小分子共价抑制剂的计算机筛选方法及其在筛选S-腺苷甲硫氨酸脱羧酶的共价抑制剂的应用与流程
本发明属于生物医学领域,尤其是涉及了小分子共价抑制剂的计算机筛选方法及其在筛选S-腺苷甲硫氨酸脱羧酶的共价抑制剂的应用。
背景技术:
长期以来,在临床和基础研究中,小分子化合物被广泛作为药物或调控分子,用于治疗疾病或研究生物系统。这些小分子化合物,可以以共价或非共价的方式与靶标蛋白进行结合。但是,由于早期的研究人员担心共价化合物会有较大的脱靶("off-target")效应,造成很大的副作用,因此在过去的合理药物筛选和设计中,共价化合物通常被避免。也正因为如此,过去的计算机辅助筛选方法,基本上都针对非共价化合物的对接(docking)进行优化,用于筛选、设计、优化和评价非共价化合物与蛋白质的结合。然而,近几年的总结研究发现,在临床上使用的小分子化合物药物中,有近1/3的药物是共价化合物,即通过共价机制(中间态或最终形式),与靶标蛋白进行结合。这类共价化合物药物包括阿司匹林、盘尼西林、磷霉素等。因此,近几年来,很多计算软件和方法进行了改善,以用于针对蛋白靶标的共价化合物(药物或调控分子)的筛选和优化,例如DOCKovalent,CovalentDock,CovDock,DOCKTITE等。但是,这些方法都涉及到很复杂的共价键形成的计算,而且预测结果也并不太理想。其中,DOCKovalent是目前唯一一个成功指导实验筛选得了到共价抑制剂的计算方法。
在我们的前期研究中(Scientific Reports,2015,vol.5:10754),我们提出了一个S-腺苷甲硫氨酸脱羧酶(AdoMetDC)的非共价抑制剂的筛选策略。通过将活性AdoMetDC(腺苷甲硫氨酸脱羧酶)的一个底物结合口袋的关键非天然氨基酸Pyruvate68,突变为Ser(丝氨酸),我们验证可以预测非共价抑制剂的结合构象,并用于非共价抑制剂的筛选。同时,我们发现,共价抑制剂的结合构象,也可以比较好的得到预测,但是由于小分子共价基团与Ser68间存在空间碰撞,所以共价基团会有较大的偏差。如图1所述,由于空间位阻,其尾部发生了明显的偏差。
技术实现要素:
基于此,本发明提供一种小分子共价抑制剂的计算机辅助筛选方法,能够解决空间位阻使其尾部发生了明显的偏差问题,有效筛选小分子共价抑制剂。
本发明技术方案:
小分子共价抑制剂的计算机筛选方法,所述方法为在计算机辅助筛选和设计共价抑制剂的过程中,首先减弱或消除蛋白受体的空间位阻,再将其用于共价抑制剂或共价结合分子的筛选、设计和改造。
所述减弱或消除蛋白受体的空间位阻的方法为,将蛋白受体的共价残基突变为较小体积的残基或部分移除或整体移除或用无空间位阻的虚原子代替。
所述减弱或消除蛋白受体的空间位阻的方法为,当参与共价键形成的残基位于蛋白链的中间,且参与共价的都为侧链原子时,将共价残基突变为侧链较小的氨基酸,例如但不限于甘氨酸。
所述减弱或消除蛋白受体的空间位阻的方法为,当参与共价键形成的残基位于N端或C端时,将共价残基完全删除。
所述方法还包括消除空间位阻的蛋白受体进行小分子-蛋白对接计算后,再通过原子间距离约束,对计算结果进行筛选,以保证共价原子能够符合形成共价键的空间位置条件。
所述方法适用于以任何形式、在任何阶段嵌入到有关药物筛选软件中,进行共价抑制剂或共价结合分子的筛选或设计或改造或检验或评价。
本发明方法称为SCAR(Steric-Clashes Alleviated Receptor)方法。
所述的方法在筛选S-腺苷甲硫氨酸脱羧酶的共价抑制剂上的应用,所述应用包括以下步骤:
1)以S-腺苷甲硫氨酸脱羧酶的晶体结构为基础,将S-腺苷甲硫氨酸脱羧酶晶体(如PDBID为3DZ5的S-腺苷甲硫氨酸脱羧酶晶体)结构中的丙酮酰68(pyruvoyl68,Pyr68)残基删除,并在Rosetta软件中优化,得到优化后的蛋白晶体结构,即SCAR蛋白或SCAR受体;
2)搜索用于对接的小分子,小分子化合物库来自ZINC(http://zinc.docking.org)对接数据库,搜索小分子,服务器返回搜索结果后,得到对应的小分子结构;
3)将所述步骤1)优化后的蛋白晶体结构和步骤2)搜索得到的小分子结构利用AutoDockVina软件进行对接计算,得到对接计算结果;
4)对步骤3)得到的计算结果进行筛选,根据打分排序,筛选得到小分子共价抑制剂;
完成所述方法在筛选S-腺苷甲硫氨酸脱羧酶的共价抑制剂上的应用。
所述步骤2)搜索小分子条件为:①SMILES表达式:C(=O)NN,并选择“substructure”子结构,以搜索含有上述基团的分子结构;②分子量大于200且小于400;③脂水分配系数(xLogP)≤3.5;④可旋转键大于等于3,且小于等于9;⑤氢键供体大于等于2,且小于等于10;⑥氢键受体大于等于2,且小于等于10。
所述步骤4)筛选条件包括:①SMILES表达式中共价结合原子即NH2中的N的位置,与原晶体结构中Pyr68的共价O原子或抑制剂共价结合后的共价N原子位置的距离小于1埃;②该构象的对接打分小于等于-8.0。
所述应用还包括光谱法和高效液相色谱方法检测小分子对S-腺苷甲硫氨酸脱羧酶的抑制作用;
所述光谱法为小分子与S-腺苷甲硫氨酸脱羧酶混合后在37℃共孵育30min后,添加底物甲硫氨酸反应5min后进行检测,DMSO为溶剂对照,S-腺苷甲硫氨酸抑制剂米托胍腙(MGBG)为阳性对照;
光谱法具体步骤为ZL201410530672.3,一种S-腺苷甲硫氨酸脱羧酶的活性检测方法及其应用。
所述高效液相色谱方法所用仪器为Waters e2695,柱子为Waters C18反相柱,所述反向柱为5um,4.6x250mm,柱温30℃,流动相0.01mol/L甲酸铵:甲醇=97:3,所述0.01mol/L甲酸铵pH为3.5,流速0.5mL/min,检测波长254nm;上样前,加入反应体系9倍体积的甲醇终止反应,12000g离心10min除去变性的蛋白后,取上清样品进行分析,通过检测底物甲硫氨酸的消耗,验证了小分子对S-腺苷甲硫氨酸脱羧酶的抑制作用。
所述应用还包括验证小分子共价抑制剂与S-腺苷甲硫氨酸脱羧酶是否是共价结合的,具体为将小分子共价抑制剂与S-腺苷甲硫氨酸脱羧酶孵育不同时间后添加底物甲硫氨酸启动催化反应,10min后终止反应检测底物的消耗,所述孵育不同时间为0min,30min,60min,90min。
所述的应用中所用的S-腺苷甲硫氨酸脱羧酶的药物口袋,其特征在于:以S-腺苷甲硫氨酸脱羧酶的晶体结构为基础,将AdoMetDC晶体结构中的第68位丙酮酰基删除 后,与底物甲硫氨酸结合口袋形成扩大的结合口袋,即与其旁边由alpha链和beta链形成结合口袋共同形成的扩大的结合口袋,为S-腺苷甲硫氨酸脱羧酶的药物筛选和设计口袋,主要用于AdoMetDC抑制剂的筛选设计和改造。
本发明优点:
1、本发明方法操作简单,不需要对已有小分子对接算法进行复杂修改,且效果良好,该方法适用于快速进行共价抑制剂的筛选与设计;
2、本发明方法与其他预测方法比,具有相当或更好的预测效果。
3、本发明的计算筛选方法,由于不需要根据实验体系的不同而修改对接算法,所以与其它已知共价抑制剂筛选方法相比,具有更广泛的适用性,可以适用于所有共价抑制剂的筛选和设计体系。
4、本发明可以广泛利用已经发展完善的众多非共价抑制剂的对接计算方法,因此可以大大加速共价抑制剂的筛选、优化和发现。
5、本发明的实验对象是人源AdoMetDC,但是由于不同来源的AdoMetDC之间具有同源性,因此我们的抑制剂通用能够作用于其它非人源的AdoMetDC,以及其它的AdoMetDC的同源蛋白酶。
附图说明
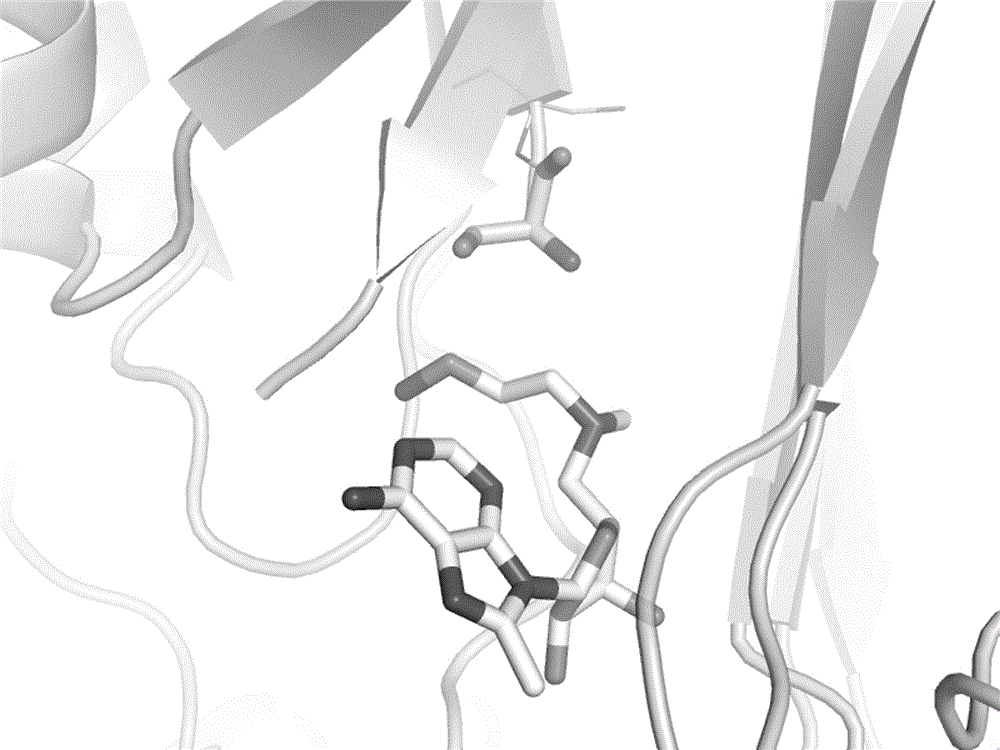
图1:PDB代码3DZ5中对应的AdoMetDC共价抑制剂分子,对接到Ser68突变的AdoMetDC中的构象。
图2:PDB代码3DZ5中对应的AdoMetDC共价抑制剂分子,对接到AdoMetDC的SCAR蛋白结构中的构象,灰色棒状为晶体结构构象,灰白棒状为对接构象。
图3:PDB代码1I72中对应的AdoMetDC共价抑制剂分子,对接到AdoMetDC的SCAR蛋白结构中的构象,灰色棒状为晶体结构构象,灰白棒状为对接构象。
图4:PDB代码1I79中对应的AdoMetDC共价抑制剂分子,对接到AdoMetDC的SCAR蛋白结构中的构象,灰色棒状为晶体结构构象,灰白棒状为对接构象。
图5:PDB代码1I7B中对应的AdoMetDC共价抑制剂分子,对接到AdoMetDC的SCAR蛋白结构中的构象,灰色棒状为晶体结构构象,灰白棒状为对接构象。
图6:光谱法检测小分子共价抑制剂对AdoMetDC的活性抑制作用对比效果。
图7:HPLC分析结果验证小分子对AdoMetDC的抑制作用;
图8:小分子与AdoMetDC共孵育不同时间对小分子抑制能力的影响;
图9:小分子共价抑制剂MCULE-4717492978分子结构;
图10:小分子共价抑制剂MCULE-3626300541分子结构;
图11:小分子共价抑制剂MCULE-3265041117分子结构;
图12:3个小分子共价抑制剂与AdoMetDC的对接构象。
具体实施方式
下面结合实施例来进一步说明本发明,但本发明要求保护的范围并不局限于实施例表述的范围。
实施例1
1)将S-腺苷甲硫氨酸脱羧酶(AdoMetDC)晶体结构中的丙酮酰68(Pyr)残基删除,并在Rosetta中优化,得到AdoMetDC的SCAR蛋白;
2)利用AutoDockVina,进行共价化合物的对接计算,将PDB代码3DZ5中对应的AdoMetDC共价抑制剂分子,对接到AdoMetDC的SCAR蛋白结构中,构象如图构象图2所示,对接构象与晶体结构构象有很好的匹配。
实施例2
1)将S-腺苷甲硫氨酸脱羧酶(AdoMetDC)晶体结构中的丙酮酰(Pyr)68残基删除,并在Rosetta中优化,得到AdoMetDC的SCAR蛋白;
2)利用AutoDockVina,进行共价化合物的对接计算,将PDB代码1I72中对应的AdoMetDC共价抑制剂分子,对接到AdoMetDC的SCAR蛋白结构中,构象如图构象图3所示,对接构象与晶体结构构象有很好的匹配。
实施例3
1)将S-腺苷甲硫氨酸脱羧酶(AdoMetDC)晶体结构中的丙酮酰(Pyr)68残基删除,并在Rosetta中优化,得到AdoMetDC的SCAR蛋白;
2)利用AutoDockVina,进行共价化合物的对接计算,将PDB代码1I79中对应的AdoMetDC共价抑制剂分子,对接到AdoMetDC的SCAR蛋白结构中,构象如图构象图4所示,对接构象与晶体结构构象有很好的匹配。
实施例4
1)将S-腺苷甲硫氨酸脱羧酶(AdoMetDC)晶体结构中的丙酮酰(Pyr)68残基删除,并在Rosetta中优化,得到AdoMetDC的SCAR蛋白;
2)利用AutoDockVina,进行共价化合物的对接计算,将PDB代码1I7B中对应的 AdoMetDC共价抑制剂分子,对接到AdoMetDC的SCAR蛋白结构中,构象如图构象图5所示,对接构象与晶体结构构象有很好的匹配。
实施例2本专利方法与其它共价化合物晶体构象预测方法的比较
选择一个在CovalentDock(J.Comput.Chem.2012,34,326–336)、CovDock(J.Chem.Inf.Model.2014,54,1932–1940)和DOCKTITE(J.Chem.Inf.Model.2015,55,398–406)等方法中共同使用的含76个蛋白-共价配体复合物的数据集,进行共价配体晶体结合构象的预测。由于在这些蛋白中,参与共价键形成的残基位于蛋白链的中间,不能整体移除。但由于这些残基中,参与共价的都为侧链原子,所以我们选择将其突变为甘氨酸,以消除侧链的空间位阻。我们先利用蛋白设计软件Rosetta,对上述蛋白中参与共价键形成的残基进行了突变,得到SCAR蛋白。然后,利用AutoDock Vina,进行共价化合物的对接计算。表1为本专利方法(SCAR)方法与其它共价化合物晶体构象预测方法的比较。其中表1(A)中,a:本发明方法(SCAR方法)的预测结果,SCAR_repacked是在对接前将蛋白在Rosetta中先进行整体的结构优化;b:DOCKTITE和CovDock的预测结果;c:CovalentDock、AutoDock和GOLD的预测结果;d:只考虑对接中小分子的共价结合原子与晶体结构中的位置距离在1埃内的情况。表1(B)对上述数据集中的所有β-lactams化合物的预测结果比较。a:SCAR方法的预测结果;b,c:DOCKovalent的预测结果。
由从表1可知,我们的方法与其他方法比,具有相当或更好的预测效果。
表1 为本专利方法(SCAR)方法与其它共价化合物晶体构象预测方法的比较
实施例3采用本专利方法进行S-腺苷甲硫氨酸脱羧酶新型共价抑制剂的筛选
1)以S-腺苷甲硫氨酸脱羧酶(AdoMetDC)的晶体结构为基础,将PDB ID为3DZ5的AdoMetDC晶体结构中的Pyr68残基删除,并在Rosetta中优化,得到优化后的SCAR蛋白晶体结构;
2)用于对接的小分子化合物库,来自与ZINC(http://zinc.docking.org)对接数据库,小分子搜索条件为:①SMILES表达式:C(=O)NN,并选择“substructure”子结构,以搜索含有上述基团的分子结构;②分子量大于200且小于400;③xLogP≤3.5;④可旋转键大于等于3,且小于等于9;⑤氢键供体大于等于2,且小于等于10;⑥氢键受体大于等于2,且小于等于10,服务器返回搜索结果后,得到对应的小分子的结构;
3)将所述步骤1)优化后的结构和步骤2)搜索到小分子结构利用AutoDock Vina进行对接计算,得到对接计算结果;
4)对步骤3)得到的计算结果进行筛选,筛选条件包括:①SMILES表达式中共价 结合原子(NH2中的N)的位置,与原晶体结构中Pyr68的共价O原子(或抑制剂共价结合后的共价N原子)位置的距离小于1埃;①该构象的对接打分小于等于-8.0,根据打分排序,选择可购买化合物用于实验筛选,得到3个小分子共价抑制剂MCULE-4717492978、MCULE-3626300541、MCULE-3265041117(小分子名称为销售商编号);
MCULE-4717492978(http://zinc.docking.org/substance/4387691)
MCULE-3626300541(http://zinc.docking.org/substance/80699)
MCULE-3265041117(http://zinc.docking.org/substance/41347)
5)利用专利(ZL201410530672.3)的方法,我们进行了AdoMetDC活性评价,光谱法检测所述步骤4)得到的3个小分子共价抑制剂对AdoMetDC的活性抑制作用,小分子与AdoMetDC混合后在37度共孵育30min后,添加底物反应5min后进行检测,DMSO为溶剂对照,AdoMetDC抑制剂MGBG(米托胍腙)为阳性对照,结果见图6,这3个小分子共价抑制剂都比MGBG有更好的抑制能力;
6)我们利用高效液相色谱(HPLC)方法,通过检测底物AdoMet的消耗,验证了这3个小分子对AdoMetDC的抑制作用。HPLC所用仪器为Waters e2695,柱子为Waters C18反相柱(5um,4.6x250mm),柱温30度,流动相0.01mol/L甲酸铵(pH 3.5):甲醇=97:3,流速0.5mL/min,检测波长254nm。上样前,加入9倍体积的甲醇终止反应,12000g离心10min除去变性的蛋白后,取上清样品进行分析,结果见图7,由图7可知小分子与AdoMetDC在37度混合孵育30min后,添加底物反应0min、5min或10min后终止反应,并检测底物的消耗。图7显示,这3个小分子共价抑制剂都能够抑制底物的消耗(主峰越高,表示检测体系中底物量越多);
7)利用HPLC方法,验证了这3个小分子共价抑制剂都是与AdoMetDC共价结合的,将小分子共价抑制剂与AdoMetDC孵育不同的时间(0min,30min,60min,90min)后添加底物AdoMet启动催化反应,10min后终止反应检测底物的消耗。结果显示(图8)随着共孵育时间的延长,小分子具有更高的抑制能力,随着孵育时间的延长,AdoMetDC的活性(空心圆)没有显著变化,非共价抑制剂MGBG(空心方块)的抑制能力也没有增加。而我们发现的3个小分子抑制剂,与AdoMetDC孵育时间越长,抑制效果越好(底物残余百分比越大)。底物残余百分比是指所述样品的底物峰面积与单独的底物对照样品的峰面积的比值。该比值越大,表明底物残余量越多,酶催化 反应消耗的底物越少,即表观酶活性越弱。
这3个小分子共价抑制剂的分子结构见图9、10、11。-C(=O)-NH-N*H2为共价基团,N*为共价结合原子。3个小分子共价抑制剂与AdoMetDC的对接构象见图12,其中M8M为PDB结构3DZ5中的AdoMetDC共价抑制剂。这3个小分子的共价结合原子N*均位于共价抑制剂M8M的共价N原子位置(球状点显示其原子半径),因此能够与Pyr68的O原子形成共价键。周围的关键相互作用残基在M8M对应图中进行了标明。
另外,共价抑制剂与AdoMetDC共价结合后,在一定条件下,小分子可以脱去氨基使AdoMetDC的Pyr68转变为丙氨酸后,从AdoMetDC上脱离下来。由于这种情况下,小分子依旧与AdoMetDC产生了共价结合,而且生成的AdoMetDC也是没有酶催化活性的,因此这种机制也属于共价抑制剂范畴。
上述的实施例仅为本发明的优选技术方案,而不应视为对于本发明的限制,本申请中的实施例及实施例中的特征在不冲突的情况下,可以相互任意组合。本发明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等同替换方案为保护范围。即在此范围内的等同替换改进,也在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!