有噪声的多核苷酸序列读段的高效聚类的制作方法
诸如脱氧核糖核酸(dna)的多核苷酸的测序产生错误。多核苷酸测序仪不能以100%准确性来读取dna分子上的核苷酸碱基的序列。然而,由于核苷酸碱基的序列不能被直接观察到,所以难以标识错误何时由多核苷酸测序仪产生。因此,dna分析的正确序列最好能够仅从由多核苷酸测序仪生成的数据来推测。对来自多核苷酸测序仪的输出的分析可以校正一些错误。有时,针对dna序列的中等水平的准确性是足够的。然而,在其他的一些情况下中,期望具有尽可能准确的dna序列。各种技术可用于减少序列数据中的错误。一些技术涉及校准或以其他方式改变多核苷酸测序仪的操作。其他技术涉及处理由多核苷酸测序仪生成的序列数据。
技术实现要素:
提供本发明内容从而以简化的形式介绍下面在具体实施方式中进一步描述的一系列概念。本发明内容不旨在标识要求保护的技术方案的关键特征或必要特征,也不旨在用于限制要求保护的技术方案的范围。
从多核苷酸测序仪接收dna链的大量读段(read)并对其进行分析。dna读段的序列之间的差异可能由于dna读段为不同dna链的表示或者由于在测序过程中的某个点处引入了错误。表示完全不同的dna链的dna读段可能具有与彼此大不相同的序列。作为相同开始dna链的所有表示的dna读段也由于错误而不同,可能具有相当相似的序列。从多核苷酸测序仪接收的大量dna读段被聚类成分组,使得每个分组应当仅包含表示相同原始dna链的那些dna读段。换句话说,一个簇内的dna读段的变化应当仅归因于错误。
分析可以确定相应读段之间的编辑距离并基于编辑距离将读段分组到簇中。编辑距离测量将一个dna读段改变为另一个所需的插入、删除以及替代的最小数目。编辑距离可以通过dna读段的其他特性来近似。在一个实现方式中,散列值用于确定dna读段的相似性,并且因此它们用作针对编辑距离的近似。存在计算散列值的多种方式。一种计算散列值的方式是生成针对dna读段的二进制签名并从二进制签名和随机数的串来创建散列。
迭代的过程可以使用散列值之间的距离和/或编辑距离来重复地分析大量dna读段。具有低于阈值距离的距离的dna读段可以被一起分组在相同簇中。已经被放置到簇中的一些dna读段可以从后续迭代中省略,由此减少后续迭代的计算开支。
附图说明
参考附图阐述具体实施方式。在附图中,附图标记的最左边的(一个或多个)数字识别该附图标记首次出现的附图。在不同的附图中对相同的附图标记的使用指示相似或相同的项。
图1示出了创建dna读段并对dna读段进行聚类的示意性表示。
图2示出了说明性计算设备的框图。
图3示出了对dna读段进行聚类的过程。
图4a和图4b示出了对dna读段进行聚类的过程。
具体实施方式
本公开提供用于对序列数据中的读段进行聚类使得针对相同原始dna链的读段被放置在相同簇中的计算上高效的技术。对读段进行聚类本身不能校正序列数据中的错误但是其却能以使错误校正更高效和/或准确的方式来组织dna读段。在美国临时专利申请no.62/329,945中描述了使用聚类的针对序列数据的错误校正的一个示例。由于由多核苷酸测序仪生成的大量数据,对于涉及dna序列的应用期望计算效率。例如,通过多核苷酸测序仪的一次运行输出的数据可以包含超过表示数百万个不同的dna链的十亿个不同的dna读段。
术语“dna链”或简单的“链”是指dna分子。如本文中所使用的,“读段”可以是指代当多核苷酸测序仪读取dna链的序列时由多核苷酸测序仪生成的一串数据的名词。由多核苷酸测序仪产生的读段常常包含错误,并且因此不能以100%准确性表示dna链的结构。然而,dna测序技术产生相同区域dna的多个读段或相同dna链的多个不同副本的多个读段。读段被称为“有噪声的读段”,因为核苷酸的相同序列的读段的汇集可能包含具有近似随机分布的错误,即“噪声”。尽管给定的读段可能无错误,但是不能够知道哪些读段无错误或者哪些错误存在,除非序列是已知的。但是在dna链的序列已知的情况下测序通常不必要。
自然出现的dna链包括四类核苷酸:腺嘌呤(a)、胞嘧啶(c)、鸟嘌呤(g)和胸腺嘧啶(t)。dna链或多核苷酸是这些核苷酸的线性序列。dna链的两端(被称为5’和3’端)在化学上是不同的。dna序列通常以在左边的5’核苷酸端开始来表示。不同链之间的交互可基于序列来预测:两个单链可以结合到彼此并且在它们互补的情况下形成双螺旋:一个链中的a与另一个链中的t,并且对于c和g类似。双螺旋中的两个链具有相反的方向性(5’端被附接到另一链的3’端),并且因此两个序列是彼此的“反向互补”。两个链不需要完全互补以结合到彼此。核糖核酸(rna)具有与dna类似的结构并且自然出现的rna包括四种核苷酸a、c、g以及尿嘧啶(u)而非t。本公开中的讨论为简洁和可读性起见提到了dna,但是rna可以代替或组合dna来使用。附加地,本公开中提出的技术可以容易地适用于dna或包含多于或少于四种不同单体的其他聚合物。例如,与a、c、g和t一起使用合成碱基的dna将具有五种不同字母的字母表。此外,除了dna或rna之外的能够以类似的方式被扩增和测序的其他聚合物也可以与本文中公开的技术一起使用。
鉴于利用字母a、c、g和t来表示dna核苷酸的惯例,由dna测序仪生成的读段是包括字母a、c、g和t的文本串。一些读段可以包括描述读段的特性的元数据,读段的特性诸如针对读段中的个体碱基响应的准确性的置信度水平。读段还可以包含表示碱基响应中的不确定性的其他字母,例如,字母n可以表示未知的碱基响应。
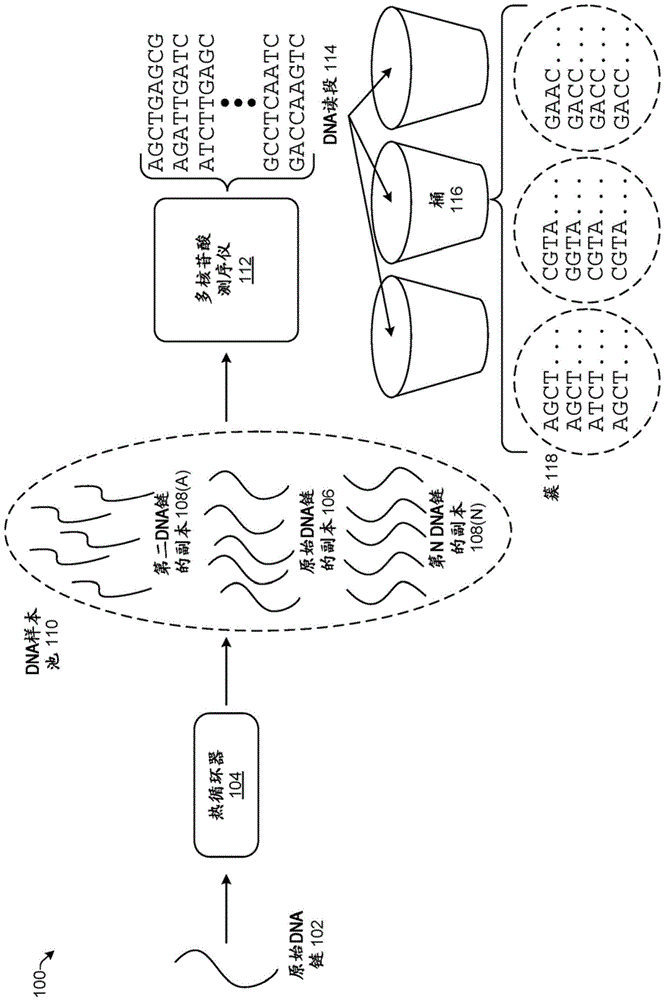
图1示出了创建dna读段114并对dna读段114进行聚类的示意性表示100。原始dna链102表示要被测序的单个分子。dna链102可以从生物样本来提取、被化学合成、或来自其他源。
原始dna链102被扩增以制作dna链的大量相同副本。用于扩增dna链的常见机器是热循环器104。尽管本文中描述了热循环器104,但是用于扩增dna链的任何其他技术可以来替代。热循环器104(也称为热循环仪、pcr机、或dna扩增器)可以利用热块来实现,热块具有保持扩增反应混合物的管可以被插入其中的孔。术语“扩增反应混合物”是指包括用于扩增目标核酸的各种试剂的水溶液。热循环器104可以然后以离散的、预编程的步骤来升高和降低块的温度。另一实现方式是扩增反应混合物经由通过微流控芯片上的热区和冷区的通道移动的小型化热循环器。包括各种技术修改的热循环器的行为和使用对于本领域普通技术人员而言是众所周知的。热循环器104用于通过聚合酶链式反应(pcr)来扩增原始dna链102。pcr是用于扩增所选择的dna序列的浓度的方法。通常是指目标核酸的副本数目的“指数”增加的术语“扩增”在本文中用于描述核酸的所选择的目标序列的数目的线性增加和指数增加两者。
pcr反应具有三个主要组成:模板、测序引物、以及酶。模板是包含将被扩增的(子)序列的单链或双链分子。dna测序引物是限定要被扩增的区域的开始和结束的短合成链。酶包括聚合酶和耐热性聚合酶,诸如dna聚合酶、rna聚合酶和反转录酶。酶从单链模板通过从引物边界开始到该模板通过核苷三磷酸的添加逐个地“填充”互补核苷酸来创建双链dna。pcr“循环”发生,其中的每一个将溶液中的模板的数目加倍。过程可以被重复直到创建了期望数目的副本。
各种pcr技术是已知的并且可以被使用在本文中描述的试验中。pcr技术通常用于扩增多核苷酸的至少部分。要被扩增的样本接触:第一和第二寡核苷酸引物;核酸聚合酶;以及与要在pcr期间添加的核苷酸相对应的核苷三磷酸。自然碱基核苷三磷酸包括datp、dctp、dgtp、dttp和dutp。非标准碱基的核苷三磷酸也可以在期望或需要的情况下被添加。用于pcr的合适的聚合酶是已知的并且包括例如耐热性聚合酶,诸如栖热菌属物种的天然的和更改的聚合酶,包括但不限于栖热水生菌(taq)、黄栖热菌(tfl)、以及嗜热菌(tth)以及dna聚合酶i和hiv-1聚合酶的克列诺(klenow)片段。
附加类型的pcr是微滴式数字tmpcr(ddpcrtm)(加拿大赫拉克勒斯的bio-rad实验室)。ddpcr技术使用微流体和表面活性剂化学的组合来将pcr样本分成油包水液滴。液滴支持目标模板分子的pcr扩增,它们包含并使用类似于用于大多数标准的基于taqman探针的试验的那些的试剂和工作流。在pcr之后,每个液滴在流式细胞分析仪中被分析或读取,以确定原始样本中的pcr阳性液滴的分数。这些数据然后使用泊松统计来分析以确定原始样本中的目标浓度。参见bio-rad液滴数字tm(ddpcrtm)pcr技术。
尽管ddpcrtm是一种pcr方法,但是也可以使用基于相同潜在原理的其他样本划分pcr方法。样本的划分的核酸可以通过可以在样本划分数字pcr内实践的任何合适的pcr方法来扩增。说明性pcr类型包括等位基因特异性pcr、组装pcr、非对称pcr、端点pcr、热启动pcr、原位pcr、序列间特异性pcr、逆pcr、指数后线性pcr、连接介导pcr、甲基化特异性pcr、微型引物pcr、多重连接依赖性探针扩增、多重pcr、嵌套pcr、重叠延伸pcr、聚合酶循环组装、定性pcr、定量pcr、实时pcr、单细胞pcr、固相pcr、热不对称交错pcr、降落式pcr、通用快速行走pcr等。也可以使用连接酶链反应(lcr)。
通过热循环器104进行的扩增创建原始dna链的许多副本106同时还保留原始dna链102本身。可以产生任何数目的副本。副本的数目部分地基于扩增的循环的数目。在一些实现方式中,大约10–20个副本可以被创建。原始dna链的所有副本106具有与原始dna链102相同的核苷酸序列,除了通过pcr扩增过程引入的任何错误。pcr扩增可以导致由于通过dna聚合酶对核苷酸的错误并入的测序错误。pcr是涉及许多(常常20-30)轮反应以合成dna的新副本的技术。在pcr期间出现的错误可以在任何轮的dna合成反应期间出现,因此在聚合酶在早前轮的合成期间错误并入碱基的情况下,pcr错误可以导致具有给定错误的大量dna片段,或者在聚合酶在稍后轮的合成期间错误并入碱基的情况下,可以导致具有错误的少量dna片段。各种pcr技术已经被报告为具有从2.4×10-6(1:416,667)到5.6×10-5(1:17,857)的范围的错误率。paulmcinerney等人的errorratecomparisonduringpolymerasechainreactionbydnapolymerase(molecularbiologyint’l.,文章id287430(2014))。因此,平均来说在十亿个dna链的集合中将存在至多约55,000个具有通过pcr引入的错误的链。这种错误水平比通过测序引入的错误低得多。pcr引发的错误总体上被均匀地且随机地分布,并且因此不会引入将改变本文中描述的聚类的结果的偏差。
第二dna链108(a)一直到任何编号“n”的其他dna链108(n)的副本可以在测序之前与原始dna链102的副本进行组合。在一些实现方式中,存在约一百万个不同的dna链的多个副本。多个不同的dna链的多个副本的组合在本文中被称为dna样本池110。dna样本池110因此包含许多不同dna链的多个大体相同的副本。原始dna链中的每一个可以与彼此大不相同或有一点相似。原始dna链之间的差异取决于dna的源的序列。假设例如一百万个不同的原始dna链,各自通过扩增创建20个副本,并且没有由于扩增的错误,则dna样本池110将包含具有每个原始dna链的20个副本的2千万个dna链。dna样本池110中的dna链可以被混合并且无差别。
dna样本池110被提供到多核苷酸测序仪112以确定存在于dna样本池110内的dna链中的核苷酸的序列。多核苷酸测序仪112读取给定dna分子中的dna碱基的顺序。dna测序包括用于确定dna的链中的四种碱基a、g、c和t的顺序的任何方法或技术。多核苷酸测序仪112使用各种技术来解读分子信息,并且可以以系统方式和随机方式两者将错误引入到读取的数据中。下文描述说明性测序技术。来自测序的错误以几个百分数到几十个百分数的比率被引入。这比来自pcr的错误大几个数量级。错误可以包括例如对标记分子的错误并入,诸如用于通过合成进行测序的荧光标记或用于通过电子显微镜进行测序的金属标记。附加地,如果在合成期间收集的输出数据具有较差质量,则核苷酸碱基可以被错误地标识。例如,在通过合成进行测序期间,荧光成像期间的发射峰可以是低分辨率,或者在离子敏感场效应晶体管(isfet)测序期间,氢发射峰可以被较差地分辨。
错误可以通常被分类为:替代错误,其中实际核苷酸被错误检测为另一核苷酸(例如a而不是g);插入,其中核苷酸当不存在时被检测到(例如agt被读取为agct);或者删除,其中从读段中省略核苷酸(例如,agta被读取为ata)。读段中的每个位置是通过多核苷酸测序仪112基于由多核苷酸测序仪112的部件感测到的性质来确定的个体碱基响应。由多核苷酸测序仪112感测到的性质取决于使用的具体测序技术而变化。碱基响应表示对dna(或rna)的链中的四种核苷酸碱基(a、g、c和t(或u))中的哪个存在于链中的给定位置处的确定。有时,碱基响应是错误的,并且这是通过测序引入的错误的源。
多核苷酸测序仪112提供原始序列数据输出,其在本文中被称为dna读段114(或读段),其包含部分地通过多核苷酸测序仪112引入的噪声。dna读段114可以具有近似相同长度(例如,全部接近于100个核苷酸或全部在95个核苷酸与105个核苷酸之间)。返回早前示例,如果2千万dna链表示一百万个不同的dna链的20个副本,则多核苷酸测序仪112将对2千万个不同的分子进行测序,并且输出将是由a、g、c和t构成的2千万个文本串(即,读段)。因此,为了对2千万个读段进行聚类,各自包含来自相同原始dna链102的20个读段的一百万个簇必须被创建。该问题由于庞大数据量而在计算上具有挑战。
该问题甚至变得更具挑战,因为原本相同的读段具有由于通过多核苷酸测序仪112和扩增而引入的错误的不同序列。然而,如以上所描述的通过多核苷酸测序仪112读取dna样本池110而生成的数据具有独特特性。对该特性的识别和合适使用使计算上高效的聚类变得可能。dna读段114包含具有比通过多核苷酸测序仪112产生的其他读段中的任何读段与彼此更加相似的序列的若干组读段。这些是来自相同原始dna链102的读段并且相对小的差异是通过测序或扩增错误而引入的那些。继续先前示例,相同原始dna链的每组20个副本具有比2千万dna读段的剩余部分中的任何与彼此更高的相似性。当然,总是存在两个不同的原始dna链可能具有相似序列并且通过测序引入的错误可能导致dna读段似乎与对应于不同原始dna链的其他读段更相似的可能性。然而,总的来说并且平均来说,对应于相同原始dna链102的dna读段114将比其他读段中的任何与彼此更加相似。
简言之,dna读段114首先被分离成被称为桶(bucket)116的许多相对大的分组。当dna读段114共享特性时,dna读段114被放置到相同桶116中。可以用于分组到桶116中的一些特性包括dna读段114的前缀、地址或散列。每个桶116内的dna读段114被进一步分组到簇118中。分组到相同簇118中基于第一读段与第二读段的序列之间的相似性或“距离”。可以用于聚类的说明性类型的距离包括汉明距离和编辑距离。下面更详细地描述用于分组到桶116中和簇118中的技术。
将dna读段114分组到桶116中和簇118中被迭代地执行。每次迭代将更多的dna读段114分组到簇118中并且重新生成桶116。在一个实现方式中,针对每个簇118的一个dna读段114被指定为代表性读段,并且其是在后续迭代中被分析的来自簇118的唯一读段。随着簇118大小增长,评估的不同dna读段114的数目减小。这在计算上比在每次迭代时评估每个dna读段114代价更低。迭代分析继续直到所有读段被充分地簇。读段可以基于预定数目的迭代被执行(例如,300、400、500等)而被认为被充分地聚类。备选地,迭代可以继续直到簇的特性满足停止准则。例如,合适的停止准则包括具有包括于簇中的至少最小数目的dna读段114的最小簇。
图2示出了可以用于形成图1中介绍的桶116和/或簇118的框图200或说明性计算设备202。计算设备202可以用一个或多个处理单元204和存储器206来实现,一个或多个处理单元204和存储器206两者可以跨一个或多个物理或逻辑位置分布。(一个或多个)处理单元204可以包括中央处理单元(cpu)、图形处理单元(gpu)、单核处理器、多核处理器、处理器集群、专用集成电路(asic)、诸如现场可编程门阵列(fpga)的可编程电路等的任何组合。在一个实现方式中,(一个或多个)处理单元204中的一个或多个可以使用单指令多数据(simd)或单程序多数据(spmd)并行架构。例如,(一个或多个)处理单元204可以包括实现simd或spmd的一个或多个gpu或cpu。(一个或多个)处理单元204中的一个或多个可以以除了硬件实现方式之外的软件和/或固件来实现。(一个或多个)处理单元204的软件或固件实现方式可以包括以任何合适的编程语言编写以执行所描述的各种功能的计算机或机器可执行指令。(一个或多个)处理单元204的软件实现方式可以整体或部分地被存储在存储器206中。
备选地或附加地,计算设备202的功能可以至少部分地由一个或多个硬件逻辑部件来执行。例如但非限制,可以被使用的说明性类型的硬件逻辑部件包括现场可编程门阵列(fpga)、专用集成电路(asic)、专用标准产品(assp)、片上系统(soc)、复杂可编程逻辑器件(cpld)等。
计算设备202的存储器206可以包括可移除存储设备、不可移除存储设备、本地存储设备和/或远程存储设备,以提供对计算机可读指令、数据结构、程序模块、以及其他数据的存储。存储器206可以被实现为计算机可读介质。计算机可读介质包括至少两种类型的介质:计算机可读存储介质和通信介质。计算机可读存储介质包括以用于存储诸如计算机可读指令、数据结构、程序模块、或其他数据的信息的任何方法或技术实现的易失性和非易失性、可移除和不可移除介质。计算机可读存储介质包括但不限于ram、rom、eeprom、闪存或其他存储器技术、cd-rom、数字多用盘(dvd)或其他光学存储设备、磁带盒、磁带、磁盘存储设备或其他磁性存储设备、或可以用于存储用于由计算设备访问的信息的任何其他非传输介质。
与之相比,通信介质可以在经调制的数据信号(诸如载波或其他传输机制)中实施计算机可读指令、数据结构、程序模块、或其他数据。如本文中所限定的,计算机可读存储介质和通信介质是相互排斥的。
计算设备202可以包括一个或多个输入/输出设备208,诸如键盘、指向设备、触摸屏、麦克风、相机、显示器、扬声器、打印机等。物理上远离(一个或多个)处理单元204和存储器206的输入/输出设备208(例如,瘦客户端的监视器和键盘)也被包括在输入/输出设备208的范围内。
网络接口210还可以被包括在计算设备202中。网络接口210是计算设备202与网络212之间的相互连接点。网络接口210可以以硬件来实现,例如被实现为网络接口卡(nic)、网络适配器、lan适配器或物理网络接口。网络接口210可以部分地以软件来实现。网络接口210可以被实施为扩展卡或母板的部分。网络接口210实现电子电路以使用特定物理层和数据链路层标准(诸如以太网、无限带宽或wi-fi)进行通信。网络接口210可以支持有线和/或无线通信。网络接口210提供针对全网络协议栈的基础,从而允许相同局域网(lan)上的计算机组之间的通信以及通过诸如互联网协议(ip)的可路由协议的大规模网络通信。
网络212可以被实现为任何类型的通信网络,诸如局域网、广域网、网状网络、adhoc网络、对等网络、互联网、线缆网络、电话网络等。
设备接口214可以是提供硬件以建立与诸如多核苷酸测序仪112、寡核苷酸合成仪等的其他设备的通信连接的计算设备202的一部分。设备接口214还可以包括支持硬件的软件。设备接口214可以被实现为不跨网络的有线或无线连接。有线连接可以包括将计算设备202物理连接到另一设备的一个或多个线或线缆。有线连接可以由耳机线缆、电话线缆、scsi线缆、usb线缆、以太网线缆、火线等来创建。无线连接可以由无线电波(例如,任何版本的蓝牙、ant、wi-fiieee802.11等等)、红外光等来创建。例如,dna读段114可以由计算设备202经由设备接口214接收。在一些实现方式中,例如如果多核苷酸测序仪112被定位为远离计算设备202,则dna读段114可以经由网络212和网络接口210被传输到计算设备202。
计算设备202包括可以被实现为存储于存储器206中以用于由(一个或多个)处理单元204执行的指令和/或整体或部分地由一个或多个硬件逻辑部件或固件来实现的多个模块。
预处理模块216执行对从多核苷酸测序仪112接收的dna读段114的预处理。预处理可以被应用到从多核苷酸测序仪112接收的多个读段中的每个读段。然而,在一些实现方式中,预处理可以仅被应用到dna读段114的所选择的子集,其可以包括从多核苷酸测序仪112接收的多个读段中的全部或少于全部。在这样的情况中,预处理将被应用到所选择的子集中的所有读段但是不一定被应用到从多核苷酸测序仪112接收的dna读段114中的全部。在聚类的上下文中对“所有”或“每一个”读段的引用是指包括于所选择的子集中的dna读段114并且不一定是指从多核苷酸测序仪112接收的全体读段。
预处理模块216可以创建包含单独的簇中的dna读段114中的每一个的数据结构。数据结构可以是并查数据结构(也称为不相交集合数据结构或合并-查找集)。并查数据结构是保持对划分成多个不相交(非交叠)子集的元素的集合的跟踪的数据结构。其支持两种有用的操作:“查找”和“并集”。查找确定特定元素处于哪个子集中。查找通常返回来自子集的用作子集的“代表”的项。通过比较两个查找操作的结果,两个元素在相同子集中的存在可以被确定。簇118可以被认为是dna读段114的子集。并集将两个子集联合成单个子集。将两个簇合并成单个簇是并集操作。最初,将每个dna读段114本身放置在簇118中的数据结构被创建。每个dna读段114被标记为针对它被包含在其内的簇118的代表性读段。当每个簇118中仅存在一个读段时,仅存在针对代表性读段的一个选择。
签名模块218(其可以是预处理模块216的一部分)计算针对每个dna读段114的签名。签名是读段的二进制表示(即,0和1)。签名不会独特地标识读段,但是提供计算上快速的方式来标识两个读段是相似还是不同。对于读段x,签名被表示为s(x)。读段x被拆分成m个子读段。例如,如果读段是100个核苷酸(nt)长并且m=5,则x被拆分成五个子读段x1、x2、x3、x4和x5。子读段可以是相等长度(例如,20nt)或不同长度的。接下来,针对子读段查找所有k元组(k-gram)。例如,如果子读段xi=“ctagcagca”并且k=3(k可以是任何整数),则长度为3的所有独特子串的集合包括{cta,tag,agc,gca}。重复的子串仅被计数一次。在该示例中,子串agc和gca仅被计数一次,即使每个出现两次。将独特串与包括长度为k的所有可能子串的比较串进行比较。如果在dna“字母表”中存在四个字母,则可能子串的数目是4k,其对于k=3是64。子读段的长度应当小于4k。如果子读段的长度更长,则子读段包含长度为k的所有或几乎所有可能子串的概率增大。因此,如果几乎所有子读段的签名将为1’s的串并且将难以使用签名来区分子读段。继续先前示例,比较串包括:{aaa,aag,aac,aat,aga,agg,agc,agt,aca,acg,acc,act,ata,atg,atc,att,gaa,gag,gac,gat,gga,ggc,ggg,ggt,gca,gcg,gcc,gct,gta,gtg,gtc,gtt,caa,cag,cac,cat,cga,cgg,cgc,cgt,cca,ccg,ccc,cct,cta,ctg,ctc,ctt,taa,tag,tac,tat,tga,tgg,tgc,tgt,tca,tcg,tcc,tct,tta,ttg,ttc,ttt}。比较串中的作为与子串之一的匹配的每个位置由1表示并且比较串中的不具有匹配的每个位置由0表示(备选地,0可以用于指示匹配)。因此,在该示例中,xi的二进制表示是长度为64的串,其具有对应于在比较串中发现cta、tag、agc和gca的位置的四个1并且剩余60个位置具有0。xi的二进制表示中的1和0的顺序当然取决于比较串中的子串的顺序。在该示例中,xi的二进制表示是{0,0,0,0,0,0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1,0,0,0,0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0}。这将先前k元组的编码提供为位串。
针对读段x的完整签名s(x)是子读段的位串的级联。因此,如果子读段的数目(m)为5并且k元组的长度为3,则每个位串的长度为64,并且签名的总长度(l)为320。因此,l=m4k。
对于以这种方式计算的签名,读段之间的编辑距离(下面描述的)在并且仅在读段的签名之间的汉明距离小(例如,小于阈值t′h)的情况下才小(例如,小于阈值t)。阈值t和t′h两者可以在数学上被估计以使作为相同原始dna链的有噪声副本的两个读段结束于相同桶中的概率最大化同时保持每个桶的期望大小较小。所估计的阈值可以使用测试数据集来实验地调整。签名是将编辑距离空间确定性嵌入到汉明空间中的一个示例。相等长度的两个串之间的汉明距离是在其处的对应符号不同的位置的数目。汉明距离测量用以将一个串改变成另一个所需要的替代的最小数目。对于二进制串a和b,汉明距离等于a异或b中的一的数目(群体计数)。签名汉明距离通常指示编辑距离,但是例外在数学上是可能的。直观上这种关系可以被认识到,因为读段的序列中的任何单个变化仅影响读段的签名中的几个k元组。
聚类模块220对已经被预处理的dna读段114进行聚类。聚类基于多个读段的子集从相同原始dna链102得到的可能性。尽管在dna读段114中的许多中可能存在错误,但是相同原始dna链102的副本的读段通常比它们是来自不同dna链的读段与彼此更相似。如果读段的簇包括源自于不同dna链的读段,则对dna序列数据的进一步分析(未包括在本公开中)可能受到妨碍,诸如对通过多核苷酸测序引入的错误的校正可能受到妨碍。因此,聚类试图将仅由于测序(或pcr)中的错误而不同的那些读段分组并分离来自不同dna链的那些。
较差地形成的簇可以由于过包含或欠包含而被“较差地”形成。过包含的较差地形成的簇是将多于一个链的读段分组在单个簇中的簇。欠包含的较差地形成的簇包括应当被分组到单个大簇中但是代之被划分成多个更小簇的读段。聚类模块220基于dna读段114之间的编辑距离来创建簇。
编辑距离模块222计算读段的对之间的编辑距离。“编辑距离”等于用以将一个dna读段转变为另一dna读段所需要的插入、删除和替代的最小数目。例如,第一dna读段x具有序列acgttac,并且第二dna读段y具有序列cgtctag。读段x与读段y之间的编辑距离ed(x,y)是3。下面是示出是三个步骤的说明性转换。
acgttac→cgttac(删除第一个a)
cgttac→cgtatac(在t之间添加a)
cgtatac→cgtatag(用g替代最后一个c)
长度为l的两个读段之间的编辑距离可以使用标准动态编程算法(诸如wagner-fischer算法)在时间o(l2)内被计算。动态编程是指通过以递归方式将复杂问题分解成更简单的子问题来简化复杂问题。给定dna读段114的源,任何读段对之间的编辑距离将在读段对是相同原始dna链102的两个读段的情况下相对小(例如,小于阈值距离t),或者在读段对是不同dna链的读段的情况下相对大(例如,大于阈值距离t)。因此,编辑距离小于t的所有dna读段114可以被一起放置在相同簇118中。
在一个实现方式中,在所有dna读段114之间成对地计算编辑距离并且具有小于t的距离的读段的那些分组被一起分组在簇118中。然而,该方法在计算上代价高,因为要比较的dna读段对的数目随着要被分析的dna读段114的数目的平方而增长。例如,如果存在十亿个读段要分析,则1,000,000,0002个编辑距离必须被计算并且这种计算量可能是不实际的或者在计算时间方面代价过高。然而,本公开中提出的其他技术可以用于以相对较少的计算开支来对读段进行聚类。
散列模块224计算针对每个代表性读段的散列。回想在聚类的开始,dna读段114中的每一个是代表性读段并且每个dna读段114本身在簇中。在一个实现方式中,第一次迭代使用基于dna读段114的前缀(例如,头1-8个核苷酸)的散列码。散列码可以将核苷酸中的每一个解读为四碱基系统中的不同数字并且然后将核苷酸串转换成数字串。在该实现方式中,如下面所描述的那样计算针对稍后迭代的散列码。在一个实现方式中,散列码如下面所描述的那样在所有迭代中被计算。可以被使用的一种类型的散列是随机化的局部敏感散列(lsh)。散列可以仅针对代表性读段被计算,因此在后续迭代中,散列针对少于全部的读段被计算。
在一个实现方式中,散列以生成签名的长度(l)的数字的随机排列(r)开始。随机排列是从1到l的整数的列表,其按随机顺序并且包括每个整数仅一次。因此,r=(r1,…,rl)从1到l。
散列是对来自r的数字的选择。回想s是0同时包括一些1的串。r和s是相同长度。在下面的示例中,s和r两者都具有9个位置。算法从r1到rl逐个地考虑来自r的数字:对于每个i=1...l,检查是否
s=(0,0,1,0,0,1,0,1,0)
r=(4,7,9,1,8,5,3,2,6)
则r中的对应位置处于s=1的位置是3、6和8。按数字顺序(从左到右:4,7,9,1,8,5,3,2,6)检查r中的每个整数以确定s中的对应位置具有0还是1。如果是1,则s中的对应位置被添加到散列。在以上示例中,r中的位置5中的数字8是对应于s中的1的第一个数字(即,s8=1)。因此,散列中的第一个值是8——r中具有与s中的1相对应的第一个数字的位置。r中的对应于s中的1的下一个最低数字是在位置7处的3。当限制于头2个数字时,hr(s)=(8,3)。给定特定r,具有相同签名的读段将具有相同散列。回想签名的定义,具有不同碱基序列的读段可以具有相同签名。q的减小增加共享相同散列码的读段的数目。在以上示例中,如果q=2,则具有hr(s)=(9,5)的任何读段将共享相同散列,但是如果q=3,则仅具有hr(s)=(9,5,2)的读段将共享相同散列。在一些实现方式中,q可以在稍后迭代中增大,由此增大稍后迭代中的散列的特异性。
在r的随机选择上针对两个签名s’和s”的hr(s’)=hr(s”)的概率近似等于j(s’,s”)q,其中j(s’,s”)是如下定义的针对s’和s”的杰卡德(jaccard)相似系数:
并且如果s’和s”被认为是{1,…,l}的子集,则:
如果s1=s2则j(s’,s”)=1。如果s1与s2之间的汉明距离小,则j(s’,s”)接近于1。如果s1与s2之间的汉明距离大,则j(s’,s”)<<1。如果s1和s2不相交,则j(s’,s”)=0。因此,对于任何任意阈值t,两个读段x和y具有相同散列码的概率对于在至多t的编辑距离处的x和y比在大于t的编辑距离处的x和y大得多。因此,散列码的这种设计以所有dna读段114中仅相对少的读段共享相同签名并且具有相同散列的两个读段可能具有至多t的编辑距离的方式来划分由签名占据的汉明空间。
在一个实现方式中,针对读段x的散列可以在不使用签名的情况下被计算并且这种类型的散列被表示为hr(x)。这种散列是遵循“锚定”串的q个核苷酸的串。“锚定”串是长度为p的随机串r。多个随机的“锚定”串可以被独立地生成,使得存在一系列“锚定”串r1,…,rk(例如,k=5)。散列然后被设置为与锚定串r在读段x中的第一次出现相邻(即,之前或之后)的q个核苷酸的串。如果第一个锚定串r1未在读段中找到,则检查第二个锚定串r2和每个后续锚定串被检查直到找到匹配。例如,如果p=3并且如果长度为三的核苷酸序列的随机选择生成r1=“acg”且x=“acgtacgac”,则r1在x中的第一次出现是头三个核苷酸。接下来的q个核苷酸(如果q=6的话)是“tacgac”。因此,在该示例中,hr(x)是tacgac。
在每次迭代的开始,r=r1,…,rk可以被随机地生成。然后,使用相同r针对所有读段计算hr(x)。由桶模块226(下面)在前一迭代中创建的所有桶116基于来自用于将读段分组到给定桶116中的先前顺序的散列hr(x)以词典式顺序来排序。然后,针对当前迭代的新“锚定”串(其将可能具有由于随机生成的不同值)用于将给定桶116中的读段与词典式顺序中相邻的桶中的读段进行比较。具有相同散列的读段之后被分组在新桶中。
桶模块226将dna读段114划分到桶116中。以上描述了用于基于散列值来生成桶的一种技术。在每次迭代中,代表性读段被分离到桶116中。代表性读段的数目随着簇形成进展(下面更详细地讨论的)而减小,因此针对每轮桶116创建考虑的项的数目逐渐地减小。在一个实现方式中,具有相同散列的所有dna读段114被放置到相同桶116中。桶大小,单个桶内的不同读段的数目,可以通过对q的选择来调谐(这对于q表示来自二进制串的1的数目的情况和对于q表示与“锚定”串相邻的核苷酸的数目的情况下两者都适用)。在一个实现方式中,桶大小可以开始相对大,并且之后随着q在后续迭代中的增大而减小(即,在每个桶中具有更少读段)。在一个实现方式中,桶模块226还可以从一个桶116中移除簇118并且将其与另一桶116中的簇118合并。在一个实现方式中,dna读段114基于前缀或地址被分组到相同桶116中。前缀或地址是在dna读段114非常相似的情况下可能相同的dna读段114的部分。在一个实现方式中,前缀或地址可以被分析为确定性散列函数。因此,具有相同前缀或相同地址的dna读段114可以被放置在相同桶116中。
划分模块228基于两个dna读段114之间的编辑距离小于阈值距离将处于相同桶116中的dna读段114分配给相同簇118。两个dna读段114之间的编辑距离可以基于两个dna读段114的签名之间的汉明距离来估计。出于以上描述的原因,如果两个签名之间的汉明距离小,则编辑距离也可能小。相反,如果两个签名之间的汉明距离大,则编辑距离可能大。
划分模块228可以使用dna读段x的签名sx与dna读段y的签名sy之间的汉明距离hdistance,以在汉明距离相对小时将dna读段114分组在相同桶116中。因此,hdistance=||sx-sy||1。如果hdistance小于第一阈值t′h,则dna读段x和dna读段y被合并到相同簇118中。然而,如果hdistance大于第二更大的阈值t″h,则dna读段x和dna读段y被维持在不同簇118中。划分模块228可以执行桶116中的每对dna读段114之间的成对比较。两个阈值t′h和t″h可以被实验地选择、由用户设置、或者基于从已知数据集校准的结果。
因此,在该实现方式中,许多dna读段114可以通过使用仅签名而被分组到簇118中。计算两个二进制串之间的汉明距离在计算上比计算编辑距离代价低,并且因此,使用签名代替编辑距离允许以高效的方式对许多dna读段114进行聚类。然而,对于具有t′h与t″h之间的汉明距离的一对dna读段114,划分模块228可以使用编辑距离来确定dna读段114是否被分组在相同簇118中。对于相同桶116中不能通过使用签名而被放置于簇中的dna读段114中的每一个,划分模块228可以执行编辑距离的成对比较。将针对dna读段x和dna读段y的编辑距离ed(x,y)与阈值t进行比较,并且如果ed(x,y)≤t,则dna读段x和dna读段y被放置在相同簇118中。计算dna读段114之间的编辑距离比计算签名之间的汉明距离计算代价高。通过使用签名之间的汉明距离来近似表示编辑距离,能够减少编辑距离计算的数目同时仍然以合理的准确性对dna读段114进行聚类。
说明性过程
为便于理解,本公开中讨论的过程被描绘为表示为独立框的单独操作。然而,这些单独描绘的操作不应当被理解为与它们的执行相关的必需顺序。过程被描述的顺序不旨在被理解为限制,并且任何数目的所描述的过程框可以以任何顺序被组合以实施过程、或备选的过程。此外,还能够修改或省略所提供的操作中的一个或多个。
图3示出了用于对dna读段进行聚类的过程300。过程300可以整体或部分地由图2中示出的计算设备202来实现。
在302处,将多个dna读段分离到多个桶中。分离可以部分地由桶模块226执行。dna读段可以部分地基于多个dna读段的前缀而被分离到桶中。因此,具有相同前缀的dna读段可以被放置在相同桶中。在一个实现方式中,散列可以从前缀生成,并且具有相同散列(基于前缀)的dna读段可以被放置在相同桶中。在一个实现方式中,如以上所描述的,散列可以从以数字的随机排列的dna读段的二进制签名来生成。在一个实现方式中,散列可以由“锚定”串和随后的核苷酸来生成。具有相同散列(然而生成)的dna读段可以被放置在相同桶中。因此,给定桶中的每个dna读段可以具有相同散列。过程300的全部或部分可以被迭代地重复。将dna读段放置到桶中的方法可以在不同迭代中不同。例如,散列值可以在一个迭代中基于dna读段的前缀,并且在不同迭代中基于签名和随机数的组合。用于生成散列的随机数可以在不同迭代中被更改。用于生成散列的q的值可以在迭代之间改变。
在304处,至少部分地基于编辑距离来对桶中的一个桶内的dna读段进行聚类。簇可以由聚类模块220形成。因为每个桶仅包含从多核苷酸测序仪接收的dna读段的总数的一部分,所以单个桶内的簇的创建在计算上比在没有首先标记桶的情况下创建针对所有dna读段的簇代价更低,因为使用桶限制了在每个簇分析中必须被分析的dna读段的数目。使用桶还使过程300可适用于在多核处理系统上的实现方式,其中每个处理器或核接收创建一个桶内的簇的任务。
在304内,编辑距离可以被近似或计算。在306处,如果编辑距离未被近似,则过程300前进到308,其中编辑距离由用以将第一dna读段转变成第二dna读段的插入、删除和替代的最小数目来计算。编辑距离可以由编辑距离模块222计算。直接计算编辑距离是确定编辑距离的最准确方式,但是其可能比备选的技术在计算上代价高。
如果在306处,编辑距离被近似,则过程300前进到310。在310处,编辑距离通过第一dna读段的二进制签名与第二dna读段的二进制签名之间的汉明距离来近似。二进制签名可以由签名模块218计算。在一些实现方式中,二进制签名被预先计算,使得在过程300中的该阶段处,二进制签名从存储器被检索而不是被计算。
返回304,如果编辑距离被近似,则签名之间的汉明距离可以被确定为小于第一阈值汉明距离,并且第一dna读段和第二dna读段可以然后被放置到相同簇中。在出于以上解释的原因的一些情况下,签名之间的汉明距离是针对编辑距离的合理近似。备选地,汉明距离可以被确定为大于第二阈值距离,在这种情况下,第一dna读段和第二dna读段将被放置到不同簇中。如果汉明距离大于第一阈值距离但小于第二阈值距离,则使用针对编辑距离的近似可能是对那些dna读段进行聚类的不可靠方式。因此,对于具有在该范围内的汉明距离的dna读段,编辑距离可以被计算。
因此,编辑距离可以在决定使用编辑距离而非近似的情况下被使用,或者备选地在近似可能不能够准确地对给定对的dna读段进行聚类的情况下被使用。当编辑距离被使用时,具有小于编辑距离阈值的编辑距离的dna读段可以被放置到相同簇中。具有大于编辑距离阈值的编辑距离的dna读段可以被放置到不同簇中。将dna读段分组或分离到相同或不同簇中可以由划分模块228执行。
图4a和图4b示出了用于对dna读段进行聚类的过程400。过程400可以整体或部分地通过图2中示出的计算设备202来实现。
在402处,从多核苷酸测序仪接收多个读段。多核苷酸测序仪可以是图1和图2中示出的多核苷酸测序仪112。经由设备接口214接收多个读段。当前测序技术可以从大量(即,1,000,000或更多)dna链产生非常大量的读段(即,1,000,000,000或更多)。
在404处,可以针对来自多个读段的至少第一读段计算签名。签名可以是任何类型的签名,但是在一个实现方式中签名是由第一读段内的一组k元组(例如,3元组、4元组、5元组等)生成的位串。签名可以通过将第一读段分成两个或更多个子读段来计算。例如,第一读段可以被分成相等长度的五个子读段。然后,可以针对子读段中的每个子读段查找所有k元组。k元组可以被编码为位串,使用1来指示k元组存在于子串中并且使用0来指示k元组不存在(当然1和0的使用可以被交换)。这创建针对子串中的每个子串的位串,并且这些位串可以被级联以创建针对第一读段的签名。签名可以由签名模块218创建。
在406处,基于以上描述的技术中的任何来生成针对第一读段的散列。例如,散列可以至少部分地基于签名和随机数的串。随机数的串可以被生成为包含整数的数字随机排列,其中每个整数仅被包含一次。来自随机数的串的不同随机数被分配给签名中的个体位。个体位可以基于位值来选择。例如,值1的所有位可以被选择;备选地,值0的所有位可以被选择。散列然后被设置为被分配给签名中的个体位的随机数的串的子集。散列可以包括被分配给签名中的个体位的所有数或者仅头几个,诸如头2、3、4、5、6、7、8、9、10、11、12、13、14或15个等。散列可以由散列模块224生成。作为另一示例,散列可以从“锚定”串和随后的q个核苷酸来生成。“锚定”串是长度为p的k个不同的备选随机串,其如果存在于读段中则指代散列开始的地方。散列是与“锚定”串相邻(即,在其之后或之前)的长度为q的核苷酸的串。在一个实现方式中,p=3,q=8,并且k=5。
在408处,读段可以基于散列而被分组到桶中。具有相同散列值的读段被分组到相同桶中。读段可以由桶模块226分组到桶中。
在410处,确定是否完成针对所有桶中的读段的聚类。桶中的每个桶可以继而被分析以确定是否存在应当被执行的附加聚类。在存在包含小于阈值数目的dna读段(例如,五个或更少dna读段的)的小簇的情况下,附加聚类可以被执行。聚类的迭代也可以被重复设置的次数。次数可以基于对其他序列数据的先前聚类而被实验地确定。如果不再需要针对桶中的任何桶的聚类,则过程400前进到412并且结束。然而,如果未完成针对一个或多个桶的聚类,则过程400前进到414并选择未被完全聚类的桶以进行附加聚类。
在416处,从在412处选择的桶中选择一对代表性读段。该对读段可以以任何方式来选择,诸如获取读段的列表中的头两个读段、随机地选择等。
在418处,将所选择的该对代表性读段之间的签名距离与第一签名距离阈值进行比较。在一个实现方式中,签名距离可以是汉明距离。签名距离可以例如使用以上描述的技术来设计,使得具有低于第一签名距离阈值的签名距离的任何两个读段都非常可能具有将导致两个代表性读段被放置在相同簇中的编辑距离。如果确定签名距离低于第一签名距离阈值,则过程400前进到420,并且包含代表性读段的簇被合并到一个簇中。然而,如果签名距离不低于第一签名距离阈值,则过程400前进到422。
在422处,将两个代表性读段之间的签名距离与第二签名距离阈值进行比较。高于第二签名距离阈值的签名距离指示两个代表性读段具有将两个读段放置在不同簇中的编辑距离的高概率。如果确定两个代表性读段之间的签名距离高于第二签名距离阈值,则过程400前进到424,并且包含两个代表性读段的簇被维持为单独的簇。然而,如果确定签名距离小于第二签名距离阈值(并且回想在过程400中的此时,签名距离也大于第一签名距离阈值),则过程400前进到426。
在426处,计算所选择的该对代表性读段之间的编辑距离。编辑距离由编辑距离模块222计算。因为仅针对具有高于第一签名距离阈值并且低于第二签名距离阈值的签名距离的读段对来计算编辑距离,所以没有必要计算针对从多核苷酸测序仪接收的读段的一部分的编辑距离。如果来自多核苷酸测序仪的读段是多个dna链(即,由于不同原始序列而不同)的多个副本(即,由于错误而不同)的读段,则大多数读段对将足够相似使得签名距离低于第一签名距离阈值(例如,仅由于错误而不同的读段),或者足够不同使得签名距离高于第二签名距离阈值(例如,是不同dna链的读段)。因此,在许多实现方式中,仅针对从多核苷酸测序仪接收的读段的一小部分来计算编辑距离。这产生了计算效率,因为签名距离比计算编辑距离在计算上代价更低。
在428处,将在426处计算的编辑距离与阈值进行比较。如果编辑距离高于阈值,则过程400前进到424,并且两个代表性读段被维持在单独的簇中。如果编辑距离高于阈值,则过程400前进到420,并且两个读段被合并到相同的簇中。
在420处确定两个读段以及它们所代表的簇应当被合并到一个簇中之后,过程400前进到430,并且将新形成的簇中的一个读段标记为针对该簇的代表性读段。读段的每个簇可以包括被指定为针对整个簇的代表性读段的一个读段。将多个不同的读段分组到相同簇中是那些读段的序列由于编辑距离小于阈值而相同或相似的指示。回想在初始迭代中,每个读段可以本身在簇中并且每个读段可以被指示为代表性读段。一旦多个不同读段被一起合并在相同簇中,则那些读段中的一些读段,例如除了一个以外的所有读段,在进一步分析中被忽略;仅代表性读段在评估簇时被考虑。代表性读段可以通过任何合适的技术来选择。在一个实现方式中,针对簇的代表性读段可以被随机地选择。在一个实现方式中,代表性读段可以从被合并的簇的代表性读段中选择。例如,针对新簇的代表性读段可以是来自两个合并的簇中的包含大量读段的一个簇的代表性读段。
在430处将一个读段标记为代表性或者在424处确定一对代表性读段将要被维持在单独的簇中之后,过程400前进到432。
在432处,确定当前选择的桶是否被完全聚类。如果不是,则过程400返回到416,并选择桶中的不同对的活动读段以用于分析。随着所选择的桶中的簇的数目减少,活动读段的数目也将减少,因此对要比较的活动读段对的选择将减少。最终,可以被聚类在一起的所有的活动读段对将被聚类并且然后仅彼此远离多于编辑距离阈值的活动读段将剩下。
如果在432处,确定当前选择的桶中的所有读段被完全聚类,则过程400前进到408,并基于代表性读段的散列来创建新桶。因为仅代表性读段在该分析中被使用,所以形成新桶不会打破现有的簇,但是却能提供使簇的不同组合被放置到相同桶中的机会。一旦新桶被形成,则过程400就如以上所描述的继续。在一些实现方式中,过程400的全部或部分可以被迭代100、200、300、400、500、600、700、800、或900次。
示例
以下示例说明对dna读段进行聚类的多种方式之间的准确性和计算开支的差异。在该示例中,所有结果在具有w3670@3.20ghz、6核的
六种不同的方法用于对一组人工生成的dna读段进行聚类。该组人工生成的dna读段被复制以得出每个读段的20个副本,并且然后利用5%随机错误(2%替代;1.5%删除;1.5%插入)来修改以模拟寡核苷酸测序仪错误读取的效果。四种不同数据集合大小中的原始开始读段的数目被测试,如由列“读段的数目”所指示的。数据集合中的读段的总数目是列出的数目的20倍:200,000;2,000,000;20,000,000;以及40,000,000。计算设备对读段进行聚类的以秒计的时间由“t”表示。错误百分数“e”是未恢复的簇的百分数。如果识别到包含至少r个读段的真实簇的子簇,则认为簇恢复(recovered)。第一错误值是针对恢复阈值r=10的未恢复簇的百分数,并且第二错误值是针对r=15的未恢复簇的百分数。因此,对于r=15的e=50%指示能够被形成的20个读段的簇中仅一半簇能够在至少15个读段正确读段的情况下被形成。下面表1中的空白单元格指示簇未被确定,因为处理未在合理的时间量内完成。
表1.通过编辑距离进行的聚类
“暴力(bruteforce)”方法计算每对读段之间的编辑距离(如以上所描述的)并且在编辑距离低于阈值的情况下合并读段。这种成对的比较方法要求对编辑距离的n2次计算,其中n是读段的总数目。暴力法是准确的但是缓慢的。“sgn”指示针对所有读段计算签名(如以上所描述的)并且签名之间的汉明距离当汉明距离低于第一阈值或高于第二阈值时用于聚类。暴力+sgn是准确的并且比单独暴力法快得多。
“前缀4”使用读段的四字符前缀来首先将读段分组到桶中,然后通过编辑距离的成对比较来在桶中形成簇。分组到桶中减少处理时间,但是用于制作桶的这种基础降低准确性。添加以上描述的签名方法进一步减少时间,但是不影响准确性。
“lsh”代表局部敏感散列,其是以上公开的散列技术。在该示例中,散列长度为10,q=10。“lse70”方法通过创建桶并计算簇70次的过程而迭代。“lse250”迭代250次。lsh70和lsh250两者都比“前缀4+sgn”慢。然而,准确性增大几倍。lsh250比lsh70更慢且更准确。因此,lse方法得到比前缀4方法更准确并且比暴力方法快得多的聚类。
说明性测序技术
多核苷酸测序仪112可以使用以下测序技术或除了本文具体提到的那些之外的另一技术来实现。
可以被使用的测序技术是边合成边测序(
可以被使用的测序技术的另一示例是纳米孔测序。纳米孔是直径为1纳米量级的小孔。将纳米孔浸入传导流体中和跨纳米孔施加电位导致由于离子通过纳米孔的传导引起的轻微电流。流过纳米孔的电流的量对纳米孔的大小敏感。当dna分子穿过纳米孔时,dna分子上的每个核苷酸在不同程度上阻塞纳米孔。因此,当dna分子穿过纳米孔时穿过纳米孔的电流的变化表示dna序列的读取。
可以被使用的测序技术的另一示例太平洋生物科学的单分子实时(smrttm)技术。在smrttm中,四种dna碱基中的每种被附接到四种不同荧光染料中的一种。这些染料是磷酸链接的。单个dna聚合酶利用模板单链dna的单个分子固定在零模式波导(zmw)的底部。zmw是使得能够针对快速(在微秒内)扩散入zmw中并从zmw中扩散出的荧光核苷酸的背景观察通过dna聚合酶对单个核苷酸的并入的限制结构。其花费几毫秒来将核苷酸并入增长的链中。在此期间,荧光标记被激发并且产生荧光信号,并且荧光标签被分裂掉。对染料的对应荧光的检测指示哪个碱基被并入。过程被重复。
可以被使用的另一测序技术是helicos真实单分子测序(tsms)。在tsms技术中,dna样本被分裂成大约100到200个核苷酸的链,并且polya序列被添加到每个dna链的3’端。每个链通过荧光标记的腺苷核苷酸来标记。dna链然后被混合到流细胞,其包含固定到流细胞表面的数百万个寡t捕获部位。模板可以具有约1亿个模板/cm2的密度。流细胞然后被加载到仪器(例如heliscopetm测序仪)中,并且激光照射流细胞的表面,从而披露每个模板的位置。ccd相机可以将模板的位置映射在流细胞表面上。模板荧光标记然后被分裂并被清洗掉。测序反应通过引入dna聚合酶和荧光标记的核苷酸来开始。寡t核酸用作引物。聚合酶以模板引导的方式将标记的核苷酸并入到引物。聚合酶和未并入的核苷酸被移除。通过对流细胞表面进行成像来检测已经引导对荧光标记的核苷酸的并入的模板。在成像之后,分裂步骤移除荧光标签,并且利用其他荧光标记的核苷酸重复过程直到实现期望的读取长度。序列信息利用每个核苷酸添加步骤来收集。
可以被使用的dna测序技术的另一示例是solidtm技术(应用生物系统)。在solidtm测序中,dna被剪成片段,并且接头被附接到片段的5’和3’端以生成片段文库。备选地,内部接头可以通过以下来引入:将接头捆绑到片段的5’和3’端、将片段制成圆形、消化制成圆形的片段以生成内部接头、以及将接头附接到得到的片段的5’和3’端以生成末端配对文库。接下来,克隆磁珠群体在包含磁珠、引物、模板、以及pcr成分的微型反应器中制备。在pcr之后,模板变性并且磁珠被浓缩以将磁珠与延伸的模板分离。所选择的磁珠上的模板经受允许结合到载玻片的3’修改。
可以被使用的测序技术的另一示例涉及使用化学敏感场效应晶体管(chemfet)阵列来对dna进行测序。在该技术的一个示例中,dna分子可以被放置到反应腔室中,并且模板分子可以被混合到结合到聚合酶的测序引物。在测序引物的3’端处将一个或多个三磷酸并入新核酸链中可以通过由chemfet引起的电流的变化来检测。阵列可以具有多个chemfet传感器。在另一示例中,单个核酸可以被附接到磁珠,并且核酸可以在磁珠上被扩增,并且个体磁珠可以被转移到chemfet阵列上的个体反应腔室,其中每个腔室具有chemfet传感器,并且核酸可以被测序。
可以被使用的测序技术的另一示例涉及离子敏感场效应晶体管(isfet)来对dna进行测序。iontorrenttm(离子激流)测序是该技术的一个示例。在该技术中,不需要标记分子,并且在dna合成期间检测对每个核苷酸的并入。腺嘌呤、胞嘧啶、鸟嘌呤或胸腺嘧啶连续地流过dna腔室,并且如果核苷酸变得并入到新生链中,则反应发射氢离子。氢离子发射被检测到,并且这指示哪个碱基变得被并入在给定位置处。
可以被使用的测序技术的另一示例涉及使用电子显微镜。在该技术的一个示例中,个体dna分子使用金属标记来标记,金属标记可使用电子显微镜来区分。这些分子然后在平坦表面上被拉伸并且使用电子显微镜被成像以测量序列。
用于对dna进行测序的所有技术与某种错误水平相关联,并且错误的类型和频率通过测序技术而不同。例如,边合成边测序创建碱基响应中的约2%的错误。这些错误中的大多数是替代错误。纳米孔测序具有约15%至40%的高得多的错误率,并且由这种测序技术造成的大多数错误是删除。具体测序技术的错误分布可以描述错误的总体频率以及各种类型错误的相对频率。
说明性实施例
以下条款描述了用于实现本公开中描述的特征的多个可能的实施例。本文中描述的各种实施例不是限制性的,并且来自任何给定实施例的每一个特征不需要存在于另一实施例中。实施例中的任何两个或更多个可以被组合在一起,除非上下文清楚地另行指出。如本文中所使用的,在本文档中,“或”意指和/或。例如,“a或b”意指a而没有b,b而没有a,或者a和b两者。如本文中所使用的,“包括”意指包括所有列出的特征并且可能包括未列出的其他特征的添加。“基本上由…构成”意指包括列出的特征和不会实质上影响列出的特征的基本和新颖特性的那些附加特征。“由…构成”意指仅列出的特征以排除未列出的任何特征。
条款1.一种系统,包括:
至少一个处理单元;
与处理单元通信的存储器;以及
聚类模块,被存储在存储器中并且在处理单元上可执行以至少部分地基于以下项将多个dna读段划分成簇:(i)将编辑距离空间确定性地嵌入到汉明空间中的签名,和(ii)随机化的局部敏感散列(lsh)。
条款2.根据条款1的系统,其中至少一个处理单元包括具有相同指令多数据(simd)或单程序多数据(spmd)架构的中央处理单元(cpu)。
条款3.根据条款1-2的系统,其中聚类模块包括编辑距离模块,编辑距离模块被存储在存储器中并且在处理单元上可执行以:基于将多个dna读段中的第一读段改变为多个dna读段中的第二读段的插入、删除和替代的最小数目来计算多个dna读段中的第一读段与多个dna读段中的第二读段之间的编辑距离。
条款4.根据条款1-3的系统,其中聚类模块包括散列模块,散列模块至少部分地基于以下项来确定随机化的lsh:(i)数字的随机排列并且其中签名包括二进制签名,或者(ii)与dna读段内的随机选择的串的出现相邻的核苷酸。
条款5.根据条款4的系统,其中聚类模块包括桶模块,桶模块将具有相同散列的dna读段分组到相同桶中。
条款6.根据条款5的系统,其中聚类模块包括划分模块,划分模块至少部分地基于相同桶中的两个dna读段之间在汉明空间中的差小于阈值距离来将该两个dna读段分配给相同簇。
条款7.根据条款1-6的系统,还包括签名模块,签名模块被存储在存储器中并且在处理单元上可执行以:
查找针对多个dna读段的k元组;
将k元组编码为位串;以及
将位串级联成签名。
条款8.根据条款1-7的系统,还包括设备接口,设备接口被配置为从多核苷酸测序仪接收多个dna读段。
条款9.一种方法,包括:
从多核苷酸测序仪接收多个读段;
计算针对来自多个读段的第一读段的签名,签名是部分地由第一读段内的一组k元组生成的位串;
生成针对第一读段的散列,散列至少部分地基于第一读段的序列;
将第一读段与具有相同散列的第二读段分组到相同桶中;
计算第一读段与第二读段之间的编辑距离;
确定编辑距离低于阈值;以及
将包含第一读段的第一簇与包含第二读段的第二簇合并成第三簇。
条款10.根据条款9的方法,其中多个读段包括表示多于百万个不同的dna链的多于十亿个读段。
条款11.根据条款9-10的方法,其中计算签名包括:
将第一读段划分成两个或更多个子读段;
查找针对两个或更多个子读段中的每个子读段的所有k元组;
将k元组编码为位串;以及
将位串级联成签名。
条款12.根据条款9-11的方法,其中生成散列包括:
生成随机数的串;
将来自随机数的串的不同随机数分配给签名的至少部分中的个体位;以及
将散列设置为被分配给个体位的随机数的串的子集。
条款13.根据条款12的方法,还包括基于位值来选择个体位。
条款14.根据条款9-11的方法,其中生成散列包括:
生成随机核苷酸的串;
标识随机核苷酸的串在第一读段中的出现;以及
将散列设置为与随机核苷酸的串在第一读段中的出现相邻的核苷酸的序列。
条款15.根据条款9-14的方法,其中计算编辑距离包括对将第一读段改变为第二读段的插入、删除和替代的最小数目进行计数。
条款16.根据条款9-15的方法,其中确定编辑距离低于阈值包括确定针对第一读段的签名与针对第二读段的签名之间的签名距离低于签名距离阈值。
条款17.根据条款16的方法,其中签名距离是汉明距离。
条款18.根据条款9-17的方法,还包括:
将第一读段标记为针对第三簇的代表性读段;
将代表性读段和在相同桶中的第四簇内的第三读段标识为具有低于阈值的第二编辑距离;以及
将包含代表性读段的第三簇与包括第三读段的第四簇合并。
条款19.一种方法,包括:
将多个dna读段分离到多个桶中;以及
至少部分地基于dna读段的相应对之间的编辑距离,将多个桶中的一个桶中的dna读段聚类到簇中。
条款20.根据条款19的方法,其中将多个dna读段分离到多个桶中至少部分地基于多个dna读段的前缀。
条款21.根据条款19-20的方法,其中将多个dna读段分离到多个桶中至少部分地基于多个dna读段的散列。
条款22.根据条款21的方法,其中针对多个dna读段中的一个dna读段的散列至少部分地基于多个dna读段中的该一个dna读段的二进制签名和数字的随机排列。
条款23.根据条款19-22的方法,其中编辑距离通过用以将第一dna读段转变为第二dna读段的插入、删除和替代的最小数目来计算。
条款24.根据条款19-23的方法,其中编辑距离通过多个dna读段中的第一dna读段的二进制签名与多个dna读段中的第二dna读段的二进制签名之间的汉明距离来近似。
条款25.根据条款24的方法,还包括:
确定汉明距离小于第一阈值;以及
将多个dna读段中的第一dna读段和多个dna读段中的第二dna读段放置在相同簇中。
条款26.根据条款24的方法,还包括:
确定汉明距离大于第二阈值;以及
将多个dna读段中的第一dna读段和多个dna读段中的第二dna读段放置在不同簇中。
条款27.根据条款24的方法,还包括:
确定汉明距离在第一阈值与第二阈值之间;
计算针对多个dna读段中的第一dna读段和多个dna读段中的第二dna读段的编辑距离;
确定编辑距离小于编辑距离阈值;以及
将多个dna读段中的第一dna读段和多个dna读段中的第二dna读段放置在相同簇中。
条款28.一种编码指令的计算机可读介质,指令当由处理单元执行时使计算设备执行根据条款9-27中的任一项的方法。
条款29.一种系统,包括一个或多个处理单元和存储器,系统被配置为实现根据条款9-27中的任一项的方法。
条款30.一种系统,包括:
至少一个处理单元;
与处理单元通信的存储器;
用于至少部分地基于以下项将多个dna读段划分成簇的装置:(i)将编辑距离空间确定性地嵌入到汉明空间中的签名,和(ii)随机化的局部敏感散列(lsh);
用于基于将多个dna读段中的第一读段改变为多个dna读段中的第二读段的插入、删除和替代的最小数目来计算多个dna读段中的第一读段与多个dna读段中的第二读段之间的编辑距离的装置;
用于至少部分地基于以下项来确定随机化的lsh的装置:(i)数字的随机排列并且其中签名包括二进制签名,或者(ii)与dna读段内的随机选择的串的出现相邻的核苷酸;以及
用于至少部分地基于相同桶中的两个dna读段之间在汉明空间中的差小于阈值距离来将该两个dna读段分配给相同簇的装置。
结论
尽管已经以对结构特征和/或方法动作特定的语言描述了本技术方案,但是应理解在权利要求中限定的技术方案不必限于以上描述的特定特征或动作。相反,具体特征和动作被公开为实施权利要求的示例形式。
在描述本发明的上下文中使用的术语“一”、“一个”、“该”和类似的指称要被理解为涵盖单数和复数两者,除非本文中另外指示或由上下文明确地发生矛盾。
本文中描述了某些实施例,包括对发明人已知的用于执行本发明的最好模式。当然,这些描述的实施例的变型将在本领域普通技术人员阅读前述描述后而变得明显。技术人员将知道如何合适地采用这样的变型,并且本文中公开的实施可以以除具体描述的之外的其他方式来实践。因此,本文随附的权利要求中记载的技术方案的所有修改和等价方案被包括在本公开的范围内。此外,其所有可能变型中的上述元件的任何组合由本发明涵盖,除非本文中另外指示或另外由上下文明确地发生矛盾。
- 还没有人留言评论。精彩留言会获得点赞!