一种贵金属@石墨层核壳结构电催化剂的制备方法及应用与流程
本发明属于贵金属电催化剂领域,具体涉及一种贵金属@石墨层核壳结构电催化剂的制备方法及应用。
背景技术:
随着时代的发展,能源和环境问题已经成为了文明前进道路上不可避免的阻碍。近年来,化石能源已成为全球消耗的最主要能源,占消耗能源总比重的80%甚至90%以上。一方面,随着化石燃料的不断开采,化石能源的枯竭是不可避免的;另一方面,化石能源使用过程中所产生的温室气体及一些污染气体对生态环境不利。所以,寻找清洁的可再生能源势在必行。
质子交换膜燃料电池作为将化学能转变为电能的电化学装置,具有能量转化效率高、运行温度低、能量密度高、启动快等特点,因此受到广泛关注。与阳极氢氧化反应相比,阴极氧还原反应动力学是限制燃料电池能量转化效率、成本和稳定性的主要因素之一,因此,对氧还原反应催化剂的研究具有重要意义。高性能的氧还原反应催化剂需要满足以下两个条件:低过电位和长寿命。铂基电催化剂具有良好的活性及稳定性,但价格昂贵、资源有限且易被毒化,低铂和非铂电催化剂的研究就变得很有意义和价值。
王衡东等将碳纳米管加入到表面活性剂溶液中分散,向形成的分散液中滴加铂卤化合物溶液,配制成铂含量为0.01-10g/L的混合溶液,混合溶液用γ射线辐照,辐照剂量为0.1–100kGy,辐照时间为2-150小时,制得所述燃料电池用电极催化剂。该方法制备电催化剂时,需要辐照时间较长,成本较高,并且在反应过程中加入了表面活性剂,由于表面活性剂不易清洗且易附着在电催化剂表面,将会影响电催化剂的活性。(王衡东,邱士龙,叶寅,上海世龙科技有限公司,申请号200510024474.0)。
张兵等先在反应釜中加入乙二醇、贵金属前驱物和表面活性剂,搅拌均匀;再向反应釜中加入甲醛,搅拌均匀;将反应釜密封后,置于130–200℃烘箱中,反应6-10小时;最后将反应釜自然冷却至室温,离心洗涤,制备三维网络结构贵金属纳米电催化材料。该方法制备的催化剂贵金属颗粒较大,影响了贵金属电催化剂的利用效率,并且采用反应釜,需要高温、高压,耗能高,不利于工业化生产。(张兵,张晋,崔建华,张华,侯双霞,许友,天津大学,申请号201110067916.5)
陈敬艳利用射频磁控溅射设备,在多晶钛片上沉积纯Fe薄膜,使用高纯度的铁靶,当背景压强达到1×10-4Pa时,通入Ar气体,并调节气体流量为20sccm,等待一段时间。调节反应气压为0.5Pa,待气压稳定。开射频电源,射频功率为150W,开始溅射,溅射时间为40min。溅射结束后,将样品取出待下一步备用。将样品放入高真空热处理炉中,当背景压强达到1×10-4Pa时,通入保护气体Ar,升温时间为2h,热处理温度为400℃,保温时间为5min。热处理结束后,继续通保护气体Ar,冷却至室温;可得到纯Fe薄膜催化剂。该方法制备的非贵金属电催化剂活性依然较低,限制了其在质子交换膜燃料电池中的应用。(陈敬艳,长春师范大学,申请号201410140463.8)
安丽等人将含钼前驱体、含钴前驱体及载体分散在邻二甲苯溶液中,在140-155℃的条件下反应2.5-4.5h,洗涤干燥后得到MoCo/载体合金的前驱体,然后在还原气氛下进行低温热处理,最终得到负载型MoCo/载体合金和/或MoCo-N/载体合金非贵金属电催化剂。该方法制备得到的电催化剂在碱性中的活性较好,但是由于该非贵金属电催化剂为负载型催化剂,即合金颗粒担载在载体上,在酸性条件下非贵金属并不稳定,限制了其在质子交换膜燃料电池中的应用。(安丽,夏定国,张楠林,陈鑫,鲁元军,北京大学,申请号:201310170148.5)
综上所述,已报道的贵金属和非贵金属电催化剂主要存在以下几个问题:(1)贵金属电催化剂中贵金属颗粒尺寸较大,贵金属的利用效率较低,造成了电催化剂的成本过高,并且其活性和耐久性还有待提高;(2)已报到的非贵金属电催化剂在氧还原活性的测试过程中,使用的电解液是氢氧化钾,但是质子交换膜燃料电池为酸性介质,而非贵金属电催化剂通常在酸性工况下,活性较低,若表面含有过渡金属,则会影响电催化剂的寿命,限制了其在质子交换膜燃料电池中的应用。
技术实现要素:
针对现有技术存在的问题,本发明提供一种贵金属@石墨层核壳结构电催化剂的制备方法及应用,本发明制得的贵金属@石墨层核壳结构电催化剂结合了贵金属电催化剂和非贵金属电催化剂的优点,并以氮掺杂的石墨层固定贵金属纳米颗粒,使得在贵金属载量较低时,电催化剂依然具有较高的活性和稳定性。
为了达到上述目的,本发明的技术方案为:
一种贵金属@石墨层核壳结构电催化剂的制备方法,包括以下步骤:
(1)制备担载在碳上的贵金属电催化剂
将表面活性剂溶解在憎水的有机溶剂A中,其中,表面活性剂在憎水的有机溶剂中的摩尔浓度为0.1-1000mmol/L;然后加入贵金属盐水溶液,其中,贵金属盐水溶液的浓度为0.1-500mmol/L,在10–50℃下搅拌0.2-2h,静置分层,取有机相溶液加入碳载体,超声分散,加入去离子水稀释2~30倍,然后加入还原剂水溶液,其中,还原剂水溶液的浓度为0.1–5000mmol/L,在室温下搅拌2-60min,并在50-200℃下回流搅拌0.5h-3h,然后将沉淀洗涤至中性、烘干,最终得到担载在碳上的贵金属电催化剂。
(2)制备贵金属@石墨层核壳结构电催化剂
将质量比为0.01-10:1的金属大环化合物和担载在碳上的贵金属电催化剂分散在有机溶剂B中,其中,金属大环化合物在有机溶剂中的浓度为0.001-100g/L,旋转蒸发,蒸干有机溶剂后,取出剩余物质,在惰性气氛中200-1200℃条件下热处理0.5-12h,得到碳化物;然后将得到的碳化物加入到酸性水溶液中,在30-100℃的水浴中回流0.5-24h后,抽滤,水洗至中性,干燥后得到贵金属@石墨层核壳结构电催化剂,所述的贵金属@石墨层核壳结构电催化剂中贵金属在碳载体上的载量为5-60wt%。
所述的表面活性剂为十六烷基三甲基溴化铵、十八烷基三甲基氯化铵、十二烷基苯磺酸钠、十六烷基磺酸钠、硬脂酸钾、油酰基多缩氨基酸钠、十二烷基氨基丙酸钠、月桂基硫酸钠、聚环氧乙烯月桂酰醚、月桂酸失水山梨醇酯、油酸二乙醇酰胺、十四烷基二甲基磺乙基甜菜碱、硬脂酸、油胺、油酸和它们的衍生物及类似物中的一种或两种以上的混合物;
所述的憎水的有机溶剂A为甲苯、二甲苯、己烷、环己烷、环己酮、四氯化碳、氯仿、甲基异丁酮、醋酸异丙酯和它们的衍生物及类似物中的一种或二种以上的混合物。
所述的贵金属盐为氯铂酸、氯亚铂酸、氯铂酸钾、氯亚铂酸钾、氯铂酸钠、氯亚铂酸钠、氯铂酸铵、氯亚铂酸铵、三氯化钌、氯钌酸、氯钌酸钾、氯钌酸钠、氯钌酸铵、氯亚钌酸铵、三氯化铑、氯铑酸、氯铑酸钾、氯铑酸钠、氯铑酸铵、二氯化钯、氯钯酸钾、氯钯酸钠、氯钯酸铵、氯亚钯酸钾、氯亚钯酸钠、氯亚钯酸铵、四氯化铱、氯铱酸、氯铱酸钠、氯铱酸钾、氯铱酸铵、三氯化金、氯金酸、氯金酸钠、氯金酸钾、氯金酸铵中的一种或两种以上的混合物;
所述的碳载体为炭黑、活性碳、碳纳米管、碳纤维、石墨烯中的一种或两种以上的混合物。
所述的还原剂为硼氢化锂、硼氢化钠、硼氢化钾、甲醇、乙醇、水合肼、乙二酸、甲醛、甲酸、苹果酸、柠檬酸、葡萄糖、抗坏血酸中的一种或两种以上的混合物。
所述的有机溶剂B为氯仿、二氯甲烷、四氯甲烷、苯、甲苯、二甲苯、四氯化碳、乙醚、乙酸乙酯、正己烷中、乙醇、甲醇、丙三醇、丙酮中的一种或两种以上混合。
所述的金属大环化合物为氯化血红素(Hemin)、亚铁血红素(Heme B、Heme C)、meso-四(4-甲氧基苯基)卟啉铜、meso-四(4-甲氧基苯基)卟啉铁、meso-四(4-甲氧基苯基)卟啉钴、meso-四(4-甲氧基苯基)卟啉锌、meso-四(4-甲氧基苯基)卟啉锰、meso-四(4-甲氧基苯基)卟啉镍和其类似物及衍生物中的一种或两种以上的混合物;
所述的惰性气氛为氮气、氨气、氦气、氩气中的一种或两种以上的混合物。
所述的酸性水溶液为硼酸、硫酸、苯磺酸、盐酸、氢溴酸、氢碘酸、磷酸、氢氟酸、硝酸、高氯酸中的一种或两种以上的混合物;酸性水溶液的浓度为0.1‐10mol/L。
采用上述方法制备得到的的贵金属@石墨层核壳结构电催化剂能够应用于燃料电池。
本发明制备的电催化剂结合了贵金属电催化剂在酸中的活性高和稳定性好的优势以及非贵金属电催化剂(氮掺杂的石墨层电催化剂)稳定性较好的优势,与普通的贵金属电催化剂及非贵金属电催化剂相比,主要具有以下优势:(1)活性位点增加,与普通的贵金属电催化剂相比,存在贵金属纳米颗粒活性位点的同时还具有非贵金属电催化剂活性位点;(2)氮掺杂的石墨层与贵金属纳米颗粒之间存在协同作用,使电催化剂的活性和稳定性显著提高;(3)由于氮掺杂的石墨层对原位的贵金属纳米颗粒存在一定的固定作用,抑制了奥斯瓦尔德熟化,限制了贵金属纳米颗粒粒径的增长以及组分的迁移和流失,从而可以提高催化剂的稳定性。此电催化剂可用于燃料电池。
附图说明
图1为本发明实施例1制备产物的TEM照片;
图2为本发明实施例2制备产物的TEM照片;
图3为本发明实施例2制备得到的产物和商业Pt/C(20wt%)在氮气饱和的0.1mol/L KOH电解液中的循环伏安曲线;
图4为本发明实施例2制备得到的产物和商业Pt/C(20wt%)在氧气饱和0.1mol/L KOH电解液中的氧还原曲线;
图5为本发明实施例4制备得到的产物和商业Pt/C(20wt%)在氮气饱和的0.1mol/L HClO4电解液中的循环伏安曲线;
图6为本发明实施例4制备得到的产物和商业Pt/C(20wt%)在氧气饱和的0.1mol/L HClO4电解液中的氧还原曲线;
图7为本发明实施例5制备得到的产物和商业Pt/C(20wt%)在氮气饱和的0.1mol/L HClO4电解液中的循环伏安曲线;
图8为本发明实施例5制备得到的产物和商业Pt/C(20wt%)在氧气饱和的0.1mol/L HClO4电解液中的氧还原曲线。
具体实施方式
下面的实施例将对本发明予以进一步的说明,但并不因此而限制本发明。
实施例1:
将十六烷基三甲基溴化铵145.8mg溶于10mL氯仿中,将氯亚铂酸钾水溶液(10mL,20mmol/L)加入到上述氯仿溶液中,在25℃下磁力搅拌1h;然后取出10mL有机相并加入156.1mg炭黑,超声分散均匀,加入90mL去离子水,再加入硼氢化钠(10mL,300mmol/L)水溶液,在室温下搅拌5min,再在80℃下冷凝回流1h,抽滤,获得沉淀物,烘干,得到黑色沉淀;将50mg meso-四(4-甲氧基苯基)卟啉铁加入35mL氯仿中,超声分散均匀后再加入50mg前述黑色沉淀,超声分散0.5h,在40℃水浴下进行旋转蒸发,转速为100rpm。将药品转移到石英舟中,在氩气气氛下进行热处理,在800℃下热处理2h,随炉自然冷却至室温。用0.5mol/L硫酸在80℃冷凝回流6h,抽滤,洗至中性,在65℃下干燥。即得到Pt载量为12wt%的低载量贵金属@石墨层核壳结构电催化剂。
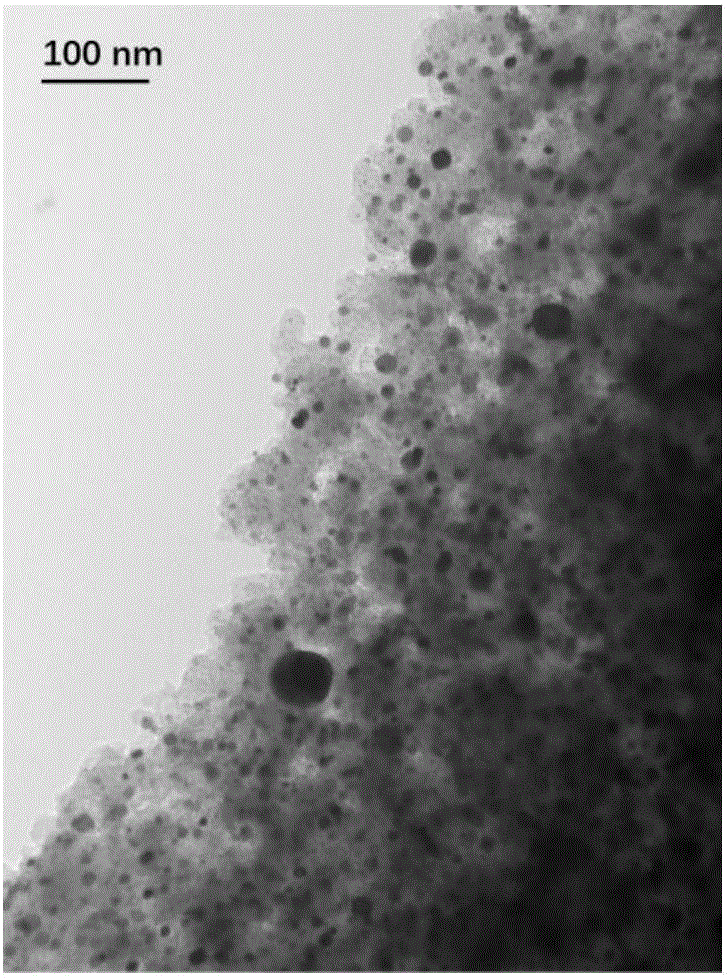
如图1,本发明实施例1制备产物的TEM照片,Pt纳米颗粒大小约为8–15nm,均匀担载到碳载体上,没有散落的Pt纳米颗粒。
实施例2:
将十六烷基三甲基溴化铵145.8mg溶于10mL氯仿中,将氯亚铂酸钾水溶液(10mL,20mmol/L)加入到上述氯仿溶液中,在25℃下磁力搅拌1h;用移液枪将10mL有机相取出并加入156.1mg炭黑,超声分散均匀,加入90mL去离子水,再加入10mL 300mmol/L硼氢化钠水溶液,在室温下搅拌5min,再在80℃下冷凝回流1h,抽滤,获得沉淀物;真空抽滤,沸水洗涤,在65℃干燥,得到黑色沉淀;将50mg meso-四(4-甲氧基苯基)卟啉铁加入44mL氯仿中,超声分散均匀后再加入75mg前述黑色沉淀,超声分散0.5h,在40℃水浴下进行旋转蒸发,转速为100rpm。将药品转移到石英舟中,在氩气氛下进行热处理,在800℃下热处理2h,随炉自然冷却至室温。用0.5mol/L硫酸在80℃冷凝回流6h,抽滤,并水洗至滤液呈中性,在65℃下干燥。即得到Pt载量为12wt%的低载量贵金属@石墨层核壳结构电催化剂。
如图2,本发明实施例2制备产物的TEM照片,Pt纳米颗粒大小约为3–10nm,均匀担载到碳载体上,未见聚集及散落的Pt纳米颗粒。
如图3,本发明实施例2制备得到的产物和商业Pt/C(20wt%)在氮气饱和的0.1mol/L KOH电解液中的循环伏安曲线,扫描速率为50mV/s,参比电极为饱和甘汞电极,碳棒为对电极。
如图4,本发明实施例2的制备得到的产物和商业Pt/C(20wt%)在氧气饱和0.1mol/L KOH电解液中的氧还原曲线,扫描速率为10mV/s,转速为1600rpm,参比电极为饱和甘汞电极,碳棒为对电极。
实施例3:
将十二烷基苯磺酸钠291.6mg溶于10mL氯仿,将氯铱酸水溶液(10mL,20mmol/L)加入到上述氯仿溶液中,在10℃下磁力搅拌0.2h;然后取出10mL有机相并加入156.1mg活性炭,超声分散均匀,加入90mL去离子水,再加入硼氢化钾(10mL,20mmol/L)水溶液,在室温下搅拌5min,再在200℃下冷凝回流0.5h,抽滤,获得沉淀物,烘干,得到黑色沉淀;将50mg meso-四(4-甲氧基苯基)卟啉铁加入35mL氯仿中,超声分散均匀后再加入50mg前述黑色沉淀,超声分散0.5h,在40℃水浴下进行旋转蒸发,转速为100rpm。将药品转移到石英舟中,在氩气气氛下进行热处理,在800℃下热处理2h,随炉自然冷却至室温。用0.5mol/L硫酸在100℃冷凝回流0.5h,抽滤,洗至中性,在65℃下干燥。即得到Pt载量为10wt%贵金属@石墨层核壳结构电催化剂。
实施例4:
将十六烷基三甲基溴化铵145.8mg溶于10mL氯仿,将氯亚铂酸钾水溶液(10mL,20mmol/L)加入到上述氯仿溶液中,在25℃下磁力搅拌1h;然后取出10mL有机相并加入156.1mg炭黑,超声分散均匀,加入90mL去离子水,再加入硼氢化钠(10mL,300mmol/L)水溶液,在室温下搅拌5min,再在80℃下冷凝回流1h,抽滤,获得沉淀物,烘干,得到黑色沉淀;将25mg meso-四(4-甲氧基苯基)卟啉铁加入35mL氯仿中,超声分散均匀后再加入100mg前述黑色沉淀,超声分散0.5h,在40℃水浴下进行旋转蒸发,转速为100rpm。将药品转移到石英舟中,在氩气气氛下进行热处理,在800℃下热处理2h,随炉自然冷却至室温。用0.5mol/L硫酸在80℃冷凝回流6h,抽滤,洗至中性,在65℃下干燥。即得到Pt载量为16wt%贵金属@石墨层核壳结构电催化剂。
如图5,本发明实施例4制备得到的产物和商业Pt/C(20wt%)在氮气饱和的0.1mol/L HClO4电解液中的循环伏安曲线,扫描速率为50mV/s,参比电极为饱和甘汞电极,Pt网为对电极。
如图6,本发明实施例4制备得到的产物和商业Pt/C(20wt%)在氧气饱和的0.1mol/L HClO4电解液中的氧还原曲线,扫描速率为10mV/s,转速为1600rpm,参比电极为饱和甘汞电极,Pt网为对电极。由于该样品的Pt载量为16wt%,与商业Pt/C中Pt的载量为20%,可以看出,在酸性条件下,所制备的样品的比质量活性要优于商业Pt/C。
实施例5:
将十六烷基三甲基溴化铵145.8mg溶于10mL氯仿,将氯亚铂酸钾水溶液(10mL,20mmol/L)加入到上述氯仿溶液中,在25℃下磁力搅拌1h;然后取出10mL有机相并加入156.1mg炭黑,超声分散均匀,加入90mL去离子水,再加入硼氢化钠(10mL,300mmol/L)水溶液,在室温下搅拌5min,再在80℃下冷凝回流1h,抽滤,获得沉淀物,烘干,得到黑色沉淀;将13.1mg meso-四(4-甲氧基苯基)卟啉铁加入35mL氯仿中,超声分散均匀后再加入112.8mg前述黑色沉淀,超声分散0.5h,在40℃水浴下进行旋转蒸发,转速为100rpm。将药品转移到石英舟中,在氩气气氛下进行热处理,在800℃下热处理2h,随炉自然冷却至室温。用0.5mol/L硫酸在80℃冷凝回流6h,抽滤,洗至中性,在65℃下干燥。即得到Pt载量为18wt%贵金属@石墨层核壳结构电催化剂。
如图7,本发明实施例5制备得到的产物和商业Pt/C(20wt%)在氮气饱和的0.1mol/L HClO4电解液中的循环伏安曲线,扫描速率为50mV/s,参比电极为饱和甘汞电极,Pt网为对电极。
如图8,本发明实施例5制备得到的产物和商业Pt/C(20wt%)在氧气饱和的0.1mol/L HClO4电解液中的氧还原曲线,扫描速率为10mV/s,转速为1600rpm,参比电极为饱和甘汞电极,Pt网为对电极。由于该样品的Pt载量为18wt%,与商业Pt/C中Pt的载量为20%,可以看出,在酸性条件下,所制备的样品的比质量活性要优于商业Pt/C。
实施例6:
将硬脂酸钾161.3mg溶于10mL四氯化碳,将三氯化铑水溶液(5mL,20mmol/L)和氯亚钯酸钾水溶液(5mL,20mmol/L)依次加入到上述四氯化碳溶液中,在30℃下磁力搅拌0.8h;然后取出10mL有机相并加入120.6mg炭黑,超声分散均匀,加入20mL去离子水,再加入硼氢化钠(10mL,5000mmol/L)水溶液,在室温下搅拌10min,再在170℃下冷凝回流1.5h,抽滤,获得沉淀物,烘干,得到黑色沉淀;将50mg meso-四(4-甲氧基苯基)卟啉铜加入35mL氯仿中,超声分散均匀后再加入50mg前述黑色沉淀,超声分散0.5h,在40℃水浴下进行旋转蒸发,转速为100rpm。将药品转移到石英舟中,在氩气气氛下进行热处理,在200℃下热处理12h,随炉自然冷却至室温。用0.5mol/L硫酸在100℃冷凝回流2h,抽滤,洗至中性,在65℃下干燥。
实施例7:
将油胺107.0mg溶于10mL氯仿,将氯亚铂酸水溶液(10mL,20mmol/L)加入到上述氯仿溶液中,在45℃下磁力搅拌0.5h;然后取出10mL有机相并加入156.1mg石墨烯,超声分散均匀,加入150mL去离子水,再加入硼氢化钠(10mL,3000mmol/L)水溶液,在室温下搅拌20min,再在150℃下冷凝回流2h,抽滤,获得沉淀物,烘干,得到黑色沉淀;将50mg meso-四(4-甲氧基苯基)卟啉镍加入35mL氯仿中,超声分散均匀后再加入50mg前述黑色沉淀,超声分散0.5h,在40℃水浴下进行旋转蒸发,转速为100rpm。将药品转移到石英舟中,在氩气气氛下进行热处理,在400℃下热处理10h,随炉自然冷却至室温。用8mol/L硫酸在100℃冷凝回流5h,抽滤,洗至中性,在65℃下干燥。
实施例8:
将十四烷基二甲基磺乙基甜菜碱300mg溶于10mL氯仿,将氯亚铂酸水溶液(10mL,20mmol/L)加入到上述氯仿溶液中,在40℃下磁力搅拌1.2h;然后取出10mL有机相并加入156.1mg碳纤维,超声分散均匀,加入200mL去离子水,再加入乙二酸(10mL,1000mmol/L)水溶液,在室温下搅拌30min,再在130℃下冷凝回流2.5h,抽滤,获得沉淀物,烘干,得到黑色沉淀;将50mg meso-四(4-甲氧基苯基)卟啉锌加入35mL丙酮中,超声分散均匀后再加入50mg前述黑色沉淀,超声分散0.5h,在30℃水浴下进行旋转蒸发,转速为150rpm。将药品转移到石英舟中,在氩气气氛下进行热处理,在600℃下热处理8h,随炉自然冷却至室温。用6mol/L苯磺酸在90℃下冷凝回流10h,抽滤,洗至中性,在65℃下干燥。
实施例9:
将月桂酸失水山梨醇酯145.8mg溶于10mL环己酮,将氯亚铂酸水溶液(10mL,20mmol/L)加入到上述氯仿溶液中,在50℃下磁力搅拌1.5h;然后取出10mL有机相并加入156.1mg碳纳米管,超声分散均匀,加入245mL去离子水,再加入葡萄糖(10mL,4000mmol/L)水溶液,在室温下搅拌40min,再在110℃下冷凝回流3h,抽滤,获得沉淀物,烘干,得到黑色沉淀;将50mg meso-四(4-甲氧基苯基)卟啉钴加入35mL乙醚中,超声分散均匀后再加入50mg前述黑色沉淀,超声分散0.5h,在40℃水浴下进行旋转蒸发,转速为100rpm。将药品转移到石英舟中,在氦气气氛下进行热处理,在1000℃下热处理0.5h,随炉自然冷却至室温。用4mol/L盐酸在70℃冷凝回流15h,抽滤,洗至中性,在65℃下干燥。
实施例10:
将油酸二乙醇酰胺500mg溶于10mL环己烷,将氯铱酸水溶液(10mL,20mmol/L)加入到上述氯仿溶液中,在20℃下磁力搅拌1.8h;然后取出10mL有机相并加入58.8mg活性碳,超声分散均匀,加入290mL去离子水,再加入柠檬酸(10mL,2000mmol/L)水溶液,在室温下搅拌50min,再在70℃下冷凝回流1h,抽滤,获得沉淀物,烘干,得到黑色沉淀;将50mg亚铁血红素加入35mL甲苯中,超声分散均匀后再加入50mg前述黑色沉淀,超声分散0.5h,在40℃水浴下进行旋转蒸发,转速为100rpm。将药品转移到石英舟中,在氨气气氛下进行热处理,在1200℃下热处理0.5h,随炉自然冷却至室温。用2mol/L高氯酸在50℃冷凝回流20h,抽滤,洗至中性,在65℃下干燥。
实施例11:
将十六烷基三甲基溴化铵182.2mg溶于10mL甲苯,将氯亚钯酸水溶液(10mL,25mmol/L)加入到上述氯仿溶液中,在15℃下磁力搅拌2h;然后取出10mL有机相并加入156.1mg炭黑,超声分散均匀,加入90mL去离子水,再加入抗坏血酸(10mL,500mmol/L)水溶液,在室温下搅拌60min,再在50℃下冷凝回流1h,抽滤,获得沉淀物,烘干,得到黑色沉淀;将75mg氯化血红素加入35mL乙醇中,超声分散均匀后再加入50mg前述黑色沉淀,超声分散0.5h,在40℃水浴下进行旋转蒸发,转速为100rpm。将药品转移到石英舟中,在氮气气氛下进行热处理,在800℃下热处理4h,随炉自然冷却至室温。用0.1mol/L硝酸在30℃下冷凝回流24h,抽滤,洗至中性,在65℃下干燥。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种核壳结构Cu-SSZ-13分子筛催化剂及其制备和应用
- 核壳结构多级孔道式钴基费托合成催化剂及其制备方法
- 一种Pt-Au/GR-RuO<sub>2</sub>核壳结构甲醇燃料电池催化剂及应用
- 核壳结构NiCo<sub>2</sub>S<sub>4</sub> @NiCo<sub>2</sub>O<sub>4</sub>纳米针复合催化电极的制备方法
- 一种核壳型铁基金属有机骨架光Fenton催化剂及其制备与应用
- 生产头孢丙烯母核用的催化剂回收装置的制造方法
- 一种核壳状CuAu催化剂及其制备方法和用图
- 一种纳米多孔铜/铂核壳结构催化电极及其制备方法
- 一种燃料电池用核壳型金钴-硼催化剂的制作方法
- 核壳催化剂催化氧化木质素制取芳香基含氧化合物的方法
- 一种纺织纤维/石墨烯/SrTiO<sub>3</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>/Bi<sub>2</sub>O<sub>3</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>/Bi<sub>2</sub>WO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>/BiPO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>MoO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>WO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>MoO<sub>6</sub>/BiPO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>GeO<sub>5</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiOBr/BiOI复合环境催化材料的制备方法