂uplc对照特征图谱(峰2:新绿原酸、峰3:绿原酸、峰4:隐绿原酸);图7为白术饮片uplc对照特征图谱(峰2:新绿原酸、峰3:绿原酸、峰4:隐绿原酸);图8为白术药材uplc对照特征图谱(峰2:新绿原酸、峰3:绿原酸、峰4:隐绿原酸);图9为麸炒白术标准汤剂uplc对照特征图谱(峰2:新绿原酸、峰3:绿原酸、峰4:隐绿原酸);图10为麸炒白术饮片uplc对照特征图谱(峰2:新绿原酸、峰3:绿原酸、峰4:隐绿原酸)。具体实施方式以下结合具体实施例对本发明作进一步详细的说明。本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明公开内容理解更加透彻全面。除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的
技术领域:
的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。一种白术药材uplc特征图谱的构建方法,包括以下步骤:参照物溶液的制备:以绿原酸为对照品,加溶剂溶解,制备对照品参照物溶液;取白术对照药材,加入提取溶剂提取,滤过,取续滤液作为对照药材参照物溶液;供试品溶液的制备:取白术药材,加入提取溶剂提取,滤过,取续滤液作为白术药材供试品溶液;取白术饮片,加入提取溶剂提取,滤过,取续滤液作为白术饮片供试品溶液;取麸炒白术饮片,加入提取溶剂提取,滤过,取续滤液作为麸炒白术饮片供试品溶液;取白术标准汤剂,加入提取溶剂提取,滤过,取续滤液作为白术标准汤剂供试品溶液;取麸炒白术标准汤剂,加入提取溶剂提取,滤过,取续滤液作为麸炒白术标准汤剂供试品溶液;测定:将所述对照品参照物溶液、对照药材参照物溶液、白术药材供试品溶液、白术饮片供试品溶液、麸炒白术饮片供试品溶液、白术标准汤剂供试品溶液和麸炒白术标准汤剂供试品溶液注入超高效液相色谱仪中测定,标定水溶性共有峰,即得所述白术药材uplc特征图谱。该方法的超高效液相色谱的色谱条件包括:固定相:十八烷基硅烷键合硅胶为填充剂的色谱柱。所述色谱柱为watersbehc18色谱柱。所述色谱柱的柱长为50±5mm,内径为2.1±0.3mm,粒径为1.7±0.3μm。流动相:流动相a为乙腈,流动相b为0.05-0.15%磷酸溶液,进行梯度洗脱。所述梯度洗脱具体为:0-4min,流动相a的体积分数由1%上升到6%,流动相b的体积分数由99%降低至94%;4-10min,流动相a的体积分数由6%上升到9%,流动相b的体积分数由94%降低至91%;10-16min,流动相a的体积分数由9%上升到18%,流动相b的体积分数由91%降低至82%;16-22min,流动相a的体积分数由18%上升到20%,流动相b的体积分数由82%降低至80%;22-23min,流动相a的体积分数由20%上升到90%,流动相b的体积分数由80%降低至10%。该方法的超高效液相色谱的色谱条件包括:柱温为25±3℃;流速为0.25±0.1ml/min;检测波长为325±40nm。具体地,该方法的提取溶剂为水、体积分数10%~80%的乙醇水溶液或体积分数10%~80%的甲醇水溶液。所述体积分数具体可为10%、20%、30%、40%、50%、60%、70%、80%。优选地提取溶剂为体积分数30%的乙醇水溶液。提取溶剂的用量为每1g白术药材加入20ml-100ml。具体地,所述提取的方法为超声提取或加热回流提取。所述提取的时间为20-40min。所述超声的功率为300w,所述超声的频率40khz。该方法的供试品特征图谱中应呈现4个特征峰,并应与对照药材参照物色谱中的4个特征峰相对应,其中峰3应与相应的对照品参照物峰保留时间相同;与绿原酸参照物峰相应的峰为s峰,各特征峰与s峰的相对保留时间应在规定值的±10%范围之内;规定值为:峰1:0.227、峰2:0.679、峰4:0.144。计算各特征峰与s峰的相对峰面积,其相对峰面积以峰1为参照峰,计算其它峰与峰1的相对峰面积,应在规定范围内,规定值为:1.00(峰1)、1.447-11.039(峰2:新绿原酸)、9.867-78.479(峰3:绿原酸)、1.750-12.511(峰4:隐绿原酸)。具体地,所述参照物溶液的制备过程为:取白术对照药材0.2~1.0g,置具塞锥形瓶中,精密加入体积分数10%~80%乙醇10ml,称定重量,超声处理(功率300w,频率40khz)或水浴回流10~40分钟,放冷,再称定重量,用30%乙醇补足减失的重量,混匀,滤过,取续滤液,作为对照药材参照物溶液;取绿原酸对照品适量,精密称定,加甲醇溶解并制成每1ml约含绿原酸500μg的对照储备溶液。精密吸取绿原酸对照储备溶液适量,加水稀释成每1ml含绿原酸100μg的溶液,作为对照品参照物溶液。优选地,取所述白术对照药材0.5g,精密加入30%乙醇水溶液。优选地,超声处理或水浴回流30min。具体地,所述白术药材、白术饮片或麸炒白术饮片供试品溶液的制备为:将白术药材、白术饮片或麸炒白术饮片粉末过三号筛,取0.2~1g,置烧瓶中,精密加入10%~80%乙醇20ml,称定重量,超声处理(功率300w,频率40khz)或水浴回流20~40分钟,放冷,再称定重量,用10%~80%乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。优选地,取1.0g过筛的白术药材粉末,精密加入30%乙醇水溶液。优选地,超声提取或水浴回流提取30min。具体地,所述白术饮片或麸炒白术饮片标准汤剂供试品溶液的制备为:将白术标准汤剂、麸炒白术标准汤剂约2g,精密称定,置10ml量瓶,加水适量,超声处理(功率500w,频率40khz)5分钟,取出,放冷,用水定容至刻度,摇匀,滤过,取续滤液,即得。具体地,所述测定方法为分别精密吸取参照物溶液和供试品溶液各1μl,注入超高效液相色谱仪,测定,即得。作为本发明进一步优选的技术方案,所述白术药材uplc特征图谱的构建方法,还包括特征峰的指认:采用uplc-ms-ms高分辨质谱仪进行测定,其中质谱条件为四级杆串联飞行时间质谱;质谱参数最终优化为:采用waterssynaptg2hdms系统。氮气作为质谱离子源的雾化、锥孔气;电喷雾电离正、负离子模式;毛细管电压:3.0kv(正离子模式)/2.5kv(负离子模式);锥孔电压:40v;萃取锥孔电压:3v;离子源温度:100℃;脱溶剂气温度:400℃(正离子模式)/350℃(负离子模式);反向锥孔气流:50l/h;脱溶剂气流速:600l/h(正离子模式)/600l/h(负离子模式);碰撞气流速:0.5ml/min;扫描时间:0.5s;扫描时间间隔:0.02s;质荷比范围:50–1200m/z;数据采集形式:continuum;灵敏性:normal;动态范围:extended;锁定质量数:[m+h]+=556.2771;[m-h]–=554.2615。具体地,所述的白术标准汤剂制备方法为:取白术饮片,拣选除去杂质,洗净,润透,切厚片,干燥;取白术饮片100g,称定重量,加水煎煮二次,第一次煎煮加9倍量水,浸泡30分钟,武火煮沸后再文火煎煮60分钟,用350目标准筛趁热过滤,滤液迅速冷水冷却;第二次煎煮加7倍量水,武火煮沸后改文火煎煮55分钟,用350目标准筛趁热过滤,滤液迅速冷水冷却;滤液减压浓缩至适量,转移至棕色西林瓶,置于冷冻干燥机中冷冻干燥,取出,轧铝盖即得。本发明该提高一种白术药材的检测方法,包括以下步骤:参照物溶液的制备:以绿原酸为对照品,加溶剂溶解,制备对照品参照物溶液;取白术对照药材,加入提取溶剂提取,滤过,取续滤液作为对照药材参照物溶液;待测溶液的制备:取待测样品,加入提取溶剂提取,滤过,取续滤液作为待测样品溶液;测定:将所述对照品参照物溶液、对照药材参照物溶液、和待测样品溶液注入超高效液相色谱仪中测定,即得所述待测样品的uplc图谱。优选地,该检测方法的超高效液相色谱的色谱条件包括:固定相:十八烷基硅烷键合硅胶为填充剂的色谱柱;流动相:流动相a为乙腈,流动相b为0.05-0.15%磷酸溶液,进行梯度洗脱;所述梯度洗脱具体为:0-4min,流动相a的体积分数由1%上升到6%,流动相b的体积分数由99%降低至94%;4-10min,流动相a的体积分数由6%上升到9%,流动相b的体积分数由94%降低至91%;10-16min,流动相a的体积分数由9%上升到18%,流动相b的体积分数由91%降低至82%;16-22min,流动相a的体积分数由18%上升到20%,流动相b的体积分数由82%降低至80%;22-23min,流动相a的体积分数由20%上升到90%,流动相b的体积分数由80%降低至10%。优选地,所述超高效液相色谱的色谱条件还包括:柱温为25±3℃。流速为0.25±0.1ml/min。检测波长为325±40nm。优选地,所述提取溶剂为体积分数10%-80%的乙醇水溶液。用量为每1g白术药材加入20ml-100ml。具体地,所述提取的方法为超声提取或加热回流提取。优选地,所述提取的时间为20-40min。以下结合具体实施例对本发明作进一步详细的说明。实施例1白术药材uplc特征图谱的构建1、仪器、试剂、试药和药材仪器见表1,试剂见表2,试药见表3,药材见表4。表1仪器信息表仪器信息型号供应厂家高效液相色谱仪h-classpda沃特世公司behc18色谱柱2.1*50mm,1.7μm沃特世公司bdshypersilc18色谱柱2.1*50mm,3μm赛默飞公司十万分之一天平ms105du梅特勒-托利多公司万分之一天平al104梅特勒-托利多公司超声清洗机kq-500de昆山超声仪器有限公司表2试剂信息表试剂信息批号供应厂家磷酸b1214004阿拉丁公司色谱级乙腈170410815b03美国bcr公司超纯水当日新制mmilliporesynergyuv表3试药信息表对照品名称来源批号纯度规格绿原酸中国食品药品检定研究院110753-201716993%20mg/瓶新绿原酸成都德思特生物技术有限公司dst180130-015≥99%20mg/瓶隐绿原酸成都德思特生物技术有限公司dst170210-035≥98%20mg/瓶药材来源:本研究共采集了15批次白术药材,如表4所示,为来自全国白术产量较大的5个道地或主要产区,其中河北省北定市3批、安徽省4批、浙江省金华市3批、河南省2批、重庆市3批,具有充分的代表性;且按《中国药典》2015年版【鉴定】项方法鉴定,均符合规定。并取上述15批白术药材按药典方法分别制成白术饮片与麸炒白术饮片。表4白术药材产地信息表2、色谱条件与供试品溶液的制备2.1色谱条件以十八烷基硅烷键合硅胶为填充剂(柱长为50mm,内径为2.1mm,粒径为1.7μm);以乙腈为流动相a,以0.1%磷酸溶液为流动相b,进行梯度洗脱;流速为每分钟0.25ml;检测波长为325nm。2.2参照物溶液的制备(1)对照品参照物溶液的制备:取绿原酸对照品适量,精密称定,加甲醇溶解并制成每1ml约含绿原酸500μg的对照储备溶液。精密吸取绿原酸对照储备溶液适量,加水稀释成每1ml含绿原酸100μg的溶液,作为对照品参照物溶液。(2)对照药材参照物溶液的制备:取白术对照药材0.5g,置具塞锥形瓶中,精密加入30%(v/v)乙醇水溶液10ml,称定重量,超声处理(功率300w,频率40khz)或水浴回流30分钟,放冷,再称定重量,用30%(v/v)乙醇水溶液补足减失的重量,混匀,滤过,取续滤液,作为对照药材参照物溶液。2.3供试品溶液的制备白术药材供试品溶液的制备:将白术药材粉末过三号筛,取1.0g,精密称定,置烧瓶中,精密加入30%(v/v)乙醇水溶液20ml,称定重量,超声处理(功率300w,频率40khz)或水浴回流30分钟,放冷,再称定重量,用30%(v/v)乙醇水溶液补足减失的重量,摇匀,滤过,取续滤液,即得白术药材供试品溶液。2.4测定方法分别精密吸取对照药材参照物溶液、对照品参照物溶液、白术药材供试品溶液各1μl,注入超高效液相色谱仪,测定,即得。测定的方法按照如下“3、色谱条件优化”确定的条件。3、色谱条件优化3.1检测波长的确定采用watersh-class(pda)对白术药材供试品溶液在200nm至400nm处进行全波长扫描,记录光谱信息。通过移动波长,在325nm处各峰的整体响应较好,故本法采用325nm波长。3.2梯度条件的优化对原始梯度条件进行优化,得到一个优化的梯度条件。见表5.表5优化梯度表时间(分钟)流动相a(%)流动相b(%)0~41→699→944~106→994→9110~169→1891→8216~2218→2082→8022~2320→9080→103.3流动相的优化①有机相的考察取白术药材(批号:bz01)约1.0g,按“2.3”项方法制备供试品溶液;按3.2色谱条件,注入超高效液相色谱仪中,分别采用甲醇和乙腈作为色谱分离的有机相。实验结果:相近的洗脱能力下,乙腈和甲醇的分离效果相差不大,因使用的是小粒径柱,甲醇产生的柱压较高且基线不平,故本法选用乙腈。②水相的选择对比水、磷酸溶液和甲酸溶液,发现0.1%磷酸作为水相时,分离效果及峰形均好,故本法水相选用0.1%磷酸。3.4不同柱温考察取白术药材(批号:bz01)约1.0g,按“2.3项”方法制备供试品溶液,按“3.2项”色谱条件,注入液相色谱仪,在20℃、25℃、30℃的柱温下分别考察。实验结果:不同柱温对整体分离效果影响不大,耐用性较好,本法选用高于室温的25℃进行研究。3.5不同流速考察取白术药材(批号:bz01)约1.0g,按“2.3项”方法制备供试品溶液,按“3.2项”色谱条件,注入液相色谱仪,在0.3ml/min、0.25ml/min、0.2ml/min的流速分别考察。实验结果:不同流速对整体分离效果影响不大,耐用性较好,本法选用0.25ml/min的流速进行研究。4、供试品溶液制备方法考察4.1提取溶媒考察取白术药材(批号:bz01)约1.0g,精密称定,平行7份,置具塞锥形瓶中,分别精密加入水、10%(v/v)甲醇水溶液、30%(v/v)甲醇水溶液、50%(v/v)甲醇水溶液、甲醇、10%(v/v)乙醇水溶液、30%(v/v)乙醇水溶液10ml,称定重量,超声处理(功率500w,频率45khz)30分钟,放冷,分别用水、10%(v/v)甲醇水溶液、30%(v/v)甲醇水溶液、50%(v/v)甲醇水溶液、甲醇、10%(v/v)乙醇水溶液、30%(v/v)乙醇水溶液补足减失的重量,摇匀,滤过,取续滤液,即得。精密吸取1μl,注入液相色谱仪,测定。实验结果:通过对比7种提取溶剂的特征图谱可发现:30%(v/v)乙醇水溶液提取所得新绿原酸、绿原酸和隐绿原酸的总峰面积与称样量的比值最高,rad值也较好,制备样品溶液时的平行性较好,因此选择30%(v/v)乙醇水溶液作为白术药材的提取溶剂。4.2提取方式考察本次实验考察不同提取方式对白术药材特征图谱的影响,选取加热回流与超声处理两种方式。取白术药材(批号:bz01)约1.0g,精密称定,平行2份,置具塞锥形瓶中,精密加入30%(v/v)乙醇水溶液10ml,称定重量,分别超声处理(功率500w,频率45khz)30分钟、加热回流30分钟,放冷,用30%(v/v)乙醇水溶液补足减失的重量,摇匀,滤过,取续滤液,即得。精密吸取1μl,注入液相色谱仪,测定。通过对比超声与回流两种提取方式对白术药材特征图谱,可发现回流提取比超声提取新绿原酸、绿原酸与隐绿原酸的总含量高将近5倍,因此,采用回流提取的方式进行白术药材样品的前处理。4.3提取时间考察本次实验考察不同提取时间对白术药材特征图谱的影响,选取超声时间分别为:10分钟、20分钟、30分钟。取白术药材(批号:bz01)约1.0g,精密称定,平行3份,置烧瓶中,精密加入30%(v/v)乙醇水溶液10ml,称定重量,水浴回流30、40、60、90、120min,取出放冷,再次称定重量,用30%(v/v)乙醇水溶液补足减失的重量,混匀,滤过,即得。精密吸取1μl,注入液相色谱仪,测定。通过对比不同提取时间的特征图谱,发现回流提取30min基本上就能将绿原酸、新绿原酸和隐绿原酸完全提取,因此选择30min的回流时间4.4特征峰的确定取15批白术标准汤剂样品约2g,精密称定,置10ml量瓶,加水适量,超声处理(功率500w,频率40khz)5分钟,取出,放冷,用水定容至刻度,摇匀,滤过,取续滤液,即得。按照上述制备方法和色谱条件测定样品特征图谱;参见图1。取15批白术饮片样品,每份1.0g,按照上述制备方法和色谱条件测定样品特征图谱;参见图2。取15批白术药材样品,每份1.0g,按照上述制备方法和色谱条件测定样品特征图谱;参见图3。取15批麸炒白术标准汤剂样品约2g,精密称定,置10ml量瓶,加水适量,超声处理(功率500w,频率40khz)5分钟,取出,放冷,用水定容至刻度,摇匀,滤过,取续滤液,即得。按照上述制备方法和色谱条件测定样品特征图谱;参见图4。取15批麸炒白术饮片样品,每份1.0g,按照上述制备方法和色谱条件测定样品特征图谱;参见图5。实验结果表明,15批白术药材均具有4个特征峰,且4个特征峰均能从白术药材稳定转移到白术饮片、麸炒白术饮片、白术标准汤剂、麸炒白术标准汤剂中;即白术药材特征图谱与白术饮片、麸炒白术饮片、白术标准汤剂、麸炒白术标准汤剂的特征图谱中的4个特征峰相对应,因此,选择此4个特征峰为白术药材的共有峰;并以峰3(绿原酸)为参照峰进行标准研究。参见图6-图10。5、方法学考察(1)专属性考察取白术药材供试品溶液(药材批号:bz01)、空白溶剂(30%(v/v)乙醇水溶液)、参照物溶液,按已确定的色谱条件进样测定,记录色谱图。结果:白术药材4个色谱峰不受阴性空白溶剂的干扰,其中白术药材中绿原酸与参照物溶液的保留时间一致,本方法具有良好的专属性。(2)精密度考察取白术药材(批号:bz01),按“2.3项”方法制备供试品溶液,按“3.2项”色谱条件,连续进样6次测定,每次1μl,以绿原酸峰(峰3)为参照峰,计算相对保留时间;以峰1为参照峰,计算相对峰面积,实验结果见表6、表7。表6精密度实验结果(相对保留时间)编号峰1峰2峰3峰410.2370.6981.0001.15020.2360.6981.0001.15030.2360.6981.0001.15040.2370.6981.0001.15050.2370.6981.0001.15060.2370.6981.0001.150均值0.2370.6981.0001.150rsd(%)0.16%0.03%0.00%0.03%表7精密度实验结果(相对峰面积)结果表明:在连续进样6次的精密度考察中白术药材4个色谱峰相对保留时间rsd<2%,表明该特征图谱方法精密度良好。(3)稳定性考察取白术药材(批号:bz01),按“2.3项”方法制备供试品溶液,按“3.2项”色谱条件,分别在0,2,4,8,18,24,30小时进样,以绿原酸峰为参照峰,计算相对保留时间与相对峰面积,结果见表8、表9。表8稳定性实验结果(相对保留时间)放置时间峰1峰2峰3峰40h0.2390.6981.0001.1502h0.2380.6971.0001.1494h0.2380.6981.0001.1498h0.2370.6991.0001.15018h0.2380.6981.0001.15024h0.2390.6981.0001.15030h0.2380.6981.0001.150均值0.2380.6981.0001.150rsd(%)0.34%0.10%0.00%0.03%表9.稳定性实验结果(相对峰面积)放置时间峰1峰2峰3峰40h1.0000.98616.4971.1562h1.0000.93915.9621.1164h1.0000.96516.2161.1418h1.0000.95016.1011.13318h1.0000.95816.3281.14924h1.0000.97916.7391.17430h1.0000.98316.7421.178均值1.0000.96616.3691.150rsd(%)0.00%1.84%1.86%1.91%结果表明:4个色谱峰,30小时内相对保留时间的rsd<2%,表明该方法下特征峰的4个峰在30小时内稳定。(4)重复性考察取白术药材(批号:bz01),按“2.3项”方法制备供试品溶液,按“3.2项”色谱条件,考察特征峰相对保留时间的一致性,结果见表10、表11。表10重复性实验结果(相对保留时间)编号峰1峰2峰3峰410.2370.6971.0001.14920.2380.6981.0001.15030.2380.6971.0001.14940.2370.6981.0001.15050.2360.6971.0001.14960.2360.6981.0001.150均值0.2370.6971.0001.150rsd(%)0.37%0.07%0.00%0.05%表11重复性实验结果(相对峰面积)编号峰1峰2峰3峰411.0000.96116.2321.13821.0000.96016.2191.20131.0000.93215.9961.11741.0000.96416.2241.21251.0000.92515.4191.09061.0000.93916.0191.118均值1.0000.94716.0181.146rsd(%)0.00%1.77%1.95%4.32%结果表明:白术药材4个特征峰的相对保留时间rsd<2%,表明该方法的重复性良好。(5)中间精密度考察由不同分析人员,在不同时间,不同仪器上,取白术药材(批号:bz01),按“2.3项”方法制备供试品溶液,按“3.2项”色谱条件,考察特征峰相对保留时间的一致性,结果见表12、表13。表12中间精密度结果(相对保留时间)表13中间精密度结果(相对峰面积)编号峰1峰2峰3峰411.0000.95715.6841.19221.0000.97316.0631.23531.0000.93615.3361.16941.0000.93015.3941.18551.0000.93715.5481.18961.0000.98816.4231.247均值1.0000.95315.7411.203rsd0.00%2.46%2.69%2.57%结果表明:在中间精密度考察中4个特征峰的相对保留时间rsd<2.0%,表明该方法的中间精密度良好。6、白术药材特征图谱的测定6.1取白术药材样品15批,每份1.0g,按照上述制备方法和色谱条件测定样品特征图谱,测定结果见表14、表15,图谱如附图3所示。表1415批白术药材特征图谱(相对保留时间)表1515批白术药材特征图谱(相对峰面积)结果:15批白术药材特征图谱的4个特征峰相对峰面积的rsd在0.0%~76.4%,结果表明不同产地白术药材的各特征峰峰面积存在一定差异。峰2相对峰面积范围为1.447~11.039,峰3相对峰面积范围为9.867~78.479,峰4相对峰面积范围为1.750~12.511。为严格控制白术药材的质量,为白术的临床汤剂提供质优且稳定的原料药材,根据15批次不同产地白术药材的峰2、峰3、峰4的相对峰面积波动范围,考虑15批次不同产地样品的代表性,综合取各特征峰相对峰面积的最小、最大值,或均值的70%~130%、±3sd为范围,规定:与峰1为s峰,各特征峰与s峰的相对峰面积,规定各峰相对峰面积范围为:1.447~11.039(峰2),9.867~78.479(峰3),1.750~12.511(峰4)。特征图谱标准的拟定将15批白术药材uplc特征图谱使用《中药色谱指纹图谱相似度评价系统》匹配,生成对照图谱,建立白术药材对照特征图谱;结果如图8。相对保留时间确定:依据15批次白术药材特征图谱相对保留时间均值的±10%;相对峰面积范围的确定:依据15批次白术药材的相对峰面积均值的70%~130%范围。供试品色谱中应呈现4个特征峰,并应与对照药材参照物色谱中的4个特征峰相对应,其中峰3应与相应的对照品参照物峰保留时间相同;与绿原酸参照物峰相应的峰为s峰,计算各特征峰与s峰的相对保留时间,其相对保留时间应在规定值的±10%之内,规定值为:0.227(峰1)、0.679(峰2)、1.144(峰4);另以峰1为参照峰,计算各特征峰与峰1的相对峰面积,其相对峰面积应在规定范围内,规定值为:1.447~11.039(峰2),9.867~78.479(峰3),1.750~12.511(峰4)。7.白术药材特征图谱特征峰的指认7.1仪器与试药仪器:synaptg2hdms超高效液相飞行时间高分辨质谱联用系统(waterscorporation,milford,ma,usa),数据处理系统为markerlynx4.1工作站(waters,manchester,u.k.),ab135-s电子分析天平(梅特勒-托利多),kq-118b超声振荡仪(昆山市超声仪器有限公司)。试剂:色谱乙腈和甲醇购于j.t.baker(phillipsburg,nj,usa),色谱级甲酸、亮氨酸脑啡肽购于sigmaaldrich(mo,usa),实验超纯水(18.2mω)用milli-q水净化系统制备(millipore,france),其他所用试剂均为分析纯。7.2色谱条件和质谱条件色谱条件:同实施例1项下“色谱条件”。质谱条件:质谱采用waterssynaptg2hdms系统。氮气作为质谱离子源的雾化、锥孔气;电喷雾电离正、负离子模式;毛细管电压:3.0kv(正离子模式)/2.5kv(负离子模式);锥孔电压:40v;萃取锥孔电压:3v;离子源温度:100℃;脱溶剂气温度:400℃(正离子模式)/350℃(负离子模式);反向锥孔气流:50l/h;脱溶剂气流速:600l/h(正离子模式)/600l/h(负离子模式);碰撞气流速:0.5ml/min;扫描时间:0.5s;扫描时间间隔:0.02s;质荷比范围:50–1200m/z;数据采集形式:continuum;灵敏性:normal;动态范围:extended;锁定质量数:[m+h]+=556.2771;[m-h]–=554.2615。7.3参照物溶液的制备:同实施例1项下“对照品参照物溶液的制备”。7.4供试品溶液的制备:同实施例1项下“供试品溶液的制备”。7.5测定法分别精密吸取对照品参照物溶液与供试品溶液各1μl,注入uplc-ms高分辨质谱仪,测定,即得。7.6结果分析根据一级和二级质谱及相关数据库分析以及文献检索结果,采用相应标准品对照指认,分析各特征峰物质分别为:峰2为新绿原酸,峰3为绿原酸,峰4为隐绿原酸。结果见表16。表16峰2~4物质及结构峰号分子量(m/z)化合物峰2353新绿原酸峰3353绿原酸峰4353隐绿原酸实施例2:白术饮片特征图谱的测定取实施例1中表4所述的15批白术药材按药典方法分别制成白术饮片。白术饮片供试品溶液的制备:将白术饮片粉末(过三号筛),取1.0g,精密称定,置烧瓶中,精密加入30%(v/v)乙醇水溶液20ml,称定重量,超声处理(功率300w,频率40khz)或水浴回流30分钟,放冷,再称定重量,用30%(v/v)乙醇水溶液补足减失的重量,摇匀,滤过,取续滤液,即得白术药材供试品溶液。取白术饮片样品15批,每份1.0g,按照上述白术饮片供试品溶液的制备方法和实施例1中参照样溶液的制备方法和色谱条件测定样品特征图谱,测定结果见表17、表18,图谱如附图2所示。表1715批白术饮片特征图谱(相对保留时间)表1815批白术饮片特征图谱(相对峰面积)结果:如表17、18所示,15批白术饮片特征图谱的4个特征峰相对峰面积的rsd在0.00%-50.20%。结果表明不同产地白术药材制备成的白术饮片的特征峰峰面积存在一定差异,峰2范围为0.746~4.197;峰3范围为6.072~20.575;峰4范围为0.876~4.709。为严格控制白术饮片的质量,为白术饮片的临床汤剂提供优质且稳定的原料,根据15批次不同产地白术药材制备的白术饮片的峰2、峰3、峰4的相对峰面积波动范围,考虑15批次不同产地样品的代表性,综合各特征峰相对峰面积的最小、最大值,或均值的70%-130%sd为范围,规定:峰1为s峰,个特征峰与s峰的相对峰面积,规定各峰相对峰面积范围为:0.746~4.197(峰2);6.072~20.575(峰3);0.876~4.709(峰4)。特征图谱标准的拟定:将15批白术饮片uplc特征图谱使用《中药色谱指纹图谱相似度评价系统》匹配,生成对照图谱,建立白术饮片对照特征图谱;结果如图7。相对保留时间确定:依据15批次白术饮片特征图谱相对保留时间均值的±10%;相对峰面积范围的确定:依据15批次白术饮片的相对峰面积均值的70%~130%范围。供试品色谱中应呈现4个特征峰,并应与对照药材参照物色谱中的4个特征峰相对应,其中峰3应与相应的对照品参照物峰保留时间相同;与绿原酸参照物峰相应的峰为s峰,计算各特征峰与s峰的相对保留时间,其相对保留时间应在规定值的±10%之内,规定值为:0.239(峰1)、0.698(峰2)、1.144(峰4);另以峰1为参照峰,计算各特征峰与峰1的相对峰面积,其相对峰面积应在规定范围内,规定值为:0.746~4.197(峰2);6.072~20.575(峰3);0.876~4.709(峰4)。实施例3:麸炒白术饮片特征图谱的测定取实施例1中表4所述的15批白术药材按药典方法分别制成麸炒白术饮片。麸炒白术饮片供试品溶液的制备:将麸炒白术饮片粉末过三号筛,取1.0g,精密称定,置烧瓶中,精密加入30%(v/v)乙醇水溶液20ml,称定重量,超声处理(功率300w,频率40khz)或水浴回流30分钟,放冷,再称定重量,用30%(v/v)乙醇水溶液补足减失的重量,摇匀,滤过,取续滤液,即得麸炒白术药材供试品溶液。取批麸炒白术饮片样品15批,每份1.0g,按照麸炒白术饮片供试品溶液的制备方法和实施例1中参照样溶液的制备方法和色谱条件测定样品特征图谱,测定结果见表19、表20,图谱如附图5所示。表1915批麸炒白术饮片特征图谱(相对保留时间)表2015批麸炒白术饮片特征图谱(相对峰面积)结果:15批麸炒白术饮片特征图谱的4个特征峰相对峰面积的rsd在0.0%~105.4%,结果表明不同产地白术药材的各特征峰峰面积存在一定差异,峰2范围为0.045~0.221;峰3范围为0.100~2.517;峰4范围为0.041~0.250。为严格控制麸炒白术饮片的质量,为麸炒白术饮片的临床汤剂提供质优且稳定的原料,根据15批次不同产地白术药材制备的麸炒白术饮片的峰2、峰3、峰4的相对峰面积波动范围,综合取各特征峰相对峰面积的最小、最大值,或均值的70%~130%、±3sd为范围,规定:与峰1为s峰,各特征峰与s峰的相对峰面积,规定各峰相对峰面积范围为:0.045~0.221(峰2),0.100~2.517(峰3),0.041~0.250(峰4)。特征图谱标准的拟定将15批麸炒白术饮片uplc特征图谱使用《中药色谱指纹图谱相似度评价系统》匹配,生成对照图谱,建立麸炒白术饮片对照特征图谱;结果如图10。相对保留时间确定:依据15批次麸炒白术饮片特征图谱相对保留时间均值的±10%;相对峰面积范围的确定:依据15批次麸炒白术饮片的相对峰面积均值的70%~130%范围。供试品色谱中应呈现4个特征峰,并应与对照药材参照物色谱中的4个特征峰相对应,其中峰3应与相应的对照品参照物峰保留时间相同;与绿原酸参照物峰相应的峰为s峰,计算各特征峰与s峰的相对保留时间,其相对保留时间应在规定值的±10%之内,规定值为:0.236(峰1)、0.689(峰2)、1.144(峰4);另以峰1为参照峰,计算各特征峰与峰1的相对峰面积,其相对峰面积应在规定范围内,规定值为:0.045~0.221(峰2),0.100~2.517(峰3),0.041~0.250(峰4)。实施例4:白术标准汤剂特征图谱的测定标准汤剂的制备:取白术饮片,拣选除去杂质,洗净,润透,切厚片。取白术饮片100g,称定重量,加水煎煮二次,第一次煎煮加9倍量水,浸泡30分钟,武火煮沸后再文火煎煮60分钟,用350目标准筛趁热过滤,滤液迅速冷水冷却;第二次煎煮加7倍量水,武火煮沸后改文火煎煮55分钟,用350目标准筛趁热过滤,滤液迅速冷水冷却;滤液减压浓缩至适量,置于冷冻干燥机中,冷冻干燥,取出,轧铝盖即得。白术标准汤剂供试品溶液的制备:取白术标准汤剂浓缩液约2g,精密称定,置10ml量瓶,加水适量,超声处理(功率500w,频率40khz)5分钟,取出,放冷,用水定容至刻度,摇匀,滤过,取续滤液,即得。取实施例2的15批白术饮片按照上述方法分别制成15批白术标准汤剂。取白术标准汤剂15批(每份相当于白术饮片1.0g),按照上述白术标准汤剂供试品溶液的制备方法和实施例1中参照物溶液的制备方法和色谱条件测定样品特征图谱,测定结果见表21、表22,图谱如附图1所示。表2115批白术标准汤剂特征图谱(相对保留时间)表2215批白术标准汤剂特征图谱(相对峰面积)结果:15批白术标准汤剂特征图谱的4个特征峰相对峰面积的rsd在0.0%~68.63%,结果表明不同产地白术药材制备的饮片所制备成的汤剂的各特征峰峰面积存在一定差异,峰2范围为0.456~3.301;1.004~7.733(峰3);峰4范围为0.551~3.306。为严格控制白术标准汤剂的质量,为白术的临床汤剂提供质优且稳定的原料药材,根据15批次不同产地白术药材制备的饮片所制备成的汤剂的峰2、峰3、峰4的相对峰面积波动范围,综合取各特征峰相对峰面积的最小、最大值,或均值的70%~130%、±3sd为范围,规定:与峰1为s峰,各特征峰与s峰的相对峰面积,规定各峰相对峰面积范围为:0.456~3.301(峰2),1.004~7.733(峰3),0.551~3.306(峰4)。特征图谱标准的拟定将15批白术标准汤剂uplc特征图谱使用《中药色谱指纹图谱相似度评价系统》匹配,生成对照图谱,建立白术标准汤剂对照特征图谱;结果如图6。相对保留时间确定:依据15批次白术标准汤剂特征图谱相对保留时间均值的±10%;相对峰面积范围的确定:依据15批次白术药材的相对峰面积均值的70%~130%范围。供试品色谱中应呈现4个特征峰,并应与对照药材参照物色谱中的4个特征峰相对应,其中峰3应与相应的对照品参照物峰保留时间相同;与绿原酸参照物峰相应的峰为s峰,计算各特征峰与s峰的相对保留时间,其相对保留时间应在规定值的±10%之内,规定值为:0.238(峰1)、0.691(峰2)、1.147(峰4);另以峰1为参照峰,计算各特征峰与峰1的相对峰面积,其相对峰面积应在规定范围内,规定值为:0.456~3.301(峰2),1.004~7.733(峰3),0.551~3.306(峰4)。实施例5:麸炒白术标准汤剂特征图谱的测定麸炒白术标准汤剂供试品溶液的制备:取麸炒白术标准汤剂浓缩液约2g,精密称定,置10ml量瓶,加水适量,超声处理(功率500w,频率40khz)5分钟,取出,放冷,用水定容至刻度,摇匀,滤过,取续滤液,即得。本实施例的麸炒白术标准汤剂,是通过如下制备方法制备的:麸炒白术饮片标准汤剂的制备:取麸炒白术饮片,拣选除去杂质,洗净,润透,切厚片。取白术饮片100g,称定重量,加水煎煮二次,第一次煎煮加9倍量水,浸泡30分钟,武火煮沸后再文火煎煮60分钟,用350目标准筛趁热过滤,滤液迅速冷水冷却;第二次煎煮加7倍量水,武火煮沸后改文火煎煮55分钟,用350目标准筛趁热过滤,滤液迅速冷水冷却;滤液减压浓缩至适量,置于冷冻干燥机中,冷冻干燥,取出,轧铝盖即得。(1)取实施例3的15批麸炒白术饮片按照上述方法分别制成15批麸炒白术标准汤剂。取麸炒白术标准汤剂15批,按照上述麸炒白术标准汤剂供试品溶液的制备方法和实施例1中参照物溶液的制备方法和色谱条件测定样品特征图谱,测定结果见表23、表24,图谱如附图4所示。表2315批麸炒白术标准汤剂特征图谱(相对保留时间)表2415批麸炒白术标准汤剂特征图谱(相对峰面积)序号批号峰1峰2峰3峰41z-cbz01b41.0000.1781.8830.2612z-cbz02b51.0000.2341.2270.3083z-cbz03b21.0000.2510.7730.3044z-cbz04b21.0000.0970.3140.1325z-cbz05b21.0000.0620.5040.0996z-cbz06b21.0000.0890.2420.0997z-cbz07b21.0000.0980.3340.1428z-cbz08b21.0000.0400.1260.0679z-cbz09b21.0000.1290.3070.15610z-cbz10b31.0000.0330.1720.06611z-cbz11b21.0000.0610.2500.09612z-cbz12b31.0000.0280.1210.06113z-cbz13b31.0000.0730.2470.10414z-cbz14b41.0000.0640.2180.08415z-cbz15b31.0000.0580.2040.089均值1.0000.1000.4610.138最小值1.0000.0280.1210.061最大值1.0000.2511.8830.308rsd(%)0.0069.86105.9261.07结果:15批白术药材特征图谱的4个特征峰相对峰面积的rsd在0.0%~105.92%,结果表明不同产地白术药材制备的麸炒白术饮片所制备成的汤剂的各特征峰峰面积存在一定差异,峰2范围为0.028~0.251;峰3相对峰面积范围为0.121~1.883;峰4范围为0.061~0.308。为严格控制麸炒白术标准汤剂的质量,为麸炒白术饮片的临床汤剂提供质优且稳定的原料药材,根据15批次不同产地白术药材制备的麸炒白术饮片所制备成的汤剂的峰2、峰3、峰4的相对峰面积波动范围,综合取各特征峰相对峰面积的最小、最大值,或均值的70%~130%、±3sd为范围,规定:与峰1为s峰,各特征峰与s峰的相对峰面积,规定各峰相对峰面积范围为:0.028~0.251(峰2),0.121~1.883(峰3),0.061~0.308(峰4)。特征图谱标准的拟定将15批麸炒白术标准汤剂uplc特征图谱使用《中药色谱指纹图谱相似度评价系统》匹配,生成对照图谱,建立麸炒白术标准汤剂对照特征图谱;结果如图9。相对保留时间确定:依据15批次麸炒白术标准汤剂特征图谱相对保留时间均值的±10%;相对峰面积范围的确定:依据15批次白术药材的相对峰面积均值的70%~130%范围。供试品色谱中应呈现4个特征峰,并应与对照药材参照物色谱中的4个特征峰相对应,其中峰3应与相应的对照品参照物峰保留时间相同;与绿原酸参照物峰相应的峰为s峰,计算各特征峰与s峰的相对保留时间,其相对保留时间应在规定值的±10%之内,规定值为:0.235(峰1)、0.690(峰2)、1.145(峰4);另以峰1为参照峰,计算各特征峰与峰1的相对峰面积,其相对峰面积应在规定范围内,规定值为:0.028~0.251(峰2),0.121~1.883(峰3),0.061~0.308(峰4)。从整体上讲,本发明:(1)采用了uplc法,及相关的色谱柱、流速等适应性色谱条件,与常规hplc法相比,更加高效、快速、环保。(2)本发明的原料来自全国白术产量较大的5个道地或主要产区共15批次栽培样品,具有充分的代表性;研究对象为白术药材、白术饮片、麸炒白术饮片、白术标准汤剂和麸炒白术标准汤剂。(3)本发明研究白术的uplc特征图谱,引入白术饮片、麸炒白术饮片、白术标准汤剂和麸炒白术标准汤剂的特征图谱进行研究,针对其汤剂与药材共有的水溶性成分特征成分进行研究,并作为特征图谱特征峰确定的依据;采用uplc-ms及相关对照品确认其中3个水溶性特征成分,分别为新绿原酸、绿原酸、和隐绿原酸成分。(4)本发明同时采用了对照品、对照药材双重对照,可以有效克服液相条件指纹图谱固有存在的耐用性偏差问题,比较更加全面。以未知峰1作为参照峰,规定每个特征峰的相对峰面积限量范围,实现多个特征成分的定量化,符合中药的整体作用理念。采用本发明的白术药材uplc特征图谱方法,其特征峰信息量丰富,方法重现性良好,准确可靠;可以提升白术药材的质量监控水平,为临床含白术的制剂如玉屏风颗粒提供质优、稳定的白术原料。以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1