一种基于分子动力学模拟筛分黄酮类化合物抗雄活性方法与流程
本发明属于生物领域,特别涉及一种通过分子动力学模拟方法来考察黄酮类化合物与雄激素受体的构型变化而筛分黄酮类化合物的抗雄活性的方法。
背景技术:
内分泌干扰物是指可模拟人体激素而扰乱人体内分泌干扰系统的物质,不仅影响生物的生殖系统、诱发癌症,甚至会祸及第二代、第三代。雌激素受体、雄激素受体和甲状腺受体作为核受体超家族的重要组成部分,是介导内分泌干扰效应的重要靶点。一部分内分泌干扰物可以模拟天然激素激活这些靶点,该效应称为拟-雌/雄/甲状腺激素效应,另一部分内分泌干扰物可以抑制天然激素的作用,这一效应称为抗-雌/雄/甲状腺激素效应。目前已被报道的相关的内分泌干扰物种类繁多,比如双酚A类、酞酸酯类、多氯联苯类、多溴联苯醚类等等。不仅如此,每年新增的化学品更是不计其数,需要对海量化学品的潜在内分泌干扰活性进行高效率筛分。针对这三类受体介导的内分泌干扰物,已经建立了许多实验方法用以筛选。目前主要的筛选方法有基于体内的生物标志法、组学方法,体外试验中的酵母双杂交、报告基因实验法以及竞争结合实验等等。但是这些实验方法费时费力,如果完全依靠它们来进行内分泌干扰物的筛查是不现实的。
基于这样一些挑战,计算机虚拟筛选内分泌干扰物成为了有力的工具和辅助手段,比如定量结构效应关系(quantitative structure-activity relationship,QSAR)已经被广泛使用,并且取得了很多成果。QSAR方法的基础是通过比较化合物的结构从而预测其活性,但现实中有大量的化合物结构相似,却存在活性有无的差别。也就是说,目前的QSAR方法基于结构的相似活性相似的原理仅能够预测那些已知有活性、且结构相似的化合物的活性,而不能够判断化合物活性有无。因此,在具体实践过程中,绝大多数研究者们只选择有活性化合物作为研究对象,经常忽视那些无活性化合物的存在,这是QSAR方法最大的缺陷,也是QSAR无法在预测化合物内分泌干扰活性领域得到有效应用的真正原因。为了弥补这一缺陷,必须找到一种能预判化合物活性有无的快速有效方法。
大量研究表明,化合物产生内分泌干扰效应主要通过以下几个步骤:首先,化合物要与受体相互结合(发生在配体结合域)并相互作用,达到一个稳定状态;其次,配体-受体复合物进行同源或者异源二聚化或四聚化;最后,二聚体或四聚体识别特定的染色体DNA序列,并与共调节因子(包括共激活和共抑制因子)结合,从而产生对目标基因的调控,即产生内分泌干扰效应。配体与本发明涉及的雄激素受体结合发生在配体结合域中,该区域有11个α螺旋构成(为了与其他核受体名称上的统一,把第二条螺旋直接命名为H3,第三条螺旋直接命名为H4,依次类推)。研究发现,该区域的构型变化将直接影响核受体的生物功能,其中尤为重要的是H12的位置的变化。一般认为,配体(也即小分子化合物)进入受体的配体结合域,与结合口袋内的残基发生范德华作用、电相互作用、氢键作用、疏水作用等,在这些力的共同作用下,配体和受体发生运动,最终稳定在特定位置后,才能发生转录作用。对于受体而言,运动的主要特征是其H12的变化,最终稳定在特定的位置,与其他相对稳定的螺旋提供一个与共激活因子/共抑制因子结合的表面。对于配体而言,其运动的主要特征是是否能一直存在于结合区域,并且是否能在结合区域稳定存在。此外,由于雄激素受体属于核受体家族中“类雌激素受体”,与甲状腺激素受体(Thyroid hormone receptor,TR)等不同,该受体只发生同源二聚化,不会受到其他核受体转录的影响。因此,我们可以通过模拟手段考察配体、H12的运动来预判在真实的生物体系中,对应小分子的受体活性。
分子动力学模拟(molecular dynamics simulation,MD)是一种基于计算机考察原子和分子物理运动的模拟技术。其基本原理是,给定模拟体系的初始态,依靠牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系综中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的宏观性质。该方法已经被大量用于解释药物激活靶点的机理上,但是作为一种准确筛分工具,却从来没有被应用。该技术可以作为QSAR预测化合物内分泌干扰效应的前提技术。
张爱茜等曾将分子动力学技术用于识别具有ER激动和拮抗作用的有机物(张爱茜,蔺远,彭素芬,刘磊,高常安,韩朔睽.一种有机物雌激素受体激动和拮抗作用的识别方法[P].江苏:CN101381894,2009-03-11),但是该方法仅针对ER,且未引入蛋白变构的概念。
于红霞等曾将分子动力学技术用于筛选核受体介导内分泌干扰物(于红霞,史薇,王小享.基于分子动力学模拟的核受体介导内分泌干扰物质的虚拟筛选方法[P].江苏:CN201310288617.3,213-9-25),但该方法仅能通过比对波形进行大致判断,无法进行定量预测,预测精度低,实用性差。
技术实现要素:
本发明的目的是根据雄激素受体与黄酮类化合物之间的相互作用关系,提供一种基于计算机技术、省时省力经济的筛分黄酮类化合物抗雄活性方法,为化学品安全和风险评估提供技术支撑。
为了实现上述目的,本发明采用以下技术方案:一种基于分子动力学模拟筛分黄酮类化合物抗雄活性方法,包括以下步骤:
(1)构建黄酮类化合物和雄激素受体的配体-受体复合物分子结构;
(2)对分子配体-受体复合物分子结构进行动力学模拟;
(3)建立模拟结果区分模型;
(4)对模拟结果根据模型进行判断:如果化合物位于有活性区域,则判断化合物为有抗雄活性;如果化合物位于无活性区域,则判断化合物为无抗雄活性;如果化合物位于模糊区域,则进行步骤五和六;
(5)模拟结果中配体在A环有甲氧基且受体于配体形成的氢键数量少于设定值时,判断判断化合物为无抗雄活性;
(6)观察受体和配体运动轨迹,如果配体的运动轨迹已经脱离了雄激素受体的结合口袋,判定化合物无抗雄活性;如果受体在模拟过程结束后出现“mousetrap”现象,判定化合物有抗雄活性;
(7)如化合物经步骤五和六均判断为无抗雄活性,则判断化合物为无抗雄活性,否则判断化合物为有抗雄活性;
(8)对判断有抗雄活性的化合物建立CoMSIA预测模型,预测具有抗雄活性黄酮化合物的活性大小。
所述步骤1中配体-受体复合物分子结构的构建采用以下步骤:构建黄酮类化合物的分子结构和雄激素受体初始态结构,进行能量优化,并赋予Gasteiger-Huckel电荷;利用Sybyl7.3的软件将配体对接入受体文件,范围阈值和膨胀系数分别为0.5和0,选取对接得分最高的构象作为初始构象,将之与受体合并,组成复合物;
所述雄激素受体的初始态结构采用雄激素受体的氨基酸序列文件,导入Swiss-Model网络服务器中,然后以ERα初始态的蛋白晶体结构为模板进行同源模建。
所述步骤2中进行动力学模拟,采用GROMACS软件进行,对配体和受体都赋予CHARMM27力场,其中受体的力场参数为软件自带,而配体的力场参数由Swiss-Param赋予;将整个系统放入被TIP3P水分子占据的模型立方体盒子中,复合物距离盒子边界的最短距离为1纳米,加入钠离子或氯离子保证系统的电性平衡;用最陡下降法和共轭梯度法实现整个系统的能量收敛,然后系统升温至300K并一直维持,并赋予1个标准大气压;约束受体蛋白后进行50ps的分子动力学模拟,最后撤销约束进行模拟。
所述动力学模拟中电相互作用使用particle mesh Ewald算法,范德华力以1纳米截断半径,模拟的步长2fs,一个体系进行20ns的模拟。
所述模拟结果区分模型建立采用以下步骤:选取48种典型的黄酮类作为筛选集,构建48个黄酮-雄激素受体复合物,体系经过预处理后使用Gromacs进行分子动力学模拟,体系维持在1atm,截断半径设为动力学模拟步长为2fs,每2ps保存一个构象,每个体系进行20ns的动力学模拟;然后对H12进行均方根偏差分析,有抗雄活性的化合物的H12在8ns保持稳定,而其他体系的H12一直保持波动;将H12在8~20ns的均方根偏差导入R语言,获得区分模型。
本发明的基本原理是利用分子动力学计算雄激素受体结合受试化合物后发生的构型变化,基于H12链在8~20ns时间波动情况、化合物与受体在8~20ns时间内形成氢键情况、配体小分子运动轨迹、配体小分子在模拟期间波动情况来综合判定受试化合物是否有相关的受体活性。
采用本发明方法可以模拟黄酮类化合物于雄激素受体结合后相互作用的动态过程,基于分子动力学模拟鉴别物质是否具有受体抗性,然后利用CoMSIA模型来预测这些有抗性但活性未知的黄酮类化合物活性。该方法成本低廉,操作简便,为大规模预测黄酮类化合物抗雄活性提供了用力的工具,也为进一步筛分具有抗雄活性的黄酮类药物提供支撑。
本方法首次引入定量判断,并运用定量和定性结合来提高预判的精确度,使得抗性和无抗性黄酮的区分率达到87.5%,无抗性黄酮的筛除率达到90%,不仅能够大幅削减实验室工作量,减少试验品消耗,节约资源,还能大幅提高QSAR预测的准确性,使得QSAR能够真正实现应用。
附图说明
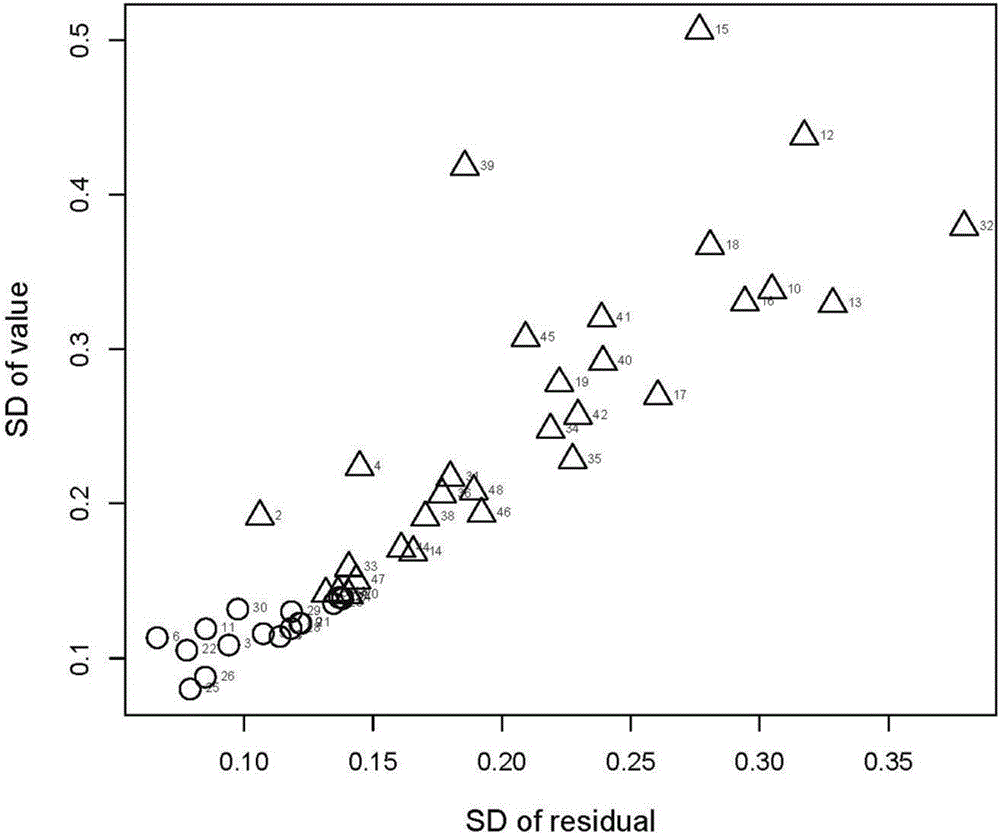
图1是实施例的区分模型,圆圈代表实测有活性化合物,三角形代表实测无活性化合物。
具体实施方式
以下通过实施例进一步说明本发明,但本发明的保护范围不仅限于实施例。
(1)在chemdraw内构建受试化合物的分子结构,在高斯09软件中用密度泛函方法b3lyp/6-311G*基组进行能量优化,然后利用Sybyl7.3软件赋予Gasteiger-Huckel电荷。本研究中需要的是受体的自由态结构,从蛋白质数据库(PDB)获得ER的自由态结构,其PDB编号是1A52,然后以1A52作为模板,利用Swiss-Model网络服务器构建AR的初始结构。构建的结果用拉氏图法进行检验,证明结果的合理性。所有结构都用spdv检查,缺失的残基用该软件自动补齐。然后利用Sybyl7.3软件的Surflex-dock模块寻找受体大分子活性口,将受试化合物分别对接入口袋。选取对接打分值最高的化合物构象作为活性构象,将之与受体合并,组成复合物用于分子动力学模拟。
(2)然后对构建好的配体-受体复合物进行分子动力学模拟。的分子动力学模拟都是基于GROMACS软件包,该软件相比于AMBER等其他分子动力学软件具有更快速的运算速度,适宜于大规模的筛选。对配体和受体都赋予CHARMM27力场,其中受体的力场参数为软件自带,而配体的力场参数有Swiss-Param赋予。将整个系统放入被TIP3P水分子占据的模型立方体盒子中,复合物距离盒子边界的最短距离为1纳米。为了保证系统的电性平衡,在体系中加入钠离子或氯离子。用最陡下降法和共轭梯度法实现整个系统的能量收敛,然后系统升温至300K并一直维持,并赋予1个标准大气压。约束受体蛋白后进行50ps的分子动力学模拟,最后撤销约束进行模拟。电相互作用使用particle mesh Ewald(PME)算法,范德华力以1纳米截断半径,模拟的步长2fs,一个体系一般进行20ns的模拟。
(3)根据模拟的结果提取受体H12(receptor-H12)空间位置的均方根偏差(root mean square deviation,RMSD),使用R语言包计算其标准偏差和标准残差:RMSD(i)=a×time(i)+b+ε(i),其中ε是残差,a和b是常数。建立标准偏差和残差之间的相关性,获得区分模型。当化合物位于有活性区域时,认为该化合物为有抗雄活性;但化合物位于无活性区域时,认为该化合物无抗雄活性;当化合物位于模糊区域时,则需要进行进一步判断。
(4)根据模拟结果提取受体-配体相互作用过程中形成的氢键(hydrogen bond),分析8-20ns间受体于配体形成的氢键数量。当配体在A环有甲氧基且在8-20ns间形成氢键数量很少,认为该化合物无抗雄活性。
(5)观察受体和配体运动轨迹,如果配体的运动轨迹已经脱离了雄激素受体的结合口袋,判定化合物无抗雄活性;如果受体在模拟过程结束后出现“mousetrap”现象,判定化合物有抗雄活性。
(6)使用技术方案(4)-(5)对技术方案(3)中无活性组合模糊组的化合物进行验证和区分:若这两组中化合物满足技术方案(4)和(5)中关于有(无)活性判定,则化合物有(无)活性。
(7)运用sybyl软件中CoMSIA模块,基于已知黄酮化合物抗雄活性,建立CoMSIA预测模型,预测未知活性但判定具有抗雄活性黄酮化合物的活性大小。
实施例
初始态受体文件构建:
AR属于“雌激素类”核受体,其初始态结构并未获得,但ERα初始态的蛋白晶体结构已经由前人通过实验获得(PDB code:1A52)。“雌激素类”核受体的特点是在激活态/抑制态的H12位置差异明显,因此AR也具有相同特性。从美国国家生物信息中心数据库(national center for biotechnology information,NCBI)获AR的氨基酸序列文件,导入Swiss-Model网络服务器中,然后以1A52为模板进行同源模建。同源模建完成后要在Pymol软件中进行叠合处理,将新构建H11-H12以H11为叠合参考系叠合至实验PDB文件,将构建的H12合并至原文件的H1-H11获得新的结构文件,也即受体的初始态结构。过程中,需要的AR的PDB文件是1T7T。
配体-受体复合物构建:
利用本发明对蛋白文件和小分子文件进行预处理,用于进一步的分子对接与分子动力学模拟。在chemdraw12.0内绘制小分子的二维结构式,然后用chem3D12.0的MM2模块进行初步优化。优化得到的结构用高斯软件进行量子优化。其中量子优化利用密度泛函方法进行,在6-311G*基组上进行。最后对所有小分子统一地赋予Gasteiger-Huckel电荷。所有用于分子动力学模拟的受体文件也都要进行预处理。先用spdbv对氨基酸缺损的支链进行修补,在Sybyl7.3内对每个蛋白文件加氢原子,同时赋予Gasteiger-Huckel电荷。
利用Sybyl7.3的Surflex-Dock模块将小分子对接入受体文件。用自动搜索方法搜寻对接口袋,范围阈值和膨胀系数分别设为0.5和0。然后将小分子对接入对接口袋,每一个小分子将得到得分最高的是个构象,我们选取得分最高的构象作为分子动力学模拟的初始构象。
建立黄酮类的抗雄激素活性区分模型:
选取48种典型的黄酮类作为筛选集,采用本发明构建48个黄酮-AR复合物,体系经过预处理后使用Gromacs进行分子动力学模拟。其中包括结构10000步最陡下降法优化、50ps升温至300K、200ps的常温常压(NPT)系综平衡和最终的模拟过程。在模拟过程中,体系维持在1atm,截断半径设为动力学模拟步长为2fs,每2ps保存一个构象用于结果分析。
每个体系进行20ns的动力学模拟,然后通过对H12的RMSD的分析发现,有抗雄活性的化合物的H12在8ns之前就保持稳定,而其他体系的H12一直保持波动。我们将H12在8-20ns的RMSD导入R语言,获得区分模型,13个黄酮化合物位于有活性区,27个位于无活性区、7个位于模糊区。分析黄酮化合物8-20ns模拟时间内生成的氢键数量,当某一化合物在A环有甲氧基但很少生成氢键的情况下,认为该化合物无活性。在活性区域中的樱黄素(prunetin)由于在A环有甲氧基且很少氢键,认为该化合物无抗雄活性;在模糊区域中的7-甲氧基黄酮也同样为无抗雄活性。此外,在活性组中,6-甲氧基黄烷酮表现出“mousetrap”现象,再一次确定活性;在非活性组中,3-羟基-6-甲氧基黄酮直接在模拟过程中从结合口袋中逃逸,再次确定无活性。通过本发明,48个黄酮化合物中的42个可以判定是否具有抗雄活性;31个无抗雄活性黄酮化合物中的27个可以被明确鉴定出来,鉴别率高达90%。鉴别结果与我们通过MDA-kb2实验结果和文献结果相一致。
以上所述,仅是本发明的较佳实施例而已,并非对本发明做任何形式的限制。凡是依据本发明的技术和方法实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明的技术和方法方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!