放热嬗变方法与流程
本发明涉及通过嬗变、更精确地通过放射性同位素的嬗变的能量产生的领域。为了满足安全能量的需求,碳燃烧必须被另一来源代替。源自50年代军事研究的使用铀裂变作为能源具有在暴露于安全隐患的同时产生大量的放射性废料的缺点。本发明还涉及减少放射性和/或毒性的废料处理的领域。
背景技术:
过去,一些研究与以晶体结构接收的氘有关。氘较昂贵且反应难以预测。
通过在被电磁辐射照射的胶体混合物中使用Li、Ni、Cu、Pd和Ti作为核燃料,进行了其它的尝试。但是,需要减速剂。
Sergio Focardi教授在90年代晚期在Ni-H生热方面公开了几份文件。进行在具有氢气的铜管中基于Ni62的尝试。能量产生低于期望。
进行了基于过渡金属向另一材料的质子发射的尝试。但是,反应器较复杂。
需要适于工作需求的安全可靠的方法。
技术实现要素:
一种用于至少部分地去活放射性材料的放热嬗变方法,包括以下步骤:
-在封闭容器外面的反应器的腔室内布置包含至少一种过渡金属的尘状化合物;
-在所述腔室内布置放射性材料,该放射性材料处于所述封闭容器中并且在其中保持被封装;
-在高于环境压力的压力下,提供与尘状化合物和放射性材料接触的氢气;
-在腔室内产生电场,该电场被施加于尘状化合物和放射性材料;
-通过超声波使尘状化合物增强活力,从而产生所述至少一种过渡金属向另一种过渡金属的嬗变以及向放射性材料的质子发射,所述放射性材料至少部分地被去活;
-从反应器去除热能。
获得连同热量生成的放射性减少。
在随后的步骤中,可从反应器去除尘状化合物。去除的尘状化合物可被处理为非放射性材料。去除的尘状化合物可在处理中被重新使用或者分为若干份,例如,分为物种,以获得与处理开始时相同的成分。在处理期间已获得的物种的一部分可被去除,在处理期间已被消耗的物种可被补全(completed)。
现在,放射性材料被指定为“被处理的材料”。可从反应器去除被处理的材料。去除的放射性材料可被处理为非放射性材料,或者通过化学处理分成非放射性部分和放射性部分。所述放射性部分,如果有的话,可在以上的方法中被重新提交。在大多数的情况下,建议具有足够强的处理以获得非放射性被处理的材料。参照诸如IAEA标准的标准,该材料可被归类为非放射性。
在实施例中,方法包括在腔室内产生电场,该电场被施加于尘状化合物和放射性材料。
在实施例中,提供一种用于至少部分地去活放射性材料的放热嬗变方法,该方法包括以下步骤:
在反应器的腔室内布置包含至少一种过渡金属的尘状化合物;
在所述腔室内布置放射性材料,该放射性材料接近尘状化合物或者与其混合;
在高于环境压力的压力下,提供与尘状化合物和放射性材料接触的氢气;
在腔室内产生电场,该电场被施加于尘状化合物和放射性材料;
通过超声波使尘状化合物增强活力,从而产生所述至少一种过渡金属向另一种过渡金属的嬗变以及向放射性材料的质子发射,所述放射性材料至少部分地被去活;
从反应器去除热能。
在实施例中,方法包括加热尘状化合物和放射性材料。
在实施例中,放射性材料是核废料。方法允许有效的放射性减少。
在实施例中,核废料是裂变产物。方法适于长寿命的裂变产物。所述长寿命裂变产物在以前回收起来最昂贵。在实施例中,核废料是医疗/工业核废料。医疗放射源用于成像。工业放射源用于非破坏性检查。大量的医疗/工业核废料被产生并且应被回收。
在实施例中,核废料是矿业废料。矿业废料较丰富并且具有各种成分。因此,已知的处理较昂贵并且/或者不实际。在一些情况下,进行简单的填埋。在其它情况下,进行与盲区(dead ground)的混合。这些不是处理,并且使得常常不远离地表且易于浸出的土壤具有放射性。由于矿业废料一般具有各种成分,因此不容易确定适当的已知的处理。本方法很好地适于矿业废料,原因是可对各种矿业废料成分使用相同的化合物成分。如果必要的话,矿业废料在去活之前被填埋,以从中去除生物产物。
在实施例中,方法包括在初始温度下加热腔室。可用电阻进行加热。初始温度可以处于100~140℃的范围内。
在实施例中,方法包括从腔室去除空气的步骤。去除空气可在引入氢气之前发生。可用真空泵进行去除空气。否则,去除空气可在引入氢气的过程中或者通过引入氢气发生。在其它条件下,进行空气冲除(air flush)。去除空气明显增加该过程的效率。
在实施例中,尘状化合物包含Ni和Fe,Ni原子嬗变为Cu,特别是嬗变为Cu的非放射性同位素。
在实施例中,尘状化合物包含按质量50%~95%的Ni和5%~50%的Fe。已对其进行实验测试。
在实施例中,尘状化合物包含按质量70%~90%的Ni和10%~30%的Fe。
在实施例中,尘状化合物包含按质量1%~10%的Cu。已发现,Cu增强放射性减少。由于当Ni嬗变为Cu时Cu量增加,因此,同一化合物可被使用几次,直到Cu百分比变得太高。
在实施例中,尘状化合物包含按质量2%~7%的Cu。优选地,初始尘状化合物包含2~3%的Cu,并且最终的尘状化合物包含6~7%的Cu。尘状化合物当在处理中被最后一次使用时为“最后”。然后,在方法中去除它。Cu可被分离以减少Cu含量并且获得重新生成的工业尘状化合物。
在实施例中,尘状化合物的Cu有至少99%的粒子的平均尺寸为在10~100μm之间、优选在10~50μm之间。Cu的选择的颗粒尺寸缩短处理的持续期并且减少要提供的能量。
在实施例中,尘状化合物的Cu有至少99.9%的粒子的平均尺寸为在10~100μm之间,优选在10~50μm之间。
在实施例中,尘状化合物的Ni有至少99%的粒子的平均尺寸不大于10μm。
在实施例中,尘状化合物的Ni有至少99.9%的粒子的平均尺寸不大于10μm。
在实施例中,尘状化合物的Fe有至少99%的粒子的平均尺寸不大于10μm。
在实施例中,尘状化合物的Fe有至少99.9%的粒子的平均尺寸不大于10μm。
在实施例中,尘状化合物的Ni有至少99%的粒子的平均尺寸不大于5μm。
在实施例中,尘状化合物的Ni有至少99.9%的粒子的平均尺寸不大于5μm。
在实施例中,尘状化合物的Fe有至少99%的粒子的平均尺寸不大于5μm。
在实施例中,尘状化合物的Fe有至少99.9%的粒子的平均尺寸不大于5μm。
在实施例中,尘状化合物包含按质量25%~40%、优选30%~40%的石墨。石墨有99%的粒子的平均尺寸不大于10μm。
在实施例中,尘状化合物包含按质量10%~15%的Fe、80%~85%的Ni和2%~5%的Cu。
在实施例中,尘状化合物包含按质量5%~10%的Fe、57%~65%的Ni、1%~3%的Cu和25%~30%的石墨。
在实施例中,尘状化合物包含按质量10%~15%的Fe、75%~80%的Ni、1%~3%的Cu和8%~15%的Cr。
优选地,尘状化合物被均质化。
在实施例中,封闭容器实质上由钢制成,该钢优选包含按质量至少1%的Cr,更优选由不锈钢制成。
在实施例中,所述腔室内的压力大于5×105Pa,所述腔室包含至少99%的H2。
在实施例中,所述腔室内的压力为在5×105Pa~20×105Pa之间,优选为在10×105Pa~15×105Pa之间。
在实施例中,氢气在加热之前被提供,并且在随后的步骤中保持处于腔室中。在从反应器去除尘状化合物之前去除氢气。
在实施例中,初始温度为在80~200℃之间、优选在100~150℃之间。
在实施例中,尘状化合物包含Cr的主动添加。
在实施例中,尘状化合物包含按质量多达15%的Cr。
在实施例中,相同的尘状化合物成分用于各种放射性材料。作为例子,相同的尘状化合物成分用于包含Co60、U235、Cs137的废料。
在实施例中,相同的尘状化合物用于多个放射性材料去活。尘状化合物在方法完成之后不具有放射性。
在实施例中,电场本质上是静态的。
在实施例中,电场为在20~30000伏特/米之间。
在实施例中,放射性材料是有至少99%、优选99.9%的粒子的平均尺寸不大于10μm的粉末。
在实施例中,放射性材料是有至少99%、优选99.9%的粒子的平均尺寸不大于5μm的粉末。
在实施例中,尘状化合物/放射性材料的原子数比例为在3/1~6/1之间。
在实施例中,氢气丧失氘和氚的主动添加。在其它条件下,使用天然氢气。不需要氢同位素分离。
在实施例中,反应器包括包含钢、不锈钢或陶瓷中的至少一种的腔室壁。优选地,腔室壁由不锈钢制成。
在实施例中,超声波具有在250~600kHz之间的频率。
在实施例中,超声波由具有在400~2000W之间的功率的发生器产生。该功率是发生器所需要的电力。
在实施例中,通过气体冷却,从反应器去除热能。
在实施例中,通过液体冷却,从反应器去除热能。
在实施例中,在在所述初始温度下加热腔室之后,产生电场和超声波,加热在电场和超声波产生时间段的第一部分中被保持,加热在所述第一部分结束时停止,去除热能在所述第一部分之后开始。
在实施例中,初始温度为在100~140℃之间。
在实施例中,用于99%放射性减少的以上步骤的持续期为在5~10小时之间。
在实施例中,电场和超声波产生时间段具有在5~10小时之间的持续期。
附图说明
在参照附图给出的以下的说明书中,解释本发明的特征和优点。
图1是用于本发明的方法的具有超声发生器和加热器的反应器的轴向断面图。
图2是用于本发明的方法的具有超声发生器和微波发生器的反应器的轴向断面图。
图3是具有一杯尘状化合物的图1的发生器的轴向断面图。
图4是具有一杯尘状化合物和一杯放射性材料的图1的发生器的轴向断面图。
图5是对实验1的被处理材料进行的光谱分析的示图。
图6是实验2的自然环境的伽马射线的测量的计数/能量图。
图7是实验2的裂变废料的伽马射线的测量计数/能量的示图。
图8是实验2的被处理材料的伽马射线的测量计数/能量的示图。
图9是表示图6~8的三个测量的结果的比较示图。
图10是用于实验4中的装置的透视示意图。
图11是用于实验4中的容器的透视示意图。
图12是用于实验4中的容器的分解透视示意图。
图13是用于实验4中的发生器的分解透视示意图。
图14是实验4的裂变废料和被处理材料的伽马射线的测量计数/能量的比较图。
附图不仅用于完成本发明,而且,如果必要的话,有助于其定义。
具体实施方式
为了改善能量产生和废料处理,发明人对由过渡金属辅助的低能量嬗变进行了长期的研究。以下的物种被识别为适于辅助嬗变:Sc、Ti、V、Cr、Mn、Fe、Co、Ni、Zn、Y、Zr、Nb、Mo、Tc、Ru、Rh、Ag、Cd、Hf、Ta、W、Re、Os、Ir、Pt、Au、镧系元素和锕类元素。它们可在工业上为纯的或者被合金化。在金属粉末的化合物中存在少量的Cu在实验中看起来是有利的。但是,Cu不是驱动金属。Cu具有增强嬗变的功能。
发明人寻求在低或中温度和工业可分级设备下当在安全过程中产生能量时净化排到不活动材料的核废料。
WO0129844涉及从经受电流脉冲的吸氢金属产生能量。
WO2010058288提出在强感应1~70000高斯和电场1~300000v/m下从氢气与金属之间的核反应产生能量。
WO2013/10859公开了用照射源照射胶质混合物的核反应器。
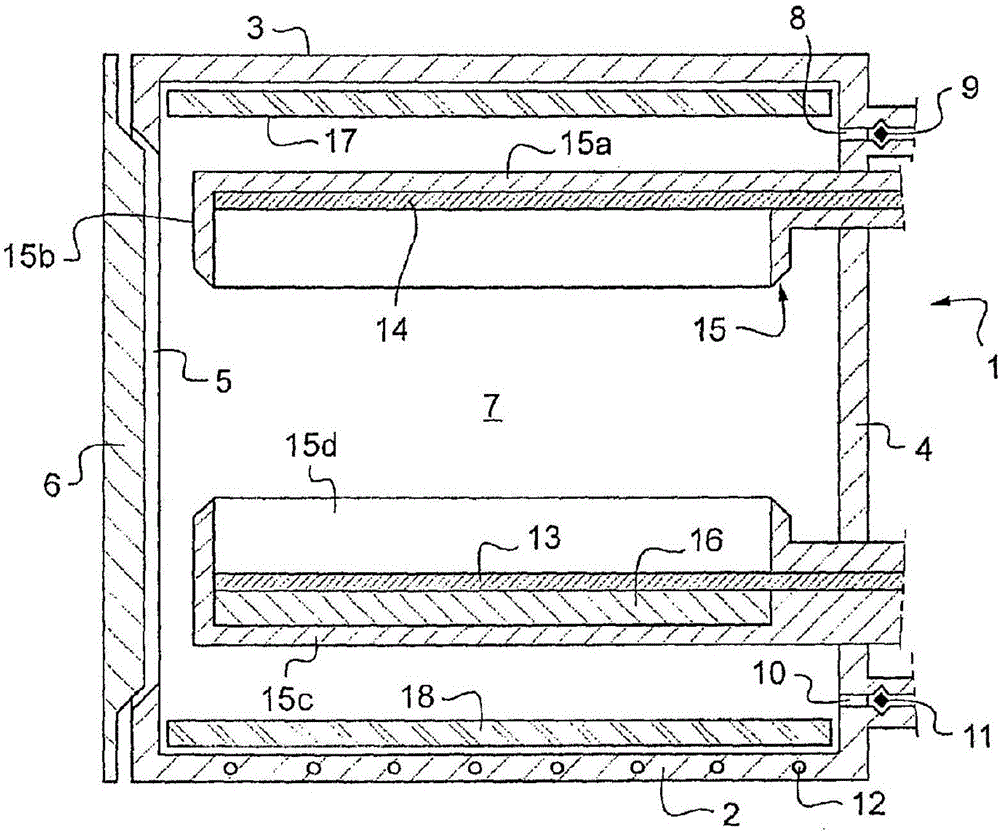
在图1中,反应器1包含下壁2、上壁3、限定孔5的周壁4和能够关闭孔5的门6。当门6被关闭时,反应器1限定密闭的腔室7。当门6打开时,能够穿过孔5在腔室7中插入固态材料。反应器1形成闭合的空间。反应壁2、3、4和门6基本上由钢制成,钢优选包含按质量至少1%(at least 1%in mass)的Cr。反应壁2、3、4和门6可由不锈钢制成。在20℃下,反应器1适于高于106Pa、优选2×106Pa的内部压力。反应器1适于100~800℃之间的平均内部温度和200~1000℃之间的局部化内部温度。处于腔室7中并且在后面描述的反应器1的多个部分能够耐受以上的温度、以上的压力和H2气氛。
反应器1具有图中未示出的与真空泵连接的第一开口8和第一阀9。第一开口钻入周壁4中。真空泵用于在门关闭之后从腔室7去除空气。反应器1具有图中未示出的与氢气源连接的第二开口10和第二阀11。氢气源可以是加压的H2容器。氢气源用于在空气去除之后在腔室7中引入氢气。氢气源被配置为在环境温度下在所述腔室7内设定高于5×105Pa、优选处于106Pa的压力。腔室7可包含至少99%的H2、优选包含至少99.9%的H2。
在变更例中,第一开口8与环境气氛连接并且配有阀。氢气源用于形成将氧气驱逐出腔室7的氢气气流。可以设置氮气源以形成氮气气流以避免混合氢气和空气。
反应器1包含冷却部件12。冷却部件12可被加入反应器1的至少一个壁中以构成至少一个冷却壁。冷却部件12可包含循环冷却剂的导管。在图1中,下壁2配有冷却部件12。
反应器1包含电场发生器。电场发生器包含配置于腔室7中的阳极13和阴极14。阳极13和阴极14具有相互面对的表面。这里,阳极13和阴极14——电极——一个被安装于腔室7的上部中并且另一个被安装于其下部中。这里,阳极13处于下部中并且阴极14处于上部中。阳极13和阴极14以及面对的表面可基本上水平。在另一实施例中,阳极13和阴极14基本上垂直。电场发生器包含包围阳极13、阴极14和阳极13与阴极14的面对表面之间的区域的绝缘部分15。绝缘部分15防止电场与反应器1的壁中的一个的短路。电场发生器包含反应器1外面的高电压源和连接电压源与阳极13以及与阴极14的绝缘导线。绝缘部分15包含配置于上壁3与阳极13之间并且与阳极13接触的上板15a和向下面突出的上圆筒缘15b。上板15a和上圆筒缘15b形成上半壳。绝缘部分15可由陶瓷制成。绝缘部分15由耐受腔室7在处理期间的温度且与H2环境相容的材料制成。
在对称配置中,绝缘部分15包含配置于下壁2与阴极14之间且与阴极14接触的下板15c和向上突出的下圆筒缘15d。下板15c和下圆筒缘15d形成下半壳。
在半壳之间即在上圆筒缘15b与下圆筒缘15d的端部之间剩余有空间。“圆筒”在其几何意思中使用,缘的横截面为圆形、正方形或多边形。所述空间足以使至少两个接收器移动通过其中,至少一个接收器用于核废料并且至少一个用于驱动化合物21。半壳的形状被配置为使得电场线尽可能平行。施加平行的电场线提高处理的均匀性并且减少核废料中的热点的出现和尺寸。可能出现通过部分熔融导致原子的结块的纳米尺寸的热点。例如为从微米到毫米的大尺寸的热点可能对处理的效率有害。在高于被处理废料的熔融温度的大热点的情况下,可能需要对被处理废料的后压碎。
反应器1包含超声发生器16。超声发生器16被配置于电场发生器的绝缘部分15的下半壳的凹处中。超声发生器16沿垂直轴被配置于下电极13与绝缘部分15的下板15c之间。超声发生器16在水平板中被绝缘部分15的下圆筒缘15d包围。超声发生器16具有400~2000W之间的标称电力。该电力是发生器所需要的电力。超声发生器16具有250~600kHz之间的频率,例如,频率为300kHz。频率可固定。
在图1的实施例中,反应器1包含两个电加热器17和18。电加热器中的一个被配置于腔室7的下部区域中。下电加热器18驻留于反应器1的下壁2上。电加热器中的另一个被配置于腔室7的上部区域中。上电加热器17与反应器1的上壁3接触。为了增强腔室7的加热,在下电加热器18与下壁2之间以及在上电加热器17与上壁3之间剩余小的空间。小的空间确保热绝缘。该小的空间能够通过设置在面向相应的壁的电加热器的表面上的间隔腿来获得。在另一实施例中,绝缘材料层被配置于电加热器的所述表面与相应的壁之间。如图1所示,电加热器17、18覆盖相应的壁的表面的大部分,例如,覆盖多于90%。获得加热均匀性。
在图2的实施例中,反应器1包含微波发射器19。微波发射器19被周壁4支撑。微波发射器19与门6相对。微波发射器19具有在腔室7中突出的波导。微波发射器19的其它部分可被配置于腔室7内。在另一实施例中,微波发射器19的其它部分被配置于腔室7外面并且通过密闭壁套管与波导连接。波导具有带有大的发射端的截头圆锥形状。波导被配置为在腔室7中向会出现核废料的接收区域发射微波。在其它条件下,核废料和驱动化合物21将在处理期间驻留于微波接收区域中。为了清楚起见,在图中没有表示用于向上述的电能接收器馈电的电缆。
如图3所示,反应器1容纳驱动化合物21的容器20。容器20驻留于下电极的表面上。容器20为杯形。容器20包含盘状底壁20a和包围底壁的圆形缘20b。缘20b是具有30~60°之间的角度的截头圆锥。容器20可制成一个零件。容器20可包含铜或黄铜。容器20可由铜或黄铜构成。在变更例中,容器20可由钢构成。容器20的厚度可选自从0.4mm到几毫米的范围。可根据其中的驱动化合物21的质量或者热传导需求选择容器20的厚度。在试验中,使用了0.5mm厚度的铜杯。除了包含以外,容器20还使驱动化合物21内的温度均匀化。
容器20容纳驱动化合物21的层。驱动化合物21的层的厚度不大于缘的高度。驱动化合物21的层具有基本上恒定的厚度。层厚度可以为在2~12mm之间。驱动化合物21在容器20的底壁的表面上均匀展开。驱动化合物21具有面对上电极的均匀表面,均匀理解为宏观的意思。驱动化合物21可被压制或者不被压制。驱动化合物21基本丧失易于与H2进行化学反应的材料,例如,氧气。
在试验中,驱动化合物21包含具有不小于99%的纯度的粉末。粉末的各金属可具有不小于99%的纯度。按质量小于1%的金属杂质可被接受。特别是在非金属杂质的情况下,粉末的纯度优选不小于99.9%。
驱动化合物21一般具有小于5μm的粒度。但是,在已进行的实验中,作为反应增强剂存在的铜粉末具有小于20μm的粒度。在变更例中,铜粒子具有10~40μm之间的直径。在其它条件下,在已进行的实验中,除铜以外的各金属晶粒具有小于5μm的粒度。
驱动化合物21为非放射活性的。在其它条件下,驱动化合物21具有放射活性,但该放射活性不高于基本天然水平。
在一个实施例中,尘状化合物包含Ni和Fe。成分可以为按质量50%~95%的Ni和5%~50%的Fe。成分可以为持续期70%~90%的Ni和10%~30%的Fe。Ni原子在处理期间嬗变为Cu。
在一个实施例中,尘状化合物包含按质量1%~10%的Cu。在一个实施例中,尘状化合物包含按质量2%~7%的Cu。Cu是尘状化合物的一部分,但不是嬗变反应的驱动剂。Cu还是从Ni的嬗变反应的产物。尘状铜增强尘状化合物的热传导性。
在一个实施例中,在尘状化合物中的Cu中,至少99.%、优选99.9%的粒子的平均尺寸为在10~100μm之间,优选为在10~50μm之间,更优选为在10~20μm之间。
在一个实施例中,在尘状化合物的Ni中,至少99%、优选99.9%的粒子的平均尺寸不大于10μm。
在一个实施例中,在尘状化合物的Fe中,至少99%、优选99.9%的粒子的平均尺寸不大于10μm。
在一个实施例中,在尘状化合物的Ni中,至少99%、优选99.9%的粒子的平均尺寸不大于5μm。
在一个实施例中,在尘状化合物的Fe中,至少99%、优选99.9%的粒子的平均尺寸不大于10μm。
可在尘状化合物中完成石墨的添加。尘状化合物可包含按质量25%~40%、优选30%~40%的石墨。当通过微波加热时,石墨是有用的。石墨可具有不大于10μm的平均尺寸。
可在尘状化合物中完成铬的添加。相同的尘状化合物成分可用于各种放射性材料。在其它条件下,尘状化合物成分在一定程度上与放射性材料成分无关。
在一个实施例中,尘状化合物包含按质量10%~15%的Fe、80%~85%的Ni、2%~5%的Cu。这种化合物已通过电加热器加热得到测试。
在一个实施例中,尘状化合物包含按质量5%~10%的Fe、57%~65%的Ni、1%~3%的Cu和25%~30%的石墨。这种化合物已通过微波加热得到测试。
在一个实施例中,尘状化合物包含按质量10%~15%的Fe、75%~80%的Ni、1%~3%的Cu和8%~15%的Cr。这种化合物已通过激光器加热得到测试。
如图4所示,反应器1容纳驱动化合物21的第一容器20和核废料23的第二容器22。第二容器22驻留于驱动化合物21的上表面上。第二容器22为杯形。第二容器22具有小于第一容器20的直径的直径。被驱动化合物21支撑的第二容器22与第一容器20分开。第二容器22包含盘形底壁23a和包围底壁23a的圆形缘23b。缘23b是具有30~60°之间的角度的截头圆锥。第二容器22可被制成一个零件。第二容器22可包含铜或黄铜。第二容器22可由铜或黄铜构成。第二容器22可由叠层的铜叶构成。第二容器22的厚度可选自从0.4mm到几毫米的范围。可根据其中的核废料23的质量或者热传导需求选择第二容器22的厚度。在试验中,使用了0.5mm厚度的铜杯。除了包含以外,第二容器22还使驱动化合物21内、核废料23内以及核废料23与驱动化合物21之间的温度均匀化。但是,减小第二容器22的厚度增强了处理的效率。
第二容器22容纳核废料23的层。核废料23的层的厚度不大于缘的高度。核废料23的层具有基本上恒定的厚度。层厚度可以为在2~12mm之间。核废料23在容器20的底壁的表面上均匀展开。核废料23具有面对上电极的均匀表面,均匀在宏观的意义上被理解。核废料23可被压制或者不被压制。在变更例中,驱动化合物21的第三容器和核废料的第四容器被设置在反应器1中,与第一和第二容器22重叠,等等。
在实施例中,第二容器22比0.4mm薄,例如,为选自0.15mm和小于0.4mm之间的厚度。为了减轻重量从而允许移动加载的第二容器22,核废料23的层可以为在2~4mm之间。在变更例中,第二容器22在被插入腔室7中时是空的,并且核废料被加载于驻留于腔室7中的第二容器22中。在另一变更例中,第二容器22的缘被增强。增强可包含形成第二容器22的铜叶的折叠的第二层,以形成双重片缘。增强可包含从缘突出并且与缘垂直的边缘。边缘可与缘一体化。增强可包含固定于缘上的钢环。在另一变更例中,中间支撑可在第二容器22的插入过程中被设置在第二容器22下面并且在插入腔室7中之后被去除;中间支撑可在其去除之前被设置在第二容器22下面。
在试验中,核废料23是尘状的。核废料23由包含一种或几种放射性元素的粉末构成。核废料23基本上丧失碳。碳尽可能地低,原因是它使反应减慢。特别是在包含有机材料的矿物废料的情况下,碳可通过燃烧被提取。核废料23基本上丧失易于与H2化学反应的材料。
核废料23一般具有小于5μm的粒度。驱动化合物21的质量和核废料23的质量为在3/1~6/1之间的比例。驱动化合物21过量延迟处理的激活。当前已检测到抑制效果。
加载有驱动化合物21的第一容器20和加载有核废料23的第二容器22可被依次插入于腔室7中。在变更例中,加载有驱动化合物21的第一容器20和加载有核废料23的第二容器22可被一起插入于腔室7中。
由于该处理,相对于自然嬗变,该嬗变被强烈地加速。该处理的基础理论在当前被完全理解。对该处理进行了实验的发明人允许强烈地增加去活速度。
通过在电场下使核废料经受处于中温下的加压的氢气气氛并且接近金属驱动剂,一起获得核废料去活与产生热。一些专家使用表达词语“中子云”以描述由驱动金属剂上的超声导致的中子可用性的效果。但是,这种表达受到其它专家的批评。
在处理的开始,提供能量以加热氢气、废料和金属驱动剂。加热可由电加热器和/或微波发生器和/或激光器提供。电场极化核废料和金属驱动剂的粒子。极化增强质子从核废料向金属驱动剂的迁移。该现象包括由超声触发的质子的迁移。在一段时间之后,停止加热并且该处理放热。保持电场。可通过冷却部件12去除热。电场被保持预设的持续期或根据测量的参数,例如为放射性、去除的能量、温度总和的持续期。持续期可以为1~10小时。停止电场发生器。从腔室7去除氢气。可进行氮气冲除以避免氢气与空气混合。如果必要的话,保持冷却,直到达到易于去除被处理的核废料的温度,例如,40℃。被处理的核废料不再具有放射性。被处理的核废料可被用作普通的金属粉末。
在应中断处理的情况下,关掉电场发生器导致嬗变的迅速减少。如果有微波发生器,最好关掉它。如果有超声发生器,也推荐关掉它。电加热器或加热器可被关掉。保持冷却。在其它条件下,输入到反应器1中的任何能量被关掉。但是,能够将电场发生器设定为具有明显低于去活阶段期间的电场的绝对值的绝对值的反向电场。可优选在低温下通过氮气冲除从腔室7去除氢气。
现在转到处理的步骤,金属驱动剂被制备为具有不低于99%的纯度和小于10μm的粒度。实验表明小于5μm的粒度具有更高的效率。具有大于金属驱动剂的意义的尘状化合物包含金属驱动剂,并且可能包含不是驱动剂本身的金属,但是增加嬗变数。不是驱动剂本身的金属可用作催化剂。已发现铜的存在是有利的。初始尘状化合物中的1~5%Cu是选定的范围。在尘状化合物几次使用之后,Cu的含量可达到7%而没有负面效果。在高于7%时,Cu含量可通过化学处理减少。已用Fe、Ni对金属驱动剂进行了实验。其它金属,如果是固态的,例如,Zn和Cr,是可能的。尘状化合物还可包含石墨的添加以增强热传导性并因此增强尘状化合物中的温度均匀性。构成尘状化合物的所有材料被混合以获得均匀的化合物。尘状化合物被灌注到第一容器20中。
同时或者不同时,核废料被制备为具有小于10μm、优选小于5μm的粒度。如果存在碳,那么从核废料去除碳。核废料可以为金属或非金属。核废料被混合以获得均匀的产物。核废料被灌注到第二容器22中。
在打开反应器1的门6之后,第一容器20被移动到腔室7中。第一容器20被放在阳极表面上。第一容器20的下表面与阳极13的上表面接触。
第二容器22被移动到腔室7中。第二容器22被放在驻留于第一容器20中的驱动化合物21上。第二容器22的下表面与驱动化合物21的上表面接触。反应器1的门6以密闭的方式关闭。氮气通过反应器1的开口被引入到腔室7中,同时通向环境气氛的另一开口保持打开。氧气含量被降低低于3%。进行氮气冲除。氮气冲除避免H2和空气的O2之间的化学反应的风险。然后,氢气通过反应器1的开口被引入到腔室7中,同时通向环境气氛的所述另一开口保持打开。氢气含量被降低低于3%,优选低于%。进行氢气冲除。氢气冲除比氮气冲除长。氢气应尽可能深地占据腔室7的可用空间。由于第一容器20比第二容器22大,因此氢气与第一容器20和第二容器22之间的尘状化合物接触。氢气渗透到核废料的粉末中并到尘状化合物的粉末中。由于H2是小分子,因此粉末可非常细。尘状化合物的氢气饱和。核废料的氢气饱和。
电场发生器被接通。电场为1000V/m或更大。根据腔室尺寸和分别在第一和第二容器中的尘状化合物和核废料的厚度选择电场。
超声发生器16被接通。超声发生器16被设定为300kHz的频率。作为替代方案,根据金属驱动剂选择频率。能量通量可不小于1.3Wm-2。超声发生器16在大于核力的Minkowski阈值的级别操作。Minkowski阈值必须被理解为使得能够与亚原子级别交互作用的机械波的值。
通过电加热器、微波发生器或激光器中的至少一个向腔室7提供热。通过电加热器,腔室7被加热到90℃。然后,超声发生器16被接通。嬗变步骤在腔室7中约180℃的平均温度开始。电加热器可被关掉。
通过微波发生器,超声发生器16被同时接通。温度的升高比电加热器慢。嬗变步骤在腔室7中约180℃的平均温度开始。微波发生器可被关掉。嬗变步骤是稳定的。
通过激光器,超声发生器16被事先接通。温度的升高比电加热器强。但是,与前面的实施例相比,氢气温度更不代表尘状化合物和核废料的温度。嬗变步骤骤然开始。激光器可被关掉。“激光器”在这里被用作“激光发射器”的同义词。
但是,加热是任选的。也可在没有专用的加热器的情况下获得嬗变步骤。在这种实施例中,嬗变步骤以利用指向核废料的电场和超声开始。超声激起核废料的颗粒与尘状化合物的颗粒之间的机械移动和温度的稍微上升。
在嬗变步骤期间,腔室7中的温度可以为约360℃。冷却可在选自180℃与360℃之间的温度下开始。更一般地,冷却在处理变得热自足之后开始。尘状化合物和核废料的温度可以为在400~600℃的范围中。尘状化合物和核废料的温度是相近的。在微观上,热点可以为更高的温度,诸如1000℃或1400℃。热点可产生粉末的金属颗粒的局部熔融。尘状化合物和容器、可能是核废料的高的热传导性减小热点的尺寸并缩短其持续期。
在嬗变步骤结束时,任选地在预设的持续期之后或者当达到相关的参数时,电气发生器被关掉。超声发生器16被关提。保持冷却以获得安全的温度。通过氮气冲除氢气。然后,打开门6。去除被去活的核废料。
尘状化合物可驻留于这里并且被重新使用几次来去活新鲜的核废料。如果Cu含量天花板水平被达到或估计,那么去除尘状化合物。富Cu尘状化合物可被化学处理以去除Cu的一部分,然后在处理中被重新使用。
一般地,不存在磁场的自发性产生。
实验1
进行实验以处理常常存在于医疗废料中的60Co。60Co嬗变成稳定的同位素61Ni或62N2的假设基于测量结果的。原来包含60Co的被处理材料的中子发射和伽马射线的等级接近零。嬗变会基于:
60Co+p+→61Ni
60Co+2p+→62Ni
用SEM EDAX仪器进行被处理材料的光谱分析。在图5上表示结果。光谱表示以指定“Ni”的三个峰为证据的镍的几乎排他地存在。与钴对应的位置由“Co”表示并且揭示非常低含量的Co。
参见图1,电阻被用作加热器。反应器内的压力为约13bar。在反应器外面测量的开始温度为约110℃。持续期为约165分钟。驱动剂包含粒子尺寸小于5微米的约13克的镍(Ni)和铁(Fe)。废料包含粒子尺寸小于5微米的约1克的钴-60(60Co)。不产生电场。
实验2
进行实验以处理裂变废料。作为参照,参见图6,在9:00进行自然环境的伽马射线的第一测量。图7的刻度为100。参见图7,在14时25分,进行被处理的裂变废料的伽马射线的测量。图7的刻度为10000。通过下面的表使得图6和室7的描述更容易。
在15:45,裂变废料被迁移到反应器并且反应器和测量装置就绪。测量装置包括:
-冷却部件12上的两个温度探针,一个处于上游,另一个处于下游;
-被配置为测量冷却剂的流动的冷却回路上的流量计;
-用于测量用于开始并保持处理的电能的电力计。
测量装置被安装以建立该处理的能量平衡。
值被连续登记。
参见图1,电阻被用作加热器。反应器内的压力为约18bar。在反应器外面测量的开始温度为约140℃。驱动剂包含约27克的粒子尺寸小于5微米的镍(Ni)和铁(Fe)和粒子尺寸小于10微米的铜(Cu)。废料包含粒子尺寸小于5微米的约1.6克的水合乙酸双氧铀(CAS n°6159-44-0)。没有产生电场。
在16:00,处理开始。进行环境气氛以及反应器的附近的放射性发射测量。在实验结束之后进行反应器内的放射性的测量。
在18:35,处理被中断。
在19:15,被处理的废料被置入用于测量伽马射线的装置中。
在19:25,测量并且在图8上报告由被处理废料发射的伽马射线。图8的刻度为100,与图6相同。
基于天然放射性的测量已得到伽马射线测量装置是可靠的。直接或者间接地通过第二代的核素的存在,裂变废料的放射性的测量表明存在放射核素的存在。被处理废料的放射性的测量表明非常明显的伽马射线减少。被处理废料的剩余伽马射线发射具有与天然放射性相同的大小。
在以对数坐标建立的图9上,比较伽马射线发射测量:
d1 09.00:天然放射性测量;
d2 14.25:处理之前的裂变废料放射性测量;
d3 19.15:处理之后的废料放射性测量。
已计算该处理的功率平衡。消耗的能量为630Whe。温度探针之间的温度差在9240秒期间为2.506℃,质量流为580kg/h冷却水,见下表:
为了简化,该表表示上游和下游的发展的温度值。通过几种近似根据表3进行计算。1.49m3的体积的水被加热2.5℃,这与4.34kWh的热对应。反应器的热损失被忽略,虽然这种热损失由于本实验期间的非密封而较明显。发送到反应器的能量为0.63kWh。
作为结论,向着自然放射性的光谱的伽马射线发射光谱变化、处理的放热性能以及点火之后的自维持表示嬗变反应。
实验3
乙酸双氧铀粉末与在处理之前添加的镍(Ni)混合,从而形成样品,并且将其置于支撑结构上。样品的总重量为20.306克,其中0.846g为乙酸双氧铀。在试验结束时,样品的重量为21.290克,即,比之前增加0.984克。重量增加可能由与脏手套接触导致。在任何情况下,看起来反应器内的乙酸双氧铀的一致(consistent)损失可被排除。反应器是根据图2的。
在插入反应器之前,在确保不可能有材料的取代的情况下,将样品置于定位于装料管周围的铜管内,从而在视觉上隐藏样品,但是允许以下描述的放射性测量。
通过Lantanium Tribromide光谱计记录来自定位的含铜样品的伽马射线发射。在下表中报告能量间隔为1.8-1534keV的600秒的总计数和85.8与97.8keV之间(即,在约93keV的234Th-doublet周围)的通道中的部分计数:
然后,在约93keV的234Th-doublet被明确检测。
在插入反应器中之后,将同一光谱计定位为尽可能地接近样品,但是没有揭示高于背景的活动;可能是由于反应器内的一些屏蔽。因此,在以下的处理过程中,没有检测到放射性变动的可能性。反应器被密闭。处理通过电场和加热在19:30开始。通过直流电流产生电场。电场为约10000V/m。在3小时之后即在约22:30,由于发热自维护,因此关掉反应器的加热电阻。电场稳定化并且反应的速度增加。电场可被用于设定源自嬗变的材料。作为例子,从U开始,高的电场允许获得大的Ba量并且低的电场允许获得大的Pb量。在40分钟之后,反应器壁折断,可能从冷却系统发出的热流泄漏。因此,氢气被放出并且处理中断。
在该活动的短时间段期间,发热十分可观:在冷却系统上,在△T=40℃进行测量,流量为与大于30kW的功率对应的650kg/h。第二天,用Germanium腔室分析处理的样品。为了比较,已分析类似的几何结构中的类似量的裸露乙酸双氧铀的γ射线光谱。处理样品的放射性总的为未处理样品5%。这事实上是由于两个样品的重量和几何结构的不同。由计算机分析识别的发射线确实相同,其相对强度相同。因此,没有发现通过识别的核素的γ射线发射的实际变动。但是,在来自处理样品的最微弱的线中,存在一些新的:特别是揭示正电子湮灭的511keV的线。
实验4
这里报告的实验的目的是阐明和展示处理之后的样品的放射性的减少。减少是可重复的,并且它与此时没有完全理解的处理有关。如图11和图12所示,样品是气密容器,内有放射性材料。
电阻被用作加热器。通过两个相应的压力控制系统,反应器内的压力在反应器内为约12bar并且在容器内为7bar。在反应器外面测量的开始温度为约140℃。驱动剂包含约36克的粒子尺寸小于5微米的约70%的镍(Ni)、20%的铁(Fe)和3%的钴(Co)和粒子尺寸小于10微米的7%的铜(Cu)。驱动剂被配置于容器外面和周围。废料包含约1.3克的粒子尺寸小于5微米的(UO2(CH3COO)2(CAS n°6159-44-0)。图10中心的天平被用于废料和驱动剂的称重。“氢气板”即事先经受吸氢的烧结的氢化钯的小的芯部被布置于容器内。气密容器内的温度升高引起氢气的释放。
发生处理的图13中的反应器在处理过程中是不能从外面检测的。还没有进行处理中的放射性测量,这主要是由于放在里面的材料的很少的光子放射性,并且还由于插入的屏蔽(与反应器的两个壁中的一个对应)。还没有进行在处理期间在反应器中使用的材料的化学和物理分析。
目的是通过处理的样品的发射率与未处理的样品的发射率之间的比值的评价来阐明效果。将放射性发射的该比值与样品重量之间的比值相比较。
通过重复的结果验证以下描述的效果的可重复性。在当前的结果之前,该过程被重复5次。在任何时间都清楚地看到以下描述的活性的减少。
容器
1.1铜气密圆筒容器(如图11和图12所示,50mm长,14mmΦ,1÷2mm厚)。用作加热器的电阻被设置在容器内。
该处理的激活器或反应器
入口加压以插入容器的金属箱
用于激活和控制处理的电子设备
反应器的水冷却回路
反应成分(反应器内容物)
固态添加剂(会与必须被处理的保护材料混合):约1.3克的镍(Ni)
放射性材料:CAS n°6159-44-0
图10中心所示的解析天平
Mettler-Tolede G603-S
图10左边所示的放射性测量
单通道分析器-Ludlum 2221(静电计)
GM探针Ludlum 44-9
NaI探针(5.1×5.1cm)Ludlum 44-10
便携式光谱计-Camberra检查仪1000
在5.4的描述(addiction)中使用的LaBr3-IPROL-1-Intelligent LaBr3探针(30keV至3Mev,具有1.5英寸×1.5英寸、38.1×38.1mm),即,“HPGe Gamma Spectrometer-Canberra HPGe with Eagle Plus MCA in a low bkg lead shield”。
用于便携式检测器5cm厚的铅屏蔽
图10所示的低背景铅屏蔽中的HPGe Gamma Spectrometer-Canberra HPGe with Eagle Plus MCA。
如引言所述,该试验的目的是估计通过该处理所处理的样品的放射性的减少。相同测量条件下的相同的样品表明放射性的减少。研究着眼于光子放射性发射辐射。容器的实体质量和受限的体积是恒定的。出于这种原因,在整个气密容器的重量上,限定质量守恒。重量用于估计容器在处理前后之间的质量守恒。观察的放射性的类型是用积分式(NaI)和光谱计(HPGe)放射线检测器测量的光子型。在两种检测器中,前后的源(样品)与检测器之间的相对位置在误差内是可比较的。即使在最坏的情况下,反应器内的源的几何分布也不能是描述的效果的原因。用积分式检测器(NaI)测量每分钟计数(CPM)比值。在这些积分值上计算的比值用于展示伽马活动的减少。光谱计量检测器的使用使得结果更完全并且允许对处理进行观察。
试验暂时分成三个部分:处理之前、其间和之后。以下表示对这些部分中的每一个执行的测量和过程。
处理之前
1.将放射发射材料称重。
2.根据放射性材料在化合物制备之前将这里为约1.3克的镍(Ni)的固体添加剂称重。
3.用容器装载化合物,然后,容器被气密关闭并且被安置。
4.将制备的容器称重。为了评价统计可变性,将该测量重复10次。
5.在低背景(bkg)屏蔽中用NaI探针(CPM模式中的SCA)测量伽马发射率,以强调来自样品的伽马信号(SNR)。为了评价统计可变性,将该测量重复10次。为了处理“之后”测量,记录样品与探针之间的相对位置。
6.用HPGe光谱计分析伽马放射线发射。实时时间被设定为21600秒,该值比如用于获得主峰下的统计合理计数值。获得的光谱,见图14,仅用于比较并且不用于样品的活动的绝对测量。
处理期间
1.参见图13,将容器放入激活处理的反应器1内。所需要的时间在经验上基于经验。通过使用的量、几何结构和其它参数,能够将3h估计为合理的时间以获得达到的结果。
处理之后
1.将包含被处理材料的容器称重。如点1.4那样,将该测量重复10次。
2.在低背景屏蔽中用NaI探针(CPM模式中的SCA)测量伽马发射率,以强调来自样品的伽马信号(SNR)。如点1.5那样,将该测量重复10次。样品与探针之间的相对位置与处理“之前”测量结果相同。
3.参见图14,用HPGe光谱计分析伽马放射线发射。如点1.6那样,实时时间被设定为21.6千秒。
设置的放射性材料的质量为1克(§1.1)。具有该材料的制备的容器称重:
处理之前(§1.5):40,666±20mg
处理之后(§3.2):40,604±20mg
放射率被表达为CMP。制备的反应器(内有化合物)称重:
处理之前(§1.4):8,860±150CPM
处理之后(§3.1):1,130±100CPM
光谱计数的积分(16÷2048keV)为:
处理之前:3,925,442计数
处理之后:301,114计数
之后与之前的样品的重量差为:
DW=-62±40mg
百分比差:-0.15%
之后与之前之间的放射率发射差为:
DR=-7730±250CPM
百分比差:-87.25%
光谱可用于质量分析(即,涉及哪个能量)。主要的能量和有关的同位素为:
Th-234(63.29keV、92.5keV、112.81keV)
Pa-234M(766.36keV、1001keV)
U-235(143.76keV、163.35keV、185.71keV、205.31keV)
Pa-234(131.28keV)
它表明在气密容器内约束的活动的减少。必须在给出的方法的累积误差内解释和考虑测量:
·容器结构不允许包含的材料留下体积。很少的质量差DW被报告。重量与CsPM之间的比值表明该差值不是减少的原因。事实上,最坏情况下的(0,062/1)g可带来10%的减少,不足以解释反应器内的伽马发射核的大于90%的损失。
·由于该处理导致的反应器内的源的分布可改变检测器的效率。出于这种原因,当样品在处理之后被放在测量位置中时,估计在关于在事先过程中测量的一个的另一侧是否由于对反应器内的源的检测器的不同的接近程度而存在更多的信号。该评价表明差异在任何情况下小于20%。表示的伽马减少可在通过用具有不可检发射的稳定的材料替代放射性材料给出的光谱上被解释。包含放射性材料的受限体积仅使得我们将结果解释为由于处理导致的所含核的放射性能的变化的结果。
根据进行的实验和基本理论评估,系统的输出能量明显依赖于尝试的引导和存在于反应器中的产物(放射性核素和驱动剂)的量。
1.能量产量
更详细地,处理的温度越高(即处理强度越大),则揭示的COP越高。COP(性能系数)测量系统或处理的输入与输出能量之间的比值。例如,COP=30意味着一定的处理提供是激活它并且支撑它所需要的30倍的能量测量。
类似地,处理的持续期增加能量过剩,这主要-但不唯一地-根据这样一种事实,即,在启动过程中向系统提供最多的能量,而发热迅速增加,因而然后在整个处理中保持基本上恒定。
存在于装置中的材料的量至少在理论上确定处理的可能的持续期。但是,在系统的能量效率的定义上,材料的量实际上不相关。但是,这种说法应被约束性地解释,原因是,嬗变(成在处理中不激活的元素)的处理中所涉及的质量百分比或者质量损失(过剩能量的产生)太小,以确保处理(以及随之发生的过剩能量的产生)的持续期本质上依赖于装置的体积(氢气饱和)和在处理过程期间保持环境的压力。
作为例子,我们给出处理和持续期的条件不同的一些实验情况。
1.a低强度处理
在这里概括的条件下实施试验:
1.a.1试验时间:9,240秒(约2小时34分钟)
1.a.2使用的材料的量(总):7.654g(±10%),其中,
1.a.2.1放射性核素:0.819g(±10%)
1.a.2.1金属驱动剂:6.835g(±10%)
1.a.3在整个试验持续过程中供给到系统的能量:0.63kWhe。
1.a.4在整个试验持续过程中产生的能量:4.3396kWht
1.a.5COP:7.0907711。
要得到更多细节,参见实验2。
1.b中低强度处理
在这里概括的条件下实施试验:
1.b.1试验时间:22,414秒(约6小时14分钟)
1.b.2使用的材料的量(总):12.581g(±10%),其中,
1.b.2.1放射性核素:1.309g(±10%)
1.b.2.1金属驱动剂:11.272g(±10%)
1.b.3在整个试验持续过程中供给到系统的能量:1,269kWhe。
1.b.4在整个试验持续过程中产生的能量:16,893kWht
1.b.5COP:13.3120567。
要得到更多细节,参见实验4。
1.c中强度处理
在这里概括的条件下实施试验:
1.c.1试验时间:27,805秒(约7小时43分钟)
1.c.2使用的材料的量(总):17.806g(±10%),其中,
1.c.2.1放射性核素:1.804g(±10%)
1.c.2.1金属驱动剂:16.002g(±10%)
1.c.3在整个试验持续过程中供给到系统的能量:2.491kWhe。
1.c.4在整个试验持续过程中产生的能量:62.397kWht
1.c.5COP:25.0489763。
1.d理论评价
由Sergio Focardi教授实施的能量生产率的理论计算(Nuclear Physics,University of Bologna)提供了等于463(明显高于在初步实验中记录的值)的COP值。
1.e尝试性结论
通过绘制使用的产物的质量的能量生产率探索的构想只能建立最小值。
实施的实验均没有导致被处理材料(甚至氢气)的耗尽潜力:该理论在于,该项可在数月内被计算。在实验室条件下,实验的时间持续期受氢气的量限制。从约16bar下的0,5到4升,时间限制为在约20天和4个月之间。
简言之,根据实验值,通过一千克的被处理产物不能净得小于3,365kWh。通过报告最小实验理论计算,来自一千克被处理产物的能量产生的净值将达到62,186kWh。
2嬗变的可能的机制
2.a 6027Co
60Co(通过中子激发从5927Co获得的合成放射性同位素-半寿命为5.27年)向6028Co的衰变自然出现。总核反应式可表达如下:
即,(仅对于末期衰变):
通过β衰变出现嬗变,直到6028Ni*(受激镍-60),然后,镍-60通过发射伽马射线切换到其最低能量状态。即,
这里,是电子反中微子,
该第二式更好地示出静电极化的作用。
在实验阶段,通过SEM EDAX分析样品,从而获得图5所示的光谱。光谱清楚地表明,存在由三个峰突出显示的镍(标有符号Ni)-几乎是明确的。会出现钴峰的位置由符号Co表示。
但是,光谱的相同的读数表明形成镍的两个不同的稳定的同位素:
60Co+p+→61Ni
60Co+2p+→62Ni
这是用于突出显示超声的作用。解释可以是极化和“中子云”有利于这两个不寻常的同位素的出现。
2.b 13755Cs
137Co(特别是在核裂变反应器中,主要作为铀的核裂变的副产品形成的铯的放射性同位素-半寿命为30.17年)向13756Ba的衰变自然出现。核式(仅对于末期衰变)可表达如下:
通过β衰变出现嬗变,直到13756Ba*(受激钡-137),然后,钡-137通过伽马发射通向最小能量的状态。任何附加的推导和评价与已描述的对6027Co表达的那些相当类似。
2.c 23592U(+23892U)
基于使用的铀的产物(CAS n°6159-44-0)看到,具有自然衰变的非常不同的模式的235U和238U均存在。
作为例子,我们简要检查在金属经受处理235U的情况下发现的衰变链。结果是钡和氪的稳定的同位素中的铀的嬗变的衰变链。235U从处理获取质子(作为结果,在相同的环境中,电子被释放),从而变为短暂的236U(236Np)。
235U+p+→236U[236Np] (→e-)
236U几乎暂间衰变,从而形成141Ba和92Kr-均是不稳定的-以及正电子处理的释放环境。
236U→141Ba+92Kr (→e+)
电子和正电子伴随能量(被冷却系统的流体保留吸收)的发射而湮没。
e-+e+→2γ
在三个中子的两种情况下,不稳定的141Ba和92Kr瞬间衰变到它们的稳定的形式(138Ba和89Kr)。
141Ba→138Ba+3n
92Kr→89Kr+3n
6个中子(寿命≤1.100秒)最终在流体限制中被热化。
- 还没有人留言评论。精彩留言会获得点赞!