肝脏病症的组合治疗的制作方法
肝脏病症的组合治疗
1.相关申请的交叉引用
2.本技术要求2020年5月13日提交的美国临时申请第63/024,359号的优先权,所述临时申请以引用方式整体并入本文中。
技术领域
3.本发明涉及用于治疗患者的肝脏病症的方法和组合物。
背景技术:
4.脂肪肝病(fld)包括一系列疾病状态,其特征为肝脏中的脂肪过度积累,通常伴有炎症。fld可导致非酒精性脂肪性肝病(nafld),其特征可为胰岛素抵抗。如果不治疗,则nafld可进展为持续性炎症反应或非酒精性脂肪性肝炎(nash)、进行性肝纤维化,并最终进展为肝硬化。在欧洲和美国,nafld是肝移植的第二大常见原因。因此,迫切需要治疗,但由于患者没有明显的症状,患者可能缺乏维持治疗方案的动力,特别是繁重的治疗方案,例如注射药物、每天多次施用的药物或任何产生危险或刺激性副作用的药物。目前没有批准的nash治疗方法。
5.概述
6.本文提供了用于治疗有需要的患者的肝脏病症的方法和组合物。所述方法包括向患者施用法尼醇x受体(fxr)激动剂和氨基脲敏感性胺氧化酶(ssao)抑制剂。
7.在一个方面,本公开提供了减轻有需要的患者的肝脏炎症的方法,所述方法包括向所述患者施用治疗有效量的fxr激动剂和治疗有效量的ssao抑制剂。fxr激动剂和ssao抑制剂的组合的施用比单独施用任一激动剂在显著更大程度上减轻有需要的患者的肝脏炎症。肝脏炎症的减轻以肝脏中白细胞活化的减少为特征。
8.在另一方面,本公开提供治疗以肝纤维化为特征的疾病或病况的方法,所述方法包括向患者施用治疗有效量的fxr激动剂和治疗有效量的ssao抑制剂。fxr激动剂和ssao抑制剂的组合的施用比单独施用任一激动剂在显著更大程度上减轻有需要的患者的纤维化。纤维化减轻以组织学改善和肝脏中促纤维化基因的表达降低为特征。
9.在另一方面,本公开提供治疗以肝脂肪变性为特征的疾病或病况的方法,所述方法包括向患者施用治疗有效量的fxr激动剂和治疗有效量的ssao抑制剂。已发现,fxr激动剂和ssao抑制剂的组合可部分地通过调节与脂质代谢和脂肪酸转运有关的基因来减少肝脂肪变性。令人惊讶的是,fxr激动剂增强了ssao抑制剂调节与脂质代谢和脂肪酸转运相关的基因的作用,从而使肝脏中的脂肪(例如甘油三酯)积累减少。因此,fxr激动剂和ssao抑制剂的组合的施用比单独施用任一药剂在显著更大程度上减轻有需要的患者的肝脂肪变性。
10.如本文所阐述,当向有需要的患者施用fxr激动剂和ssao抑制剂的组合时观察到的协同作用允许相对于作为单一疗法施用任一药剂时减少fxr激动剂和ssao抑制剂之一或两者的剂量。fxr激动剂和ssao抑制剂的较低剂量使得治疗指数改善且减轻有时伴随fxr激
动或ssao抑制的副作用。
11.在一些实施方案中,fxr激动剂和ssao抑制剂的施用不会导致患者出现严重程度为2级或以上的瘙痒。在一些实施方案中,fxr激动剂和ssao抑制剂的施用不会导致1级或以上的瘙痒。在一些实施方案中,fxr激动剂和ssao的施用不会导致瘙痒。
12.在另一方面,本公开提供了治疗或预防有需要的患者的nash的方法,所述方法包括向患者施用治疗有效量的fxr激动剂和治疗有效量的ssao抑制剂。在一个实施方案中,有需要的患者为罹患脂肪肝病例如nafld的患者。在另一实施方案中,有需要的患者为罹患胰岛素抵抗综合征的患者。
13.在一些实施方案中,fxr激动剂和ssao抑制剂同时施用。在一些此类实施方案中,fxr激动剂和ssao抑制剂作为固定剂量组合物提供于如本文所阐述的单一药物组合物中。在其它实施方案中,依次施用fxr激动剂和ssao抑制剂。在一些实施方案中,fxr激动剂和ssao抑制剂中的一者或两者是经口施用的。
14.在一些实施方案中,患者患有肝脏病症和糖尿病。在一些实施方案中,患者患有肝脏病症和心血管病症。在一些实施方案中,治疗期为患者的剩余寿命。在一些实施方案中,方法不包括施用抗组胺、免疫抑制剂、类固醇、利福平、阿片类拮抗剂或选择性血清素再摄取抑制剂(ssri)。
15.在一些实施方案中,每天一次地施用fxr激动剂。在一些实施方案中,每天两次地施用fxr激动剂。在一些实施方案中,每天一次地施用ssao抑制剂。在一些实施方案中,每天两次地施用ssao抑制剂。在一些实施方案中,施用包括每天施用fxr激动剂持续一周或多周的治疗期。在一些实施方案中,施用包括每天施用ssao抑制剂持续一周或多周的治疗期。在一些实施方案中,施用包括每天施用fxr拮抗剂和每天施用ssao抑制剂持续一周或多周的治疗期。
16.多种不同的fxr激动剂和ssao抑制剂可用于实现如本文所论述的对肝脏疾病观察到的有益效果。例如,在一些实施方案中,向有需要的患者施用的fxr激动剂为奥贝胆酸。在一些实施方案中,向有需要的患者施用的fxr激动剂为西罗非索(cilofexor)。在一些实施方案中,向有需要的患者施用的fxr激动剂为托匹非索(tropifexor)。在一些实施方案中,向有需要的患者施用的fxr激动剂为eyp001(沃纳非索(vonafexor),建议的inn)。在一些实施方案中,向有需要的患者施用的fxr激动剂为met409(metacrine)。在一些实施方案中,向有需要的患者施用的fxr激动剂为met642(metacrine)。在一些实施方案中,fxr激动剂为edp-305(enanta)。在一些实施方案中,fxr激动剂为edp-297(enanta)。
17.在一些实施方案中,向有需要的患者施用的fxr激动剂为式(i)化合物:
[0018][0019]
其中:
[0020]
q为1或2;
[0021]
r1为氯、氟或三氟甲氧基;
[0022]
r2为氢、氯、氟或三氟甲氧基;
[0023]r3a
为三氟甲基、环丙基或异丙基;
[0024]
x为ch或n,条件是当x为ch时,q为1;并且
[0025]
ar1为吲哚基、苯并噻吩基、萘基、苯基、苯并异噻唑基、吲唑基或吡啶基,它们各自任选地被甲基或苯基取代,
[0026]
或其药学上可接受的盐。
[0027]
在一些实施方案中,向有需要的患者施用的fxr激动剂为式(i)化合物,其中r1为氯或三氟甲氧基。在一些实施方案中,fxr激动剂为式(i)化合物,其中r2是氢或氯。在一些实施方案中,fxr激动剂为式(i)化合物,其中r
3a
为环丙基或异丙基。在一些实施方案中,fxr激动剂为式(i)化合物,其中ar1为5-苯并噻吩基、6-苯并噻吩基、5-吲哚基、6-吲哚基或4-苯基,它们各自任选地被甲基取代。在一些实施方案中,fxr激动剂为式(i)化合物,其中q为1并且x为n。
[0028]
在一些实施方案中,fxr激动剂为
[0029]
或其药学上可接受的盐。
[0030]
在一些实施方案中,向有需要的患者施用的ssao抑制剂为式(ii)化合物
[0031][0032]
其中:
[0033]
n为1或2;并且
[0034]
r1为h或-ch3,
[0035]
或其药学上可接受的盐。
[0036]
在一些实施方案中,向有需要的患者施用的ssao抑制剂为式(ii)化合物,其中n为1,或其药学上可接受的盐。在另一实施方案中,ssao抑制剂为式(ii)化合物,其中n为2,或其药学上可接受的盐。
[0037]
在一些实施方案中,向有需要的患者施用的ssao抑制剂为式(ii)化合物,其中r1为h,或其药学上可接受的盐。在另一实施方案中,本发明提供式(ii)化合物,其中r1为-ch3,或其药学上可接受的盐。
[0038]
在一些实施方案中,向有需要的患者施用的ssao抑制剂为或其药学上可接受的盐。
[0039]
在一些实施方案中,提供了用法尼醇x受体(fxr)激动剂和氨基脲敏感胺氧化酶(ssao)抑制剂治疗有需要的患者的肝脏病症的方法,所述方法包括施用治疗有效量的fxr激动剂,其中fxr激动剂为或其药学上可接受的盐,以及施用治疗有效量的ssao抑制剂,其中ssao抑制剂为或其药学上可接受的盐,其中肝脏病症选自由以下组成的组:肝脏炎症、肝纤维化、酒精性纤维化、脂肪变性、酒精性脂肪变性、原发性硬化性胆管炎(psc)、原发性胆汁性肝硬化(pbc)、非酒精性脂肪性肝病(nafld)和非酒精性脂肪性肝炎(nash)。
附图说明
[0040]
图1a显示了在向大鼠(1mg/kg)、狗(1mg/kg)和猴子(0.3mg/kg)静脉内(iv)施用后的不同时间点化合物1的血浆浓度。
[0041]
图1b显示了在向小鼠(10mg/kg)、大鼠(10mg/kg)、狗(3mg/kg)和猴子(5mg/kg)经口施用后的不同时间点化合物1的血浆浓度。
[0042]
图2a显示了在以2mg/kg iv向斯普拉格-杜勒(sprague-dawley,sd)大鼠施用后,化合物1、奥贝胆酸(oca)、西罗非索或托匹非索的肝脏与血浆浓度比率。
[0043]
图2b显示在有或没有共同施用利福平的情况下以2mg/kg iv向sd大鼠施用化合物1后,肾脏、肺和肝脏的化合物1浓度的组织与血浆比率。
[0044]
图3显示了在向朗-埃文斯(long-evans)大鼠经口施用5mg/kg化合物1后放射性标记的化合物1在血浆、肝脏、小肠、盲肠、肾脏、肺、心脏和皮肤中的组织分布。
[0045]
图4显示了在向食蟹猴施用0.3mg/kg、1mg/kg或5mg/kg经口剂量后,如通过7-α-羟基-4-胆甾烯-3-酮(7ac4)测量的化合物1施用的药效学。
[0046]
图5a显示了在向食蟹猴施用1mg/kg经口剂量一天或连续7个每日剂量后,化合物1施用的药物动力学。
[0047]
图5b显示了在向食蟹猴施用1mg/kg经口剂量一天或连续7个每日剂量后,如通过7-α-羟基-4-胆甾烯-3-酮(7ac4)测量的化合物1施用的药效学。
[0048]
图6显示了在向c57bl/6小鼠施用10mg/kg化合物1、30mg/kg oca或媒剂对照后测量肝脏shp1、肝脏ostb、回肠shp1和回肠fgf15 rna表达的rt-qpcr结果。
[0049]
图7a显示了通过向c57bl/6小鼠施用10mg/kg化合物1(总共500个基因被调节)或30mg/kg oca(总共44个基因被调节)调节的差异表达基因的数量(相对于媒剂治疗:倍数变化》1.5倍;p《0.05),以及被两种化合物调节的差异表达基因的共有数量(总共37个基因)。
[0050]
图7b显示了在用10mg/kg化合物1或30mg/kg oca或媒剂对照治疗的c5bl/6小鼠中所选fxr相关基因的平均表达水平(如cpm值所示)。
[0051]
图7c显示了通过向c57bl/6小鼠施用10mg/kg化合物1(32条途径)或30mg/kg oca(6条途径)而富集的途径的数量(p《0.05),以及通过任一化合物富集的途径的数量(2条途径)。
[0052]
图7d显示了向c57bl/6小鼠施用10mg/kg化合物1后统计学上最富集的25条途径,且将这些途径的富集与施用30mg/kg oca后的富集进行了比较。
[0053]
图8显示了测试化合物1对nash小鼠模型的功效的研究设计。
[0054]
图9显示了对照小鼠和用10、30和100mg/kg化合物1治疗的小鼠的nafld活动评分(nas)。
[0055]
图10a显示了对照小鼠和用10、30和100mg/kg化合物1治疗的nash小鼠的脂肪变性评分。
[0056]
图10b显示了对照小鼠和用10、30和100mg/kg化合物1治疗的nash小鼠的炎症评分。
[0057]
图10c显示了对照小鼠和用10、30和100mg/kg化合物1治疗的nash小鼠的鼓胀评分。
[0058]
图11a显示了对照小鼠和用100mg/kg化合物1治疗的nash小鼠中纤维化的组织切
片。
[0059]
图11b显示了对照小鼠和用10、30和100mg/kg化合物1治疗的nash小鼠中纤维化的量。
[0060]
图12a显示了对照小鼠和用10、30和100mg/kg化合物1治疗的nash小鼠的血清丙氨酸氨基转移酶(alt)水平。
[0061]
图12b显示了对照小鼠和用10、30和100mg/kg化合物1治疗的nash小鼠的天冬氨酸氨基转移酶(ast)。
[0062]
图12c显示了对照小鼠和用10、30和100mg/kg化合物1治疗的nash小鼠的血清甘油三酯水平。
[0063]
图12d显示了对照小鼠和用10、30和100mg/kg化合物1治疗的nash小鼠的血清总胆固醇水平。
[0064]
图13a显示了对照小鼠和用10、30和100mg/kg化合物1治疗的nash小鼠的肝脏甘油三酯水平。
[0065]
图13b显示了对照小鼠和用100mg/kg化合物1治疗的nash小鼠的脂肪变性评估的代表性组织学。
[0066]
图14a显示了对照小鼠和用10、30和100mg/kg化合物1治疗的nash小鼠的肝脏中的col1a1表达。
[0067]
图14b显示了对照小鼠和用30mg/kg化合物1治疗的nash小鼠中炎症基因的表达水平。
[0068]
图14c显示了对照小鼠和用30mg/kg化合物1治疗的nash小鼠中纤维化基因的表达。
[0069]
图15a显示了在给药后4小时和168小时,与施用单次剂量的安慰剂或1、3、6或10mg化合物2的健康志愿者的基线相比的血浆ssao特异性胺氧化酶活性。图15b显示了与施用单次剂量的安慰剂或1、3、6或10mg化合物2的健康志愿者的基线相比,血浆总胺氧化酶活性的时间过程。图15c显示在健康志愿者中单次剂量的安慰剂或1、3、6或10mg后化合物2水平的时间过程。图15d显示了在健康志愿者中单次剂量的安慰剂或1、3、6或10mg化合物2后血浆甲胺水平的时间过程。
[0070]
图16显示了通过单样本基因集富集分析确定的treg和m2巨噬细胞肝浸润水平。对施用nano2并用化合物1、化合物2或化合物1和化合物2的组合治疗的cdhfd大鼠的肝rna定序数据进行分析(*p值《0.05;***p值《0.001)。
[0071]
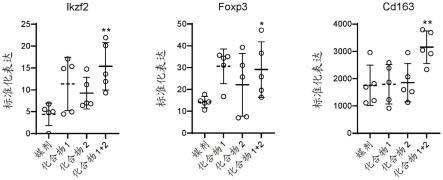
图17显示了通过rna定序对施用nano2并用化合物1、化合物2或化合物1和化合物2的组合治疗的cdhfd大鼠肝脏中的treg和m2巨噬细胞标志物的表达分析。ikzf2,ikaros家族锌指2(treg标志物);foxp3,叉头框p3(treg标志物);cd163(m2巨噬细胞标志物)。(*p值《0.05;**p值《0.01)。
[0072]
图18显示了分别使用≥1.5的倍数变化和0.01的p值截止值,通过rna定序分析在施用nano2并用化合物1、化合物2或化合物1和化合物2的组合治疗的cdhfd大鼠肝脏中鉴定的相对于媒剂nash对照的差异表达基因(deg)的数量和重叠。
具体实施方式
[0073]
定义
[0074]
如本文所用,除非另有说明,否则应适用以下定义。此外,如果本文中使用的任何术语或符号未如下所阐述地定义,则应具有其在本领域中的普通含义。
[0075]“包含”意图指组合物和方法包括所述要素,但不排除其它要素。“基本上由
……
组成”在用于定义组合物和方法时将意味着排除对组合具有任何重要意义的其它要素。例如,基本上由本文定义的要素组成的组合物不排除不会实质影响要求保护的发明的基本和新颖特征的其它要素。“由
……
组成”应指排除超过痕量的,例如所列举的其它成分和实质性方法步骤。由这些过渡术语各自定义的实施方案在本公开的范围内。
[0076]“组合疗法”或“组合治疗”是指在治疗中使用两种或多种药物或药剂,例如本文所用的式(i)或(ii)化合物与另一种可用于治疗肝脏病症(例如nafld、nash)的药剂一起使用,且其各自的症状和表现是组合疗法。“组合”施用是指两种药剂(例如本文所用的式(i)或(ii)化合物,和另一种药剂)以任何方式施用,其中两种药剂的药理作用在患者中同时表现出来。因此,组合施用不需要将单一药物组合物、相同剂型或甚至相同施用途径用于两种药剂的施用或两种药剂恰好同时施用。两种药剂也可配制成单一药学上可接受的组合物。此类单一组合物的非限制性实例为经口组合物或经口剂型。例如,且非限制性地,预期式(i)或(ii)的化合物可与根据本发明的另一药剂一起在组合疗法中施用。
[0077]
如本文所用,术语“赋形剂”意指可用于生产药物或医药的惰性或非活性物质,例如含有本发明化合物作为活性成分的片剂。术语赋形剂可涵盖各种物质,包括但不限于用作粘合剂、崩解剂、包衣、压缩/囊封助剂、乳膏或洗剂、润滑剂、肠胃外施用溶液、咀嚼片材料、甜味剂或调味剂、悬浮/胶凝剂或湿法造粒剂的任何物质。粘合剂包括例如卡波姆、聚维酮、黄原胶等;包衣包括例如乙酸邻苯二甲酸纤维素、乙基纤维素、结冷胶、麦芽糖糊精、肠溶包衣等;压缩/囊封助剂包括例如碳酸钙、右旋糖、果糖dc(dc=“可直接压缩”)、蜂蜜dc、乳糖(无水物或一水合物;任选与阿斯巴甜、纤维素或微晶纤维素组合)、淀粉dc、蔗糖等;崩解剂包括例如交联羧甲基纤维素钠、结冷胶、羟基乙酸淀粉钠等;乳膏或洗剂包括例如麦芽糖糊精、角叉菜胶等;润滑剂包括例如硬脂酸镁、硬脂酸、硬脂富马酸钠等;咀嚼片材料包括例如右旋糖、果糖dc、乳糖(一水合物,任选与阿斯巴甜或纤维素组合)等;悬浮/胶凝剂包括例如角叉菜胶、羟基乙酸淀粉钠、黄原胶等;甜味剂包括例如阿斯巴甜、葡萄糖、果糖dc、山梨糖醇、蔗糖dc等;并且湿法造粒剂包括例如碳酸钙、麦芽糖糊精、微晶纤维素等。
[0078]“患者”是指哺乳动物,且包括人和非人哺乳动物。患者的实例包括但不限于小鼠、大鼠、仓鼠、豚鼠、猪、兔、猫、狗、山羊、绵羊、牛和人。在一些实施方案中,患者是指人。
[0079]“药学上可接受的”是指安全且无毒的,优选用于体内施用,更优选用于人体施用。
[0080]“药学上可接受的盐”是指在药学上可接受的盐。本文所述的化合物可作为药学上可接受的盐施用。
[0081]“盐”是指酸与碱之间形成的离子化合物。当本文提供的化合物含有酸性官能团时,此类盐包括但不限于碱金属、碱土金属和铵盐。如本文所用,铵盐包括含有质子化氮碱基和烷基化氮碱基的盐。可用于药学上可接受的盐的示例性和非限制性阳离子包括基于天然存在的氨基酸的na、k、rb、cs、nh4、ca、ba、咪唑鎓和铵阳离子。当本文所用的化合物含有碱性官能团时,此类盐包括但不限于有机酸,例如羧酸和磺酸的盐,以及无机酸,例如卤化
氢、硫酸、磷酸等的盐。可用于药学上可接受的盐中的示例性和非限制性阴离子包括草酸根、马来酸根、乙酸根、丙酸根、琥珀酸根、酒石酸根、氯离子、硫酸根、硫酸氢根、一价、二价和三价磷酸根、甲磺酸根、甲苯磺酸根等。
[0082]
化合物或组合物的“治疗有效量”或剂量是指减轻或抑制患者症状或延长患者存活的化合物或组合物的量。结果可能需要多次剂量的化合物或组合物。
[0083]“治疗(treatment)”或“治疗(treating)”是指获得有益或所需结果,包括临床结果的方法。出于本发明的目的,有益或所需结果包括但不限于以下一项或多项:减少由疾病或病症引起的一种或多种症状,减轻疾病或病症的程度,稳定疾病或病症(例如,预防或延缓疾病或病症的恶化),延缓疾病或病症的发生或复发,延缓或减缓疾病或病症的进展,改善疾病或病症状态,提供疾病或病症的缓解(无论是部分或全部),减少治疗疾病或病症所需的一种或多种其它药物的剂量,增强用于治疗疾病或病症的另一种药物的效果,延缓疾病或病症的进展,提高生活质量,和/或延长患者的存活期。“治疗”还包括减少疾病或病症的病理后果。本发明的方法考虑了这些治疗方面中的任何一者或多者。
[0084]
如本文所用,“延缓”疾病的发展意指推迟、阻碍、减缓、延缓、稳定和/或延迟疾病的发展和/或一旦发展,减缓其进展或改变潜在的疾病过程和/或病程。此延缓可能有不同的时间长度,具体取决于疾病的历史和/或正在接受治疗的个体。正如本领域技术人员显而易见的,足够或显著的延缓实际上可包括预防,因为个体不会出现与疾病相关的临床症状。“延缓”疾病发展的方法是与不使用所述方法相比时,在给定时间范围内降低疾病发展概率和/或在给定时间范围内降低疾病程度的方法,包括稳定由疾病引起的一种或多种症状。
[0085]
有患疾病的“风险”的个体在本文所述的治疗方法之前可能患有或可能未患可检测的疾病,并且可能已经或可能尚未显示出可检测的疾病。“有风险”表示个体具有一个或多个所谓的风险因素,所述风险因素是与疾病发展相关的可测量参数。具有这些风险因素中的一者或多者的个体比没有这些风险因素的个体具有更高的患上疾病的可能性。这些风险因素包括但不限于年龄、性别、种族、饮食、既往病史、前驱疾病的存在和基因(即遗传)考虑因素。在一些实施方案中,化合物可向有患疾病或病况的风险或具有疾病或病况家族史的受试者(包括人)施用。
[0086]“立体异构体(stereoisomer/stereoisomers)”是指组成原子的立体异构性,例如但不限于一个或多个立体中心的手性或关于碳-碳或碳-氮双键的顺式或反式构型不同的化合物。立体异构体包括对映异构体和非对映异构体。
[0087]“烷基”是指具有1至12个碳原子、优选1至10个碳原子、更优选1至6个碳原子的单价饱和脂族烃基。此术语包括例如直链和支链烃基,例如甲基(ch
3-)、乙基(ch3ch
2-)、正丙基(ch3ch2ch
2-)、异丙基((ch3)2ch-)、正丁基(ch3ch2ch2ch
2-)、异丁基((ch3)2chch
2-)、仲丁基((ch3)(ch3ch2)ch-)、叔丁基((ch3)3c-)、正戊基(ch3ch2ch2ch2ch
2-)和新戊基((ch3)3cch
2-)。c
x
烷基是指具有x个碳原子的烷基。
[0088]“亚烷基”是指具有1至12个碳原子、优选1至10个碳原子、更优选1至6个碳原子的二价饱和脂族烃基。此术语包括例如直链和支链烃基,例如亚甲基(-ch
2-)、亚乙基(-ch2ch
2-或
–
ch(me)-)、亚丙基(-ch2ch2ch
2-或
–
ch(me)ch
2-或
–
ch(et)-)等。
[0089]“烯基”是指具有2至6个碳原子且优选2至4个碳原子并且具有至少1个且优选1至2个乙烯基(》c=c《)不饱和位点的直链或支链单价烃基。此类基团的实例为例如乙烯基、烯
丙基和丁-3-烯-1-基。此术语包括顺式和反式异构体或这些异构体的混合物。c
x
烯基是指具有x个碳原子的烯基。
[0090]“炔基”是指具有2至6个碳原子且优选2至3个碳原子并且具有至少1个且优选1至2个炔属(-c≡c-)不饱和位点的直链或支链单价烃基。此类炔基的实例包括乙炔基(-c≡ch)和炔丙基(-ch2c≡ch)。c
x
炔基是指具有x个碳原子的炔基。
[0091]“烷氧基”是指基团-o-烷基,其中烷基如本文所定义。烷氧基包括例如甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、叔丁氧基、仲丁氧基和正戊氧基。
[0092]“芳基”是指具有单环(例如苯基(ph))或多个稠环(例如萘基或蒽基)的6至14个碳原子的单价芳族碳环基团,所述稠环可为或可不为芳族的(例如2-苯并噁唑啉酮、2h-1,4-苯并噁嗪-3(4h)-酮-7-基等),条件是连接点在芳族碳原子上。优选的芳基包括苯基和萘基。
[0093]“氰基”是指基团-c≡n。
[0094]“环烷基”是指具有3至10个碳原子、优选3至8个碳原子、更优选3至6个碳原子的饱和或不饱和但非芳族的环状烷基,其具有单个或多个环,包括稠环、桥环和螺环系统。c
x
环烷基是指具有x个环碳原子的环烷基。合适的环烷基的实例包括例如金刚烷基、环丙基、环丁基、环戊基和环辛基。一个或多个环可为芳基、杂芳基或杂环,条件为连接点是通过非芳族、非杂环饱和碳环。“取代的环烷基”是指具有1至5个或优选1至3个选自由以下组成的组的取代基的环烷基:氧代基、硫酮、烷基、取代的烷基、烯基、取代的烯基、炔基、取代的炔基、烷氧基、取代的烷氧基、酰基、酰氨基、酰氧基、氨基、取代的氨基、氨基羰基、氨基硫代羰基、氨基羰基氨基、氨基硫代羰基氨基、氨基羰基氧基、氨基磺酰基、氨基磺酰氧基、氨基磺酰基氨基、脒基、芳基、取代的芳基、芳氧基、取代的芳氧基、芳硫基、取代的芳硫基、羧基、羧基酯、(羧基酯)氨基、(羧基酯)氧基、氰基、环烷基、取代的环烷基、环烷氧基、取代的环烷氧基、环烷硫基、取代的环烷硫基、胍基、取代的胍基、卤基、羟基、杂芳基、取代的杂芳基、杂芳氧基、取代的杂芳氧基、杂芳硫基、取代的杂芳硫基,杂环基、取代的杂环基、杂环基氧基、取代的杂环基氧基、杂环基硫基、取代的杂环基硫基、硝基、so3h、取代的磺酰基、磺酰氧基、硫代酰基、硫醇、烷硫基和取代的烷硫基,其中所述取代基定义于本文中。
[0095]“卤基”或“卤素”是指氟、氯、溴和碘,且优选为氟或氯。
[0096]“羟基(hydroxy)”或“羟基(hydroxyl)”是指基团-oh。
[0097]“杂芳基”是指环内具有1至10个碳原子和1至4个选自由氧、氮和硫组成的组的杂原子的芳族基团。此类杂芳基可具有单环(例如吡啶基或呋喃基)或多个稠环(例如中氮茚基或苯并噻吩基),其中稠环可为或可不为芳族环和/或含有杂原子,条件为连接点是通过芳族杂芳基的一个原子。在一个实施方案中,杂芳基的氮和/或硫环原子任选地被氧化以提供n-氧化物(n
→
o)、亚磺酰基或磺酰基部分。优选的杂芳基包括5元或6元杂芳基,例如吡啶基、吡咯基、苯硫基和呋喃基。其它优选的杂芳基包括9元或10元杂芳基,例如吲哚基、喹啉基、喹诺酮基、异喹啉基和异喹诺酮基。
[0098]“杂环(heterocycle)”或“杂环(heterocyclic)”或“杂环烷基(heterocycloalkyl)”或“杂环基(heterocyclyl)”是指具有1至10个环碳原子、优选1至8个碳原子且更优选1至6个碳原子,和1至4个环杂原子、优选1至3个杂原子且更优选1至2个选自由氮、硫或氧组成的组的杂原子的饱和或部分饱和但非芳族的基团。c
x
杂环烷基是指具
有x个环原子,包括环杂原子的杂环烷基。杂环包括单环或多个稠环,包括稠合桥环和螺环系统。在稠环系统中,一个或多个环可为环烷基、芳基或杂芳基,条件是连接点是通过非芳族环。在一个实施方案中,杂环基的氮和/或硫原子任选地被氧化以提供n-氧化物、亚磺酰基、磺酰基部分。
[0099]
杂环基和杂芳基的实例包括但不限于氮杂环丁烷基、吡咯基、咪唑基、吡唑基、吡啶基、吡嗪基、嘧啶基、哒嗪基、吲哚嗪基、异吲哚基、吲哚基、二氢吲哚基、吲唑基、嘌呤基、喹嗪基、异喹啉基、喹啉基、酞嗪基、萘基吡啶基、喹喔啉基、喹唑啉基、噌啉基、蝶啶基、咔唑基、咔啉基、菲啶基、吖啶基、菲咯啉基、异噻唑基、吩嗪基、异噁唑基、吩噁嗪基、吩噻嗪基、咪唑烷基、咪唑啉基、哌啶基、哌嗪基、二氢吲哚基、邻苯二甲酰亚胺基、1,2,3,4-四氢异喹啉基、4,5,6,7-四氢苯并[b]噻吩基、噻唑基、噻唑烷基、噻吩基、苯并[b]噻吩基、吗啉基、硫代吗啉基(也称为噻吗啉基)、1,1-二氧代硫代吗啉基、哌啶基、吡咯烷基和四氢呋喃基。
[0100]“氧代基”是指原子(=o)或(o)。
[0101]
在说明书通篇使用的术语“任选的”或“任选地”意指随后描述的事件或情况可能但不一定发生,并且该描述包括事件或情况发生的情况和不发生的情况。例如,“氮原子任选地被氧化以提供n-氧化物(n
→
o)部分”意指氮原子可以但不需要被氧化,并且该描述包括氮原子未被氧化的情况和氮原子被氧化的情况。
[0102]
fxr激动剂
[0103]
可根据本文所述的方法使用的合适的fxr激动剂包括但不限于奥贝胆酸、西罗非索、托匹非索、eyp001(沃纳非索,建议的inn)、met409(metacrine)、met642(metachrine)、edp-305(enanta)、edp-297(enanta)和式(i)化合物或其药学上可接受的盐。式(i)化合物公开于us 2010/0152166中,其内容以引用方式整体并入,并且特别是关于式(i)化合物或其药学上可接受的盐或对映异构体,以及制备和使用前述的方法。
[0104]
在一些实施方案中,fxr激动剂为式(i)化合物
[0105][0106]
其中:
[0107]
q为1或2;
[0108]
r1为氯、氟或三氟甲氧基;
[0109]
r2为氢、氯、氟或三氟甲氧基;
[0110]r3a
为三氟甲基、环丙基或异丙基;
[0111]
x为ch或n,
representation of stereochemical configuration”,pure and appl.chem,2006,78(10)1897,1959)。式(ii)所示的结构包括具有z立体化学构型、e立体化学构型的化合物或z或e立体化学构型的化合物的混合物。本发明的优选化合物具有e立体化学构型。
[0130]
在一种形式中,式(ii)化合物以游离碱形式存在。在其它形式中,式(ii)化合物以酸加成盐形式存在,例如单或二hcl加成盐或磺酸盐,优选4-甲基苯磺酸盐(甲苯磺酸盐)。
[0131]
在一些实施方案中,ssao抑制剂为式(iia)化合物
[0132][0133]
或其药学上可接受的盐,其中:
[0134]
n为1或2;并且
[0135]
r1为h或-ch3。
[0136]
在一些实施方案中,ssao抑制剂
[0137][0138]
或其药学上可接受的盐,其中:
[0139]
n为1或2;并且
[0140]
r1为h或-ch3。
[0141]
在一些实施方案中,ssao抑制剂为式(ii)、(iia)或(iib)化合物并且n为2。
[0142]
在一些实施方案中,ssao抑制剂为式(ii)、(iia)或(iib)化合物并且r1为ch3。
[0143]
在一些实施方案中,ssao抑制剂为式2化合物:
[0144][0145]
其药学上可接受的盐。“化合物2”是指式2化合物。
[0146]
药学上可接受的组合物和配制物
[0147]
本发明包括本文详述的任何化合物的药学上可接受的组合物或简称为“药物组合物”。因此,本发明包括药物组合物,其包含fxr激动剂(例如式(i)化合物或其药学上可接受的盐)、ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)和药学上可接受的载体或赋形剂。在一些实施方案中,药学上可接受的盐为酸加成盐,例如与无机或有机酸形成的盐。根据本发明的药物组合物可采用适合于经口、经颊、肠胃外、经鼻、局部或经直肠施用的形式或适合于通过吸入施用的形式。
[0148]
一方面,本文详述的化合物可呈纯化形式,并且本文详述了包含纯化形式的化合物的组合物。提供了包含如本文详述的化合物或其盐的组合物,例如基本上纯的化合物的组合物。在一些实施方案中,含有如本文详述的化合物或其盐的组合物是基本上纯的形式。在一个变型中,“基本上纯”意指含有不超过35%杂质的组合物,其中杂质表示除了构成组合物的大部分的化合物或其盐之外的化合物。例如,基本上纯的化合物的组合物意指含有不超过35%杂质的组合物,其中杂质表示除所述化合物或其盐之外的化合物。在一个变型中,提供了一种基本上纯的化合物或其盐的组合物,其中所述组合物含有不超过25%杂质。在另一变型中,提供了一种基本上纯的化合物或其盐的组合物,其中所述组合物含有不超过20%杂质。在另一变型中,提供了一种基本上纯的化合物或其盐的组合物,其中所述组合物含有不超过10%杂质。在另一变型中,提供了一种基本上纯的化合物或其盐的组合物,其中所述组合物含有不超过5%杂质。在另一变型中,提供了一种基本上纯的化合物或其盐的组合物,其中所述组合物含有不超过3%杂质。在另一变型中,提供了一种基本上纯的化合物或其盐的组合物,其中所述组合物含有不超过1%杂质。在另一变型中,提供了一种基本上纯的化合物或其盐的组合物,其中所述组合物含有不超过0.5%杂质。在其它变型中,基本纯的化合物的组合物意指所述组合物含有不超过15%或优选不超过10%或更优选不超过5%或甚至更优选不超过3%且最优选不超过1%杂质,所述杂质可为不同立体化学形式的化合物。
[0149]
在一个变型中,本文的化合物是制备用于向个体例如人施用的合成化合物。在另一变体中,提供了包含基本上纯形式的化合物的组合物。在另一变型中,本发明包括包含本文详述的化合物和药学上可接受的载体或赋形剂的药物组合物。在另一变型中,提供了施用化合物的方法。纯化形式、药物组合物和施用化合物的方法适用于本文详述的任何化合物或其形式。
[0150]
化合物可配制用于任何可用的递送途径,包括经口、经粘膜(例如经鼻、舌下、经阴道、经颊或经直肠)、肠胃外(例如肌内、皮下或静脉内)、局部或透皮递送形式。化合物可与合适的载体一起配制以提供递送形式,包括但不限于片剂、囊片、胶囊(例如硬明胶胶囊或软弹性明胶胶囊)、扁囊剂、锭剂、糖衣锭、咀嚼剂、分散剂、栓剂、软膏、巴布剂(膏剂)、糊剂、粉剂、敷料、乳膏、溶液、贴剂、气雾剂(例如鼻喷雾剂或吸入剂)、凝胶、悬浮液(例如水性或非水性液体悬浮液、水包油乳液或油包水液体乳液)、溶液和酏剂。
[0151]
通过将作为活性成分的化合物与药学上可接受的载体(例如上述那些)组合,本文所述的化合物可用于制备配制物,例如药物配制物。取决于系统的治疗形式(例如透皮贴剂与经口片剂),载体可呈各种形式。另外,药物配制物可含有防腐剂、增溶剂、稳定剂、再润湿剂、乳化剂、甜味剂、染料、调节剂和用于调节渗透压的盐、缓冲剂、包衣剂或抗氧化剂。包含所述化合物的配制物还可含有其它具有有价值的治疗特性的物质。药物配制物可通过已知
的药物方法制备。合适的配制物可在例如remington:the science and practice of pharmacy,lippincott williams&wilkins,第21版.(2005)中找到,所述文献以引用方式并入本文中。
[0152]
本文所述的化合物可以普遍接受的经口组合物的形式施用于个体(例如人),例如片剂、包衣片剂和硬壳或软壳凝胶胶囊、乳液或悬浮液。可用于制备此类组合物的载体的实例为乳糖、玉米淀粉或其衍生物、滑石、硬脂酸盐或其盐等。用于软壳凝胶胶囊的可接受载体为例如植物油、蜡、脂肪、半固体和液体多元醇等。另外,药物配制物可含有防腐剂、增溶剂、稳定剂、再润湿剂、乳化剂、甜味剂、染料、调节剂和用于调节渗透压的盐、缓冲剂、包衣剂或抗氧化剂。
[0153]
描述了包含本文所用的两种化合物的组合物。本文所述的任何化合物都可配制成本文所述的任何剂型的片剂。
[0154]
本公开进一步包括试剂盒(例如药物包装)。提供的试剂盒可包含本文所述的药物组合物或化合物和容器(例如药瓶、安瓿、瓶子、注射器和/或分包或其它合适的容器)。在一些实施方案中,试剂盒包括包含fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如(ii)的化合物或其药学上可接受的盐)的容器。在其它实施方案中,试剂盒包括包含fxr激动剂(例如式(i)化合物或其药学上可接受的盐)的第一容器和包含ssao抑制剂(例如(ii)的化合物或其药学上可接受的盐)的第二容器。
[0155]
在一些实施方案中,组合物包含如本文所述的fxr激动剂和ssao抑制剂。在一些实施方案中,此类组合物包括式(i)化合物或其药学上可接受的盐,和式(ii)化合物或其药学上可接受的盐。在一些实施方案中,本文提供的剂型包含治疗有效量的式(i)化合物或其药学上可接受的盐,和治疗有效量的式(ii)化合物或其药学上可接受的盐。在一些实施方案中,式(i)化合物或其药学上可接受的盐为如本文所述的化合物1,并且式(ii)化合物或其药学上可接受的盐为化合物2。
[0156]
使用方法和用途
[0157]
本文所述的化合物和组合物在一些方面可用于治疗肝脏病症。在一些实施方案中,治疗有需要的患者的肝脏病症的方法包括向患者施用法尼醇x受体(fxr)激动剂和氨基脲敏感性胺氧化酶(ssao)抑制剂。在一些实施方案中,fxr激动剂为式(i)化合物或其药学上可接受的盐,且ssao抑制剂为式(ii)化合物或其药学上可接受的盐。在一个实施方案中,式(i)化合物或其药学上可接受的盐为如本文所述的化合物1,并且式(ii)化合物或其药学上可接受的盐为化合物2。不受理论束缚,据信根据本文所述的方法的fxr激动剂和ssao抑制剂的组合与单一疗法相比可有效地提供治疗,且因此减轻可能伴随单一疗法治疗的剂量依赖性副作用。
[0158]
肝脏病症包括但不限于肝脏炎症、纤维化和脂肪性肝炎。在一些实施方案中,肝脏病症选自肝脏炎症、肝纤维化、酒精性纤维化、脂肪变性、酒精性脂肪变性、原发性硬化性胆管炎(psc)、原发性胆汁性肝硬化(pbc)、非酒精性脂肪肝病(nafld)和非酒精性脂肪性肝炎(nash)。在某些实施方案中,肝脏病症选自:肝纤维化、酒精性纤维化、脂肪变性、酒精性脂肪变性、nafld和nash。在一个实施方案中,肝脏病症为nash。在另一实施方案中,肝脏病症为肝脏炎症。在另一实施方案中,肝脏病症为肝纤维化。在另一实施方案中,肝脏病症为酒精性纤维化。在另一实施方案中,肝脏病症为脂肪变性。在另一实施方案中,肝脏病症为酒
精性脂肪变性。在另一实施方案中,肝脏病症为nafld。在一个实施方案中,本文提供的治疗方法阻碍或减缓nafld向nash的进展。在一个实施方案中,本文提供的治疗方法阻碍或减缓nash的进展。nash可进展为例如肝硬化、肝癌等中的一者或多者。在一些实施方案中,肝脏病症为nash。在一些实施方案中,患者已进行了肝活检。在一些实施方案中,方法还包括获得肝活检的结果。
[0159]
在一些实施方案中,治疗有需要的患者的肝脏病症的方法,其中所述肝脏病症选自由以下组成的组:肝脏炎症、肝纤维化、酒精性纤维化、脂肪变性、酒精性脂肪变性、原发性硬化性胆管炎(psc)、原发性胆汁性肝硬化(pbc)、非酒精性脂肪肝病(nafld)和非酒精性脂肪性肝炎(nash)。
[0160]
本文提供了用fxr激动剂和ssao抑制剂治疗有需要的患者(例如人类患者)的肝脏病症的方法,所述方法包括施用治疗有效量的fxr激动剂和治疗有效量的ssao抑制剂,其中肝脏病症选自肝脏炎症、肝纤维化、酒精性纤维化、脂肪变性、酒精性脂肪变性、原发性硬化性胆管炎(psc)、原发性胆汁性肝硬化(pbc)、非酒精性脂肪肝病(nafld)和非酒精性脂肪性肝炎(nash)。在一些实施方案中,fxr激动剂为式(i)化合物或其药学上可接受的盐,且ssao抑制剂为式(ii)化合物或其药学上可接受的盐。在一些实施方案中,式(i)化合物或其药学上可接受的盐为如本文所述的化合物1,并且式(ii)化合物或其药学上可接受的盐为化合物2。
[0161]
本文还提供了阻止或减缓有需要的患者(例如人类患者)的非酒精性脂肪肝病(nafld)进展为非酒精性脂肪性肝炎(nash)的方法,所述方法包括施用fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)。在一些实施方案中,方法包括施用治疗有效量的式(i)化合物或其药学上可接受的盐,和治疗有效量的式(ii)化合物或其药学上可接受的盐。本文还提供了阻止或减缓有需要的患者(例如人类患者)的nash进展的方法,所述方法包括施用fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)。在一些实施方案中,方法包括施用治疗有效量的式(i)化合物或其药学上可接受的盐,和治疗有效量的式(ii)化合物或其药学上可接受的盐。
[0162]
此外,瘙痒是几种fxr激动剂有充分记录的不良反应,且可导致患者不适、患者生活质量下降和停止治疗的可能性增加。瘙痒对于适应症,例如本文所述的适应症,包括nash特别繁重,可能需要长期施用药物。式(i)化合物的组织特异性,特别是相比于皮肤组织更特异于肝脏是一个惊人且出人意料的观察结果,使得所述化合物更不可能引起皮肤瘙痒,这一理论已被迄今为止的人体试验证实。
[0163]
因此,本文提供了用fxr激动剂和ssao抑制剂治疗有需要的患者(例如人类患者)的肝脏病症的方法,其中fxr为式(i)化合物或其药学上可接受的盐,优先分布于肝组织而非肾脏、肺、心脏和皮肤中的一者或多者中。
[0164]
在一些实施方案中,施用使得式(i)化合物的肝脏浓度与血浆浓度比率为10或更大,例如11或更大、12或更大、13或更大、14或更大、或15或更大。
[0165]
在一些实施方案中,施用不会导致患者出现严重程度大于2级的瘙痒。在一些实施方案中,施用不会导致患者出现严重程度大于1级的瘙痒。在一些实施方案中,施用不会导致患者出现瘙痒。不良反应的分级是已知的。根据不良事件通用术语标准第5版(2017年11
月27日发布),1级瘙痒的特征为“轻度或局部;需要局部干预”。2级瘙痒的特征为“广泛和间歇性;抓挠引起的皮肤变化(例如水肿、丘疹、表皮脱落、苔藓样变、渗出/结痂);需要口头干预;限制工具性adl”。3级瘙痒的特征为“广泛且持续;限制自理adl或睡眠;需要全身性皮质类固醇或免疫抑制疗法”。日常生活活动(adl)分为两类:“工具性adl是指准备餐食、购买杂货或衣服、使用电话、理财等”,且“自理adl是指洗澡、穿衣和脱衣、自己吃饭、上厕所、服药,而不是卧床不起”。因此,本文提供了用fxr激动剂治疗有需要的患者(例如人类患者)的肝脏病症的方法,所述激动剂不会导致有需要的患者出现可检测的瘙痒。
[0166]
在一些实施方案中,本文提供了用fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)治疗有需要的患者的肝脏病症的方法,其中fxr激动剂不活化tgr5信号传导。在一些实施方案中,fxr调节基因的水平增加。在一些实施方案中,小异二聚体伴侣(shp)、胆盐输出泵(bsep)和成纤维细胞生长因子19(fgf19)的水平增加。
[0167]
在一些实施方案中,本文提供了一种减轻肝损伤的方法,所述方法包括向有需要的个体施用fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐),其中纤维化减轻。在一些实施方案中,一种或多种纤维化标志物的表达水平降低。在一些实施方案中,ccr2、col1a1、col1a2、col1a3、cxcr3、dcn、hgf、il1a、inhbe、lox、loxl1、loxl2、loxl3、mmp2、pdgfb、plau、serpine1、perpinh1、snai、tgfb1、tgfb3、thbs1、thbs2、timp2和/或timp3的表达水平降低。在一些实施方案中,胶原的水平降低。在一些实施方案中,胶原片段的水平降低。在一些实施方案中,纤维化标志物的表达水平降低至少2、至少3、至少4或至少5倍。在一些实施方案中,纤维化标志物的表达水平降低约2倍、约3倍、约4倍或约5倍。
[0168]
在一些实施方案中,本文提供了一种减轻肝损伤的方法,所述方法包括向有需要的个体施用fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐),其中炎症减轻。在一些实施方案中,一种或多种炎症标志物减少。在一些实施方案中,adgre1、ccr2、ccr5、il1a和/或tlr4的表达水平降低。在一些实施方案中,炎症标志物的表达水平降低至少2、至少3、至少4或至少5倍。在一些实施方案中,炎症标志物的表达水平降低约2倍、约3倍、约4倍或约5倍。
[0169]
在患者中,碱性磷酸酶、γ-谷氨酰转移酶(ggt)、丙氨酸氨基转移酶(alt)和/或天冬氨酸氨基转移酶(ast)水平可升高。在一些实施方案中,本文提供了一种减轻肝损伤的方法,所述方法包括施用fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐),其中在用fxr激动剂治疗之前,ggt、alt和/或ast水平高。在一些实施方案中,fxr激动剂为式(i)化合物或其药学上可接受的盐。在一些实施方案中,患者的alt水平比正常水平的上限高约2-4倍。在一些实施方案中,患者的ast水平比正常水平的上限高约2-4倍。在一些实施方案中,患者的ggt水平比正常水平的上限高约1.5-3倍。在一些实施方案中,患者的碱性磷酸酶水平比正常水平的上限高约1.5-3倍。确定这些分子的水平的方法是众所周知的。血液中alt的正常水平在约7-56单位/升范围内。血液中ast的正常水平在约10-40单位/升范围内。血液中ggt的正常水平在约9-48单位/升范围内。血液中碱性磷酸酶的正常水平对于20至50岁的男性在约53-128单位/升范围内且对于20至50岁的女性在约42-98单位/升范围内。
[0170]
因此,在一些实施方案中,式(i)化合物或其药学上可接受的盐降低具有高ast、alt和/或ggt水平的个体中的ast、alt和/或ggt水平。在一些实施方案中,alt水平降低至少2倍、至少3倍、至少4倍或至少5倍。在一些实施方案中,alt水平降低约2倍至约5倍。在一些实施方案中,ast水平降低至少2倍、至少3倍、至少4倍或至少5倍。在一些实施方案中,ast水平降低约1.5倍至约3倍。在一些实施方案中,ggt水平降低至少2倍、至少3倍、至少4倍或至少5倍。在一些实施方案中,ggt水平降低约1.5倍至约3倍。
[0171]
在一些实施方案中,本文提供了用fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)治疗有需要的患者的肝脏病症的方法,其中ssao抑制剂选择性抑制ssao。在一些实施方案中,ssao抑制剂为式(ii)化合物或其药学上可接受的盐。因此,在一些实施方案中,mao-a(单胺氧化酶a)未被抑制。在一些实施方案中,mao-b(单胺氧化酶b)未被抑制。在一些实施方案中,mao-a和mao-b未被抑制。
[0172]
在一些实施方案中,式(ii)化合物或其药学上可接受的盐对于ssao的ic
50
比对于mao-a和/或mao-b低至少100倍。在一些实施方案中,化合物对于ssao的ic
50
比对于mao-a和/或mao-b低至少1,000倍。在一些实施方案中,化合物对于ssao的ic
50
比对于mao-a和/或mao-b低至少10,000倍。在一些实施方案中,化合物对于ssao的ic
50
比对于mao-a和/或mao-b低100倍至10,000倍。在一些实施方案中,化合物对于ssao的ic
50
比对于mao-a或mao-b低100倍至1,000倍。在一些实施方案中,化合物对于ssao的ic
50
比对于mao-a和mao-b低至少100倍,或至少1,000倍,或至少10,000倍,或100至10,000倍,或100至1,000倍。
[0173]
在一些实施方案中,患者是人。肥胖与nafld和nash高度相关,但瘦人也可能受到nafld和nash的影响。因此,在一些实施方案中,患者肥胖。在一些实施方案中,患者不肥胖。肥胖也可能与其它疾病相关或引起其它疾病,例如糖尿病或心血管病症。因此,在一些实施方案中,患者还患有糖尿病和/或心血管病症。不受理论束缚,据信合并症,例如肥胖、糖尿病和心血管病症会使nafld和nash更难治疗。相反,目前唯一公认的解决nafld和nash的方法是减肥,这可能对瘦患者几乎没有影响。
[0174]
nafld和nash的风险随着年龄的增长而增加,但儿童也可能患有nafld和nash,有关于年仅2岁儿童的文献报道(schwimmer等人,pediatrics,2006,118:1388-1393)。在一些实施方案中,患者为2-17岁,例如2-10、2-6、2-4、4-15、4-8、6-15、6-10、8-17、8-15、8-12、10-17或13-17岁。在一些实施方案中,患者为18-64岁,例如18-55、18-40、18-30、18-26、18-21、21-64、21-55、21-40、21-30、21-26、26-64、26-55、26-40、26-30、30-64、30-55、30-40、40-64、40-55或55-64岁。在一些实施方案中,患者为65岁或以上,例如70岁或以上、80岁或以上、或90岁或以上。
[0175]
nafld和nash是肝移植的常见原因,但已经接受过一次肝移植的患者通常会再次出现nafld和/或nash。因此,在一些实施方案中,患者已进行了肝移植。
[0176]
在一些实施方案中,根据本文提供的方法的治疗使得患者的nafld活动(nas)评分降低。例如,在一些实施方案中,脂肪变性、炎症和/或鼓胀在治疗后减轻。在一些实施方案中,本文提供的治疗方法减轻肝纤维化。在一些实施方案中,所述方法减少血清甘油三酯。在一些实施方案中,所述方法减少肝脏甘油三酯。
[0177]
在一些实施方案中,患者在根据本文提供的方法的施用之前有发生不良反应的风
险。在一些实施方案中,不良反应为影响肾、肺、心脏和/或皮肤的不良反应。在一些实施方案中,不良反应为瘙痒。
[0178]
在一些实施方案中,患者已接受一种或多种先前疗法。在一些实施方案中,肝脏病症在疗法期间进展。在一些实施方案中,患者在一种或多种先前疗法中的至少一者期间罹患瘙痒。
[0179]
在一些实施方案中,本文所述的方法不包括治疗患者的瘙痒。在一些实施方案中,方法不包括施用抗组胺、免疫抑制剂、类固醇(例如皮质类固醇)、利福平、阿片类拮抗剂或选择性血清素再摄取抑制剂(ssri)。
[0180]
在一些实施方案中,fxr激动剂或ssao抑制剂或两者的治疗有效量低于在患者中诱发不良反应的水平,例如低于诱发瘙痒(例如2级或3级瘙痒)的水平。
[0181]
在一些实施方案中,fxr激动剂和ssao抑制剂同时施用。在一些此类实施方案中,fxr激动剂和ssao抑制剂可在单一药物组合物中提供。在其它实施方案中,依次施用fxr激动剂和ssao抑制剂。
[0182]
本文还提供了用于向有需要的个体施用fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)的给药方案。在一些实施方案中,fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)的治疗有效量独立地为500μg/天-600mg/天。在一些实施方案中,治疗有效量独立地为500μg/天-300mg/天。在一些实施方案中,治疗有效量独立地为500μg/天-150mg/天。在一些实施方案中,治疗有效量独立地为500μg/天-100mg/天。在一些实施方案中,治疗有效量独立地为500μg/天-20mg/天。在一些实施方案中,治疗有效量独立地为1mg/天-600mg/天。在一些实施方案中,治疗有效量独立地为1mg/天-300mg/天。在一些实施方案中,治疗有效量独立地为1mg/天-150mg/天。在一些实施方案中,治疗有效量独立地为1mg/天-100mg/天。在一些实施方案中,治疗有效量独立地为1mg/天-20mg/天。在一些实施方案中,治疗有效量独立地为5mg/天-300mg/天。在一些实施方案中,治疗有效量独立地为5mg/天-150mg/天。在一些实施方案中,治疗有效量独立地为5mg/天-100mg/天。在一些实施方案中,治疗有效量独立地为5mg/天-20mg/天。在一些实施方案中,治疗有效量独立地为5mg/天-15mg/天。在一些实施方案中,治疗有效量独立地为10mg/天-300mg/天。在一些实施方案中,治疗有效量独立地为10mg/天-150mg/天。在一些实施方案中,治疗有效量独立地为10mg/天-100mg/天。在一些实施方案中,治疗有效量独立地为10mg/天-30mg/天。在一些实施方案中,治疗有效量独立地为10mg/天-20mg/天。在一些实施方案中,治疗有效量独立地为10mg/天-15mg/天。在一些实施方案中,治疗有效量独立地为25mg/天-300mg/天。在一些实施方案中,治疗有效量独立地为25mg/天-150mg/天。在一些实施方案中,治疗有效量独立地为25mg/天-100mg/天。在一些实施方案中,治疗有效量独立地为500μg/天-5mg/天。在一些实施方案中,治疗有效量独立地为500μg/天-4mg/天。在一些实施方案中,治疗有效量独立地为5mg/天-600mg/天。在另一实施方案中,治疗有效量独立地为75mg/天-600mg/天。在一个实施方案中,式(i)化合物或其药学上可接受的盐为如本文所述的化合物1,并且式(ii)化合物或其药学上可接受的盐为化合物2。
[0183]
本文所述的化合物的剂量是基于化合物的游离碱确定。在一些实施方案中,向个体施用约1mg至约30mg fxr激动剂(例如式(i)化合物或其药学上可接受的盐)。在一些实施
方案中,向个体施用约1mg至约5mg化合物。在一些实施方案中,向个体施用约1mg至约3mg化合物。在一些实施方案中,向个体施用约5mg至约10mg化合物。在一些实施方案中,向个体施用约10mg至约15mg化合物。在一些实施方案中,向个体施用约15mg至约20mg化合物。在一些实施方案中,向个体施用约20mg至约25mg化合物。在一些实施方案中,向个体施用约25mg至约30mg化合物。在一些实施方案中,向个体施用约1mg化合物。在一些实施方案中,向个体施用约2mg化合物。在一些实施方案中,向个体施用约3mg化合物。在一些实施方案中,向个体施用约4mg化合物。在一些实施方案中,向个体施用约5mg化合物。在一些实施方案中,向个体施用约6mg化合物。在一些实施方案中,向个体施用约7mg化合物。在一些实施方案中,向个体施用约8mg化合物。在一些实施方案中,向个体施用约9mg化合物。在一些实施方案中,向个体施用约10mg化合物。在一些实施方案中,向个体施用约15mg化合物。在一些实施方案中,向个体施用约20mg化合物。在一些实施方案中,向个体施用约25mg化合物。在一些实施方案中,向个体施用约30mg化合物。在一个实施方案中,化合物为如本文所述的化合物1。
[0184]
在一些实施方案中,向个体施用约1mg至约30mg ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)。在一些实施方案中,向个体施用约1mg至约5mg化合物。在一些实施方案中,向个体施用约1mg至约3mg化合物。在一些实施方案中,向个体施用约5mg至约10mg化合物。在一些实施方案中,向个体施用约10mg至约15mg化合物。在一些实施方案中,向个体施用约15mg至约20mg化合物。在一些实施方案中,向个体施用约20mg至约25mg化合物。在一些实施方案中,向个体施用约25mg至约30mg化合物。在一些实施方案中,向个体施用约1mg化合物。在一些实施方案中,向个体施用约2mg化合物。在一些实施方案中,向个体施用约3mg化合物。在一些实施方案中,向个体施用约4mg化合物。在一些实施方案中,向个体施用约5mg化合物。在一些实施方案中,向个体施用约6mg化合物。在一些实施方案中,向个体施用约7mg化合物。在一些实施方案中,向个体施用约8mg化合物。在一些实施方案中,向个体施用约9mg化合物。在一些实施方案中,向个体施用约10mg化合物。在一些实施方案中,向个体施用约15mg化合物。在一些实施方案中,向个体施用约20mg化合物。在一些实施方案中,向个体施用约25mg化合物。在一些实施方案中,向个体施用约30mg化合物。在一个实施方案中,化合物为如本文所述的化合物2。
[0185]
治疗期通常可为一周或多周。在一些实施方案中,治疗期为至少1周、2周、3周、4周、5周、1个月、2个月、3个月、4个月、5个月、6个月、7个月、8个月、9个月、10个月、11个月、12个月、1年、2年、3年、4年或更长时间。在一些实施方案中,治疗期为约一周至约一个月、约一个月至约一年、约一年至约几年。在一些实施方案中,治疗期为至少约1周、2周、3周、4周、5周、1个月、2个月、3个月、4个月、5个月、6个月、7个月、8个月、9个月、10个月、11个月、12个月、1年、2年、3年、4年或更长时间中的任一者。在一些实施方案中,治疗期为患者的剩余寿命。
[0186]
fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如(ii)的化合物或其药学上可接受的盐)的施用可独立地为每天一次、每天两次或每隔一天,持续一周或多周的治疗期。在一些实施方案中,施用包括每天施用两种化合物持续一周或多周的治疗期。在一些实施方案中,施用包括每天两次施用两种化合物持续一周或多周的治疗期。在一些实施方案中,施用包括每隔一天施用两种化合物持续一周或多周的治疗期。
[0187]
在一些实施方案中,每天一次持续至少七天向个体施用fxr激动剂(例如式(i)化
合物或其药学上可接受的盐)和ssao抑制剂(例如(ii)的化合物或其药学上可接受的盐),其中每日量独立地在约1mg至约10mg、约1mg至约5mg或约1mg至约3mg范围内,或为约1、2、3、4、5、6、7、8、9或10mg中的任一者。在一些实施方案中,每天一次持续至少14天向个体施用两种化合物,其中每日量独立地在约1mg至约10mg、约1mg至约5mg或约1mg至约3mg范围内,或为约1、2、3、4、5、6、7、8、9或10mg中的任一者。在一些实施方案中,每天一次持续一周至四周的时间段向个体施用两种化合物,其中每日量独立地在约1mg至约10mg、约1mg至约5mg或约1mg至约3mg范围内,或为约1、2、3、4、5、6、7、8、9或10mg中的任一者。
[0188]
当与ssao抑制剂组合施用时,fxr激动剂和/或ssao抑制剂可在单独施用任一药剂时典型地施用的剂量施用。或者,由于所述组合观察到的协同作用,fxr激动剂和/或ssao抑制剂的施用剂量可低于任一药剂单独施用时的剂量。例如,在其中fxr激动剂为式(i)化合物(例如化合物1)或其药学上可接受的盐的实施方案中,式(i)化合物对人类患者的治疗剂量典型地为经口施用的每天约5mg至约15mg。因此,在特定实施方案中,当与ssao抑制剂组合施用时,式(i)化合物或其药学上可接受的盐可以约5mg至约15mg(例如5mg、6mg、7mg、8mg、9mg、10mg、11mg、12mg、13mg、14mg或15mg)的经口剂量施用或可以较低剂量施用。例如,当与ssao抑制剂组合施用时,式(i)化合物或其药学上可接受的盐可以如下剂量经口施用:每天约1mg至约15mg、每天约1mg至约4.9mg、每天约1mg至约4mg、每天约2mg至约4mg,或每天1、1.5、2、2.5、3、3.5、4、4.5、4.9、5、6、7、8、9、10、11、12、13、14或15mg中的任一者。
[0189]
在其中ssao抑制剂为式(ii)化合物(例如化合物2)或其药学上可接受的盐的实施方案中,化合物对人类患者的治疗剂量典型地为经口施用的每天约4mg至约40mg。在特定实施方案中,当与fxr激动剂组合施用时,式(ii)化合物或其药学上可接受的盐可以约4mg至约20mg(例如4mg、5mg、6mg、8mg、10mg、15mg或20mg)的经口剂量施用或可以较低剂量施用。例如,当与fxr激动剂组合施用时,式(ii)化合物或其药学上可接受的盐可以如下剂量经口施用:每天约1mg至约20mg、每天约1mg至约3.9mg、每天约1mg至约3mg、每天约1.5mg至约3.5mg、每天约2mg至约3mg,或每天1、1.5、2、2.5、3、3.5、3.6、3.8、3.9、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20中的任一者。
[0190]
在其中fxr激动剂为式(i)化合物(例如化合物1)或其药学上可接受的盐且ssao抑制剂为式(ii)化合物(例如化合物2)或其药学上可接受的盐的特定实施方案中,每种单独化合物的剂量可如上文所述地施用。例如,在一些实施方案中,式(i)化合物或其药学上可接受的盐以每天约1mg至约15mg的剂量与以每天约1mg至约20mg的剂量施用的式(ii)化合物或其药学上可接受的盐组合施用。在一些实施方案中,式(i)化合物或其药学上可接受的盐以每天约5mg至约15mg的剂量与以如下剂量施用的式(ii)化合物或其药学上可接受的盐组合施用:每天约1mg至约5mg、每天约1mg至约10mg、每天约4mg至约20mg或每天约10mg至约20mg。在一些实施方案中,式(i)化合物或其药学上可接受的盐以每天约1mg至约5mg的剂量与以如下剂量施用的式(ii)化合物或其药学上可接受的盐组合施用:每天约1mg至约5mg、每天约1mg至约10mg、每天约4mg至约20mg或每天约10mg至约20mg。
[0191]
在一些实施方案中,fxr激动剂(例如式(i)化合物或其药学上可接受的盐)的量和ssao抑制剂(例如(ii)的化合物或其药学上可接受的盐)在治疗期的第1天施用的量大于或等于在治疗期的所有后续天数施用的量。在一些实施方案中,在治疗期的第1天施用的量等于在治疗期的所有后续天数施用的量。
[0192]
根据本文所述的方法使用的式(ii)化合物或其药学上可接受的盐可在第一时间段内以每日一次的剂量施用于个体,随后是第二时间段,其中停止施用化合物,其中在第一和第二时间段期间保持ssao抑制活性。在一些实施方案中,第一和第二时间段各为一周时间段。例如,本文提供了对个体治疗14天时间段的方法,包括在前7天内向个体施用每日一次剂量的式(ii)化合物或其药学上可接受的盐,随后在接下来的7天内停止施用所述化合物,其中在整个14天时间段期间,个体中的ssao抑制活性得以维持。作为另一实例,本文提供了对个体治疗四周时间段的方法,包括在前两周内向个体施用每日一次剂量的式(ii)化合物或其药学上可接受的盐,随后在接下来的两周内停止施用所述化合物,其中在整个四周时间段期间,个体中的ssao抑制活性得以维持。在一些实施方案中,日剂量为约10mg。应理解,本文公开的剂量和给药方案也适用于使用式(ii)化合物或其药学上可接受的盐治疗nash的单一疗法。
[0193]
在一些实施方案中,施用调节以下中的一者或多者:代谢途径、胆汁分泌、视黄醇代谢、药物代谢-细胞色素p450、脂肪消化和吸收、甘油脂代谢、化学致癌、甘油磷脂代谢、尼古丁成瘾、亚油酸代谢、abc转运蛋白、细胞色素p450对异生物质的代谢、鞘脂代谢、谷胱甘肽代谢、叶酸生物合成、吗啡成瘾、鞘糖脂生物合成-乳和新乳系列、花生四烯酸代谢、酪氨酸代谢、青年成年发病型糖尿病、dna复制、胆固醇代谢、药物代谢-其它酶和醚脂质代谢。在一些实施方案中,施用调节以下中的一者或多者:代谢途径、视黄醇代谢、脂肪消化和吸收、甘油脂代谢、化学致癌、甘油磷脂代谢、abc转运蛋白、细胞色素p450对异生物质的代谢、鞘脂代谢、谷胱甘肽代谢、叶酸生物合成和吗啡成瘾。在一些实施方案中,施用调节以下一者或多者的表达:abcb4、apoa5、cyp7a1、cyp8b1、nr0b2和sic51b。
[0194]
在一些实施方案中,向有需要的个体fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)的组合施用引起基因的差异表达。在一些实施方案中,以所述组合施用引起与媒剂对照相比基因的差异表达。在一些实施方案中,以所述组合施用引起与脂质代谢和脂肪酸转运相关的基因的差异表达。与脂质代谢和/或脂肪酸转运相关的基因包括但不限于vldlr、fabp2、il1r2、vegfc、lrp2、irs2、vegfa、lrp1、irs1、ppara、slc27a1、ldlrap1、ldlr、ppargc1a、rxra、slc27a5。在一些实施方案中,与媒剂对照相比,以所述组合施用引起vldlr、fabp2、il1r2、vegfc、lrp2、irs2、vegfa、lrp1、irs1、ppara、slc27a1、ldlrap1、ldlr、ppargc1a、rxra和slc27a5中的至少1者、至少2者、至少3者、至少4者、至少5者、至少6者、至少7者、至少8者、至少9者、或至少10者的差异表达。
[0195]
在一些实施方案中,相对于媒剂对照,以所述组合施用提高一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达水平。在一些实施方案中,相对于未处理的对照,以所述组合施用使至少一种与脂质代谢和/或脂肪酸转运相关的基因的表达水平提高约1至约1.5倍、约1.5至约2倍、约2至约2.5倍、约2.5至约3倍、约3至约3.5倍、或大于约3.5倍。在一些实施方案中,以所述组合施用提高至少一种与脂质代谢和/或脂肪酸转运相关的基因的表达水平,其中所述至少一种与脂质代谢和/或脂肪酸转运相关的基因选自lrp2、irs2、vegfa、lrp1、irs1、ppara、slc27a1、ldlrap1、ldlr、ppargc1a、rxra和slc27a5。
[0196]
在一些实施方案中,以所述组合施用降低一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达水平。在一些实施方案中,相对于未处理的对照,一种或多种与脂质代
谢和/或脂肪酸转运相关的基因的表达水平降低约1至约1.5倍、约1.5至约2倍、约2至约2.5倍、约2.5至约3倍、约3至约3.5倍、或大于约3.5倍。在一些实施方案中,以所述组合施用降低至少一种与脂质代谢和/或脂肪酸转运相关的基因的表达水平,其中所述至少一种与脂质代谢和/或脂肪酸转运相关的基因选自vldlr、fabp2、il1r2和vegfc。
[0197]
因此应理解,在一些实施方案中,本文详述的治疗方法包括治疗有需要的个体的肝脏病症,例如肝脏炎症、肝纤维化、酒精性纤维化、脂肪变性、酒精性脂肪变性、原发性硬化性胆管炎(psc)、原发性胆汁性肝硬化(pbc)、非酒精性脂肪性肝病(nafld)和非酒精性脂肪性肝炎(nash),其中治疗包括降低一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达。在一些实施方案中,方法包括降低fabp2表达,尤其是肝fabp2表达。
[0198]
在一些实施方案中,与以fxr激动剂或ssao抑制剂的单一疗法施用相比,以所述组合施用引起一种或多种与脂质代谢和/或脂肪酸转运相关的基因的差异表达。因此,在此类实施方案中,fxr激动剂增强ssao抑制剂的抗脂肪变性作用。在一些实施方案中,与以fxr激动剂的单一疗法施用相比,以所述组合施用增加一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达。在一些实施方案中,与以fxr激动剂的单一疗法施用相比,以所述组合施用增加一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达,所述基因选自irs2、irs1、ppara、slc27a1、ldlrap1、ldlr、ppargc1a、rxra和slc27a5。在一些实施方案中,与以ssao抑制剂的单一疗法施用相比,以所述组合施用增加一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达,所述基因选自lrp2、irs2、vegfa、lrp1、irs1、ppara、slc27a1、ldr1、ppargc1a、rxra和slc27a5。在一些实施方案中,与以fxr激动剂的单一疗法施用相比,以所述组合施用降低一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达。在一些实施方案中,与以fxr激动剂的单一疗法施用相比,以所述组合施用降低一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达,所述基因选自vldlr、fabp2、il1r2和vegfc。在一些实施方案中,与以ssao抑制剂的单一疗法施用相比,以所述组合施用增加一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达,所述基因选自fabp2、il1r2和vegfc。
[0199]
因此,应理解,在一些实施方案中,如本文详述的用fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)的组合治疗的方法包括治疗有需要的个体的肝脏病症,例如肝脏炎症、肝纤维化、酒精性纤维化、脂肪变性、酒精性脂肪变性、原发性硬化性胆管炎(psc)、原发性胆汁性肝硬化(pbc)、非酒精性脂肪性肝病(nafld)和非酒精性脂肪性肝炎(nash),其中治疗包括差异表达与脂质代谢和/或脂肪酸转运相关的基因,例如vldlr、fabp2、il1r2、vegfc、lrp2、irs2、vegfa、lrp1、irs1、ppara、slc27a1、ldlrap1、ldlr、ppargc1a、rxra和slc27a5。在一些实施方案中,治疗包括增加一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达,所述基因选自lrp2、irs2、vegfa、lrp1、irs1、ppara、slc27a1、ldlrap1、ldlr、ppargc1a、rxra和slc27a5。在一些实施方案中,治疗包括与以fxr激动剂的单一疗法施用相比增加一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达,所述基因选自irs2、irs1、ppara、slc27a1、ldlrap1、ldlr、ppargc1a、rxra和slc27a5。在一些实施方案中,治疗包括与以ssao抑制剂的单一疗法施用相比增加一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达,所述基因选自lrp2、irs2、vegfa、lrp1、irs1、ppara、slc27a1、ldr1、ppargc1a、rxra和slc27a5。在一些实施方案中,治疗包括与以fxr激动剂的单一疗法施用相比降低一种或多种与脂质代谢和/或
脂肪酸转运相关的基因的表达,所述基因选自vldlr、fabp2、il1r2和vegfc。在一些实施方案中,治疗包括与以ssao抑制剂的单一疗法施用相比降低一种或多种与脂质代谢和/或脂肪酸转运相关的基因的表达,所述基因选自fabp2、il1r2和vegfc。
[0200]
应理解,本文所述的任何基因(例如fabp2)的引用包括对来自所有物种(包括人类和啮齿动物)的直系同源物的引用。
[0201]
在某些实施方案中,本文详述的治疗方法包括用fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如(ii)的化合物或其药学上可接受的盐)以约3单位的fxr激动剂对约25单位的ssao抑制剂的重量比治疗有需要的个体。
[0202]
本文还提供了使用本文所述的方法治疗有需要的个体的肝脏病症,例如肝脏炎症、肝纤维化、酒精性纤维化、脂肪变性、酒精性脂肪变性、原发性硬化性胆管炎(psc)、原发性胆汁性肝硬化(pbc)、非酒精性脂肪肝病(nafld)和非酒精性脂肪性肝炎(nash)的fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)的组合。
[0203]
本文还提供了fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)的组合的用途,其用于制造使用本文所述的方法治疗有需要的个体的肝脏病症,例如肝脏炎症、肝纤维化、酒精性纤维化、脂肪变性、酒精性脂肪变性、原发性硬化性胆管炎(psc)、原发性胆汁性肝硬化(pbc)、非酒精性脂肪肝病(nafld)和非酒精性脂肪性肝炎(nash)的药剂。
[0204]
制品和试剂盒
[0205]
本公开进一步提供了在合适的包装中包含本文所述的化合物或其盐、本文所述的组合物或本文所述的一个或多个单位剂量的制品。在某些实施方案中,制品用于本文所述的任何方法中。合适的包装(例如容器)是本领域已知的且包括例如小瓶、器皿、安瓿、瓶子、广口瓶、软包装等。制品可进一步被灭菌和/或密封。
[0206]
本公开进一步提供了用于实施本公开的方法的试剂盒,所述试剂盒包含至少两种本文所述的化合物或其药学上可接受的盐,或包含本文所述的化合物或其药学上可接受的盐的组合物。试剂盒可使用本文公开的任何化合物或其药学上可接受的盐。在一些实施方案中,试剂盒使用本文所述的fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如式(ii)化合物或其药学上可接受的盐)。所述试剂盒可用于本文所述的任何一种或多种用途,并且因此可含有本文所述的治疗说明书。
[0207]
试剂盒通常包括合适的包装。试剂盒可包含一个或多个容器,所述容器包含本文所述的任何化合物或其药学上可接受的盐。每种组分可包装在单独的容器中,或者在交叉反应性和存放期允许的情况下,一些组分可组合于一个容器中。在一些实施方案中,试剂盒包括包含fxr激动剂(例如式(i)化合物或其药学上可接受的盐)和ssao抑制剂(例如(ii)的化合物或其药学上可接受的盐)的容器。在其它实施方案中,试剂盒包括包含fxr激动剂(例如式(i)化合物或其药学上可接受的盐)的第一容器和包含ssao抑制剂(例如(ii)的化合物或其药学上可接受的盐)的第二容器。
[0208]
试剂盒可为单位剂型、散装包装(例如多剂量包装)或亚单位剂量。例如,可提供试剂盒,其含有足够剂量的本文公开的化合物或其药学上可接受的盐,和/或对本文详述的疾病有用的额外药学活性化合物,以在延长的时间段,例如一周、2周、3周、4周、6周、8周、3个
月、4个月、5个月、7个月、8个月、9个月或更长时间中的任一者内对个体提供有效治疗。试剂盒还可包括多个单位剂量的化合物和使用说明书,并且包装的数量足以在药房(例如医院药房和复方药房)中储存和使用。
[0209]
试剂盒可任选地包括关于本公开的方法的组分的使用的一组说明书,通常是书面说明书,尽管含有说明书的电子存储介质(例如磁盘或光盘)也是可接受的。试剂盒随附的说明书通常包括有关组分以及其对个体的施用的信息。
[0210]
列举的实施方案
[0211]
实施方案1.一种治疗有需要的患者的肝脏病症的方法,所述方法包括向所述患者施用法尼醇x受体(fxr)激动剂和氨基脲敏感性胺氧化酶(ssao)抑制剂,其中所述肝脏病症选自由以下组成的组:肝脏炎症、肝纤维化、酒精性纤维化、脂肪变性、酒精性脂肪变性、原发性硬化性胆管炎(psc)、原发性胆汁性肝硬化(pbc)、非酒精性脂肪肝病(nafld)和非酒精性脂肪性肝炎(nash)。
[0212]
实施方案2.如实施方案1的方法,其中fxr激动剂为奥贝胆酸、西罗非索、托匹非索、eyp001(沃纳非索,建议的inn)、met409(metacrine)或edp-305(enanta)。
[0213]
实施方案3.如实施方案1或2的方法,其中ssao抑制剂为pxs-4728a(bi-1467335)。
[0214]
实施方案4.如实施方案1的方法,其中所述fxr激动剂为式(i)化合物
[0215][0216]
其中:
[0217]
q为1或2;
[0218]
r1为氯、氟或三氟甲氧基;
[0219]
r2为氢、氯、氟或三氟甲氧基;
[0220]r3a
为三氟甲基、环丙基或异丙基;
[0221]
x为ch或n,
[0222]
条件是当x为ch时,q为1;并且
[0223]
ar1为吲哚基、苯并噻吩基、萘基、苯基、苯并异噻唑基、吲唑基或吡啶基,它们各自任选地被甲基或苯基取代,
[0224]
或其药学上可接受的盐。
[0225]
实施方案5.如实施方案4的方法,其中:
[0226]
r1为氯或三氟甲氧基;并且
[0227]
r2为氢或氯。
[0228]
实施方案6.如实施方案4或5的方法,其中:
[0229]r3a
为环丙基或异丙基。
[0230]
实施方案7.如实施方案4至6中任一项的方法,其中:
[0231]
ar1为5-苯并噻吩基、6-苯并噻吩基、5-吲哚基、6-吲哚基或4-苯基,它们各自任选地被甲基取代。
[0232]
实施方案8.如实施方案4至7中任一项的方法,其中:
[0233]
q为1;并且
[0234]
x为n。
[0235]
实施方案9.如实施方案1或4的方法,其中所述fxr激动剂为:
[0236][0237]
或其药学上可接受的盐。
[0238]
实施方案10.如实施方案1、2和4至9中任一项的方法,其中所述ssao抑制剂为式(ii)化合物
[0239][0240]
其中:
[0241]
n为1或2;并且
[0242]
r1为h或-ch3,
[0243]
或其药学上可接受的盐。
[0244]
实施方案11.如实施方案10的方法,其中ssao抑制剂为式(iia)化合物
[0245][0246]
其中:
[0247]
n为1或2;并且
[0248]
r1为h或-ch3,
[0249]
或其药学上可接受的盐。
[0250]
实施方案12.如实施方案10或11的方法,其中n为2。
[0251]
实施方案13.如实施方案10至12中任一项的方法,其中r1为ch3。
[0252]
实施方案14.如实施方案1、2和4至9中任一项的方法,其中所述ssao抑制剂为:
[0253][0254]
或其药学上可接受的盐。
[0255]
实施方案15.如实施方案1至14中任一项的方法,其中同时施用所述fxr激动剂和所述ssao抑制剂。
[0256]
实施方案16.如实施方案1至14中任一项的方法,其中依序施用所述fxr激动剂和所述ssao抑制剂。
[0257]
实施方案17.如实施方案1至16中任一项的方法,其中所述施用不会导致患者出现严重程度为2级或以上的瘙痒。
[0258]
实施方案18.如实施方案1至17中任一项的方法,其中所述施用不会导致患者出现严重程度为1级或以上的瘙痒。
[0259]
实施方案19.如实施方案1至18中任一项的方法,其中所述施用不会导致患者出现瘙痒。
[0260]
实施方案20.如实施方案1至19中任一项的方法,其中所述患者还患有糖尿病和/或心血管病症。
[0261]
实施方案21.如实施方案1至20中任一项的方法,其中治疗期为患者的剩余寿命。
[0262]
实施方案22.如实施方案1至21中任一项的方法,其中方法不包括施用抗组胺、免疫抑制剂、类固醇、利福平、阿片类拮抗剂或选择性血清素再摄取抑制剂(ssri)。
[0263]
实施方案23.如实施方案1至22中任一项的方法,其中所述fxr激动剂每天施用一
次或每天施用两次。
[0264]
实施方案24.如实施方案1至23中任一项的方法,其中所述ssao抑制剂每天施用一次或每天施用两次。
[0265]
实施方案25.如实施方案1至24中任一项的方法,其中所述施用包括每天施用所述fxr激动剂持续一周或多周的治疗期。
[0266]
实施方案26.如实施方案1至25中任一项的方法,其中所述施用包括每天施用所述ssao抑制剂持续一周或多周的治疗期。
[0267]
实施方案27.如实施方案1至26中任一项的方法,其中所述肝脏病症选自由非酒精性脂肪肝病(nafld)和非酒精性脂肪性肝炎(nash)组成的组。
[0268]
实施方案28.如实施方案1至26中任一项的方法,其中所述肝脏病症为非酒精性脂肪性肝炎。
[0269]
实施方案29.一种药物组合物,所述药物组合物包含有效量的fxr激动剂、治疗有效量的ssao抑制剂以及药学上可接受的载体、稀释剂、赋形剂或前述任一者的组合。
[0270]
实施方案30.一种剂型,所述剂型包含治疗有效量的fxr激动剂和治疗有效量的ssao抑制剂。
[0271]
实施方案31.一种试剂盒,所述试剂盒包括包含fxr激动剂和ssao抑制剂的容器。
[0272]
实施方案32.一种试剂盒,所述试剂盒包括包含fxr激动剂的第一容器和包含ssao抑制剂的第二容器。
[0273]
实施方案33.如实施方案29的药物组合物、如实施方案30的剂型、如实施方案31或32的试剂盒,其中所述fxr激动剂为
[0274][0275]
或其药学上可接受的盐,并且所述ssao抑制剂为:
[0276][0277]
或其药学上可接受的盐。
[0278]
实施例
[0279]
实施例1:体外代谢稳定性
[0280]
在冷冻保存的肝细胞中评估化合物1的肝代谢率以确定化合物的体外半衰期。将1μm化合物1与预处理的小鼠、大鼠、狗、猴子或人肝细胞混合(0.5
×
106个细胞/ml)并允许在
37℃下培育2小时,在几个时间点收集样品并且分析化合物1。使用未校正血浆蛋白的充分搅拌的肝脏模型,确定并调整了体外半衰期值以预测肝脏清除率(cl
pred
)和肝脏提取,如obach等人,the prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data,j.of pharmacology and experimental therapeutics,第283卷,第1期,第46-58页(1997)中所述。结果显示在表1中,其表明化合物1在所有测试物种的肝细胞中被适度代谢。
[0281]
表1.化合物1的体外代谢稳定性
[0282][0283]
实施例2:体外oatp转运分析
[0284]
使用在可渗透支持物上生长的极化单层mdck-ii细胞来测试有机阴离子转运多肽(oatp)1b1或oatp 1b3将化合物1转运穿过脂质双层且进入细胞的能力。mdck-ii细胞转染以下之一:(1)表达oatp 1b1的载体,(2)表达oatp 1b3的载体,或(3)对照载体。在细胞中诱导表达,随后在37℃下在5% co2气氛中培养细胞。诱导表达后,用1μm、3μm和10μm化合物1或3μm化合物1和100μm利福平处理细胞。接着测量化合物1的细胞摄取。此实验的结果表明,化合物1不是oatp 1b1或oatp 1b3底物。
[0285]
实施例3:药物动力学分析
[0286]
化合物1以1mg/kg(n=3)静脉内或以10mg/kg(n=3)经口施用于斯普拉格-杜勒(sd)大鼠,以1mg/kg(n=3)静脉内或以3mg/kg(n=3)经口施用于比格犬,以0.3mg/kg(n=6)静脉内或以5mg/kg(n=6)经口施用于食蟹猴,且以5mg/kg(n=9)经口施用于小鼠。用于sd大鼠经口施用的化合物1在含有10% dmso、10% cremophor-el和80%水溶液(10%2-羟丙基-β-环糊精)的媒剂中配制。用于比格犬经口施用的化合物1用含有1%羧甲基纤维素、0.25% tween-80和0.05%消泡剂的水溶液配制。用于食蟹猴经口施用的化合物1用10% solutol、20%peg400、0.5% tween-80和69.5%去离子水配制。收集系列血样,且测量化合物1的血浆浓度。结果示于图1a(iv施用)和图1b(经口施用)以及表2中。结果表明化合物1在体内具有低至中等清除率。大鼠和狗中化合物1的分布体积(v
dss
)大于全身水的体积(0.70l/kg)。猴子中较小的v
dss
与较高的血浆蛋白结合相关。
[0287]
表2.化合物1的药物动力学参数
[0288][0289]
实施例4:化合物1的组织分布
[0290]
确定向大鼠施用的化合物1的组织分布,且与其它法尼醇x受体(fxr)激动剂西罗非索、托匹非索和奥贝胆酸(oca)的分布进行比较。将测试化合物通过30分钟静脉内输注以2mg/kg施用于sd大鼠(每种化合物的n=3)。从大鼠收集血液、肝脏、肾脏和肺组织样品以确定组织/血浆比率。化合物的肝组织/血浆比率显示于图2a中,其表明与其它测试化合物相比,显著更多的化合物1定位于肝组织。化合物1与100μm利福平的共同施用不会导致化合物1在肝脏中的分布发生显著变化(图2b)。这些结果共同表明,化合物1优先分布于肝脏,且在啮齿动物物种中表现出高肝脏/血浆比率,比正在研究的用于治疗nash的其它fxr激动剂(西罗非索、托匹非索和oca)高约3至20倍。
[0291]
放射性标记的(
14
c)化合物1也以5mg/kg(100μci/kg)的经口剂量施用于朗-埃文斯大鼠。收集血浆、肝脏、小肠、盲肠、肾脏、肺、心脏和皮肤组织样品长达168小时,且测量不同时间点的放射性物质的量。结果示于图3中。肝脏、小肠和盲肠的放射性物质最多。
[0292]
实施例5:化合物1的代谢
[0293]
放射性标记的(
14
c)化合物1以5mg/kg经口或以2mg/kg静脉施用于胆管完整或插管的sd大鼠(四个群组中的每一者的n=3),总放射性剂量为100μci/kg。从每只大鼠收集血液、胆汁、粪便和尿液样品长达168小时。化合物1在胆汁排泄之前被代谢为酰基葡糖苷酸代谢物,这被确定为所述化合物的主要消除途径。
[0294]
实施例6:药物动力学/药效学概况
[0295]
食蟹猴的药物动力学/药效学(pk/pd)概况通过以0(媒剂)、0.3、1或5mg/kg的剂量施用化合物1悬浮液的经口剂量,且收集血样长达24小时来确定。药效学作为如通过lc-ms/ms所定量的7-α-羟基-4-胆甾烯-3-酮(7ac4)减少的函数进行测量(图4)。药物动力学数据列于表3中,且通过非隔室分析确定。
[0296]
表3.化合物1的药物动力学参数
[0297]
[0298]
化合物1也以1mg/kg的剂量连续7天经口施用于食蟹猴(n=6),以确定多个剂量后的pk/pd概况。此研究的结果显示于图5a(pk概况)和图5b(pd概况)以及表4中,且证明化合物1的血浆暴露在第1天和第7天相当,且在重复经口给药后实现了药效学生物标志物7ac4的持续抑制。
[0299]
表4.化合物1的药物动力学参数
[0300][0301]
实施例7:作用机制
[0302]
向c57bl/6小鼠施用单次经口剂量的10mg/kg化合物1(n=6)、30mg/kg oca(n=6)或媒剂对照(n=6),且在剂量施用后6小时收集组织rna样品。通过rt-qpcr和rnaseq来分析rna。
[0303]
对于rt-qpcr,基因特异性引物用于使用2-ddct方法定量肝脏和回肠中的fxr调节基因表达。结果示于图6中(数据呈现为平均值
±
sem;****表示p《0.0001且*表示p《0.05(与媒剂相比),统计数据由单向方差分析后跟图基检验(tukey)确定)。此数据表明化合物1优先在小鼠肝脏中诱导fxr特异性基因。
[0304]
对于rnaseq分析,从总肝脏中提取mrna且使用标准illumina文库制备和定序方案进行定序。使用rsem和edger软件包确定差异表达基因(deg),且使用advaita bio的ipathwayguide软件进行分析。结果示于图7a-7d中,其表明与oca相比,化合物1调节显著更多数量的与nash相关的基因和代谢途径。图7a显示,相对于媒剂对照,施用化合物1调节500种nash相关基因的表达,oca调节44种nash相关基因的表达,包括由化合物1和oca两者调节的37种共同nas相关基因(倍数变化≥1.5;q值《0.05)。图7b显示了在媒剂、oca和化合物1治疗的小鼠中所选fxr相关基因的平均表达水平(如cpm值所示)。图7c显示化合物1的施用引起32条全局途径的富集且oca的施用引起6条全局途径的富集,包括化合物1和oca施用两者的2条共同全局途径。图7d显示了施用化合物1后统计学上最富集的25条途径,且将这些途径的富集与施用oca后的富集进行了比较。总体而言,与oca治疗相比,用化合物1治疗的小鼠肝脏的rnaseq分析显示与非酒精性脂肪肝病相关的fxr相关基因和代谢途径的调节更为稳健。
[0305]
实施例8:临床研究
[0306]
第一研究。健康人类志愿者受试者每天经口给药5mg(n=9)、75mg(n=9)、200mg或400mg(n=18)化合物1,或接受安慰剂(n=12),持续14天。在此研究期间,没有观察到瘙痒的发生。
[0307]
第二研究。化合物1每天以25mg(n=11)、75mg(n=10)或150mg(n=10)的经口剂量施用于人类受试者,持续7天,或人类受试者接受安慰剂(n=5)。定期测量患者的7-α-羟基-4-胆甾烯-3-酮(7ac4)水平,如表5中所示,其表明化合物1抑制了所述水平。在独立小组发
表的另一研究中,据报道,每天以20mg、40mg、50mg、80mg、100mg或150mg的剂量向健康人类志愿者施用fxr激动剂met409(metacrine),且7ac4水平的测量结果如表5中所示。参见chen等人,met409,an optimized sustained fxr agonist,was safe and well-tolerated in a 14-day phase 1 study in healthy subjects,the international liver congress,vienna,austria,2019年4月10-14日。虽然在接受100mg或更大剂量的met409的受试者中观察到瘙痒,但在服用最高剂量的化合物1的受试者中未观察到瘙痒。已知其它fxr激动剂,例如西罗非索、托匹非索、oca、edp-305(enanta)在长期研究中都会导致瘙痒。
[0308]
表5.met409和化合物1的比较
[0309][0310]
实施例9:nash小鼠模型
[0311]
使用小鼠模型评估化合物1对nash的影响,其中nash由高脂肪饮食与ccl4施用的组合诱导。
[0312]
向小鼠c57/bl6j小鼠喂食高脂肪饮食(d12492,研究饮食,脂肪/蛋白质/碳水化合物60/20/20kcal%,10w)以诱导肥胖(》36g小鼠),然后每日口服化合物1以及每两周进行一次腹膜内四氯化碳(ccl4)治疗,持续四周。图8。化合物1以10、30和100mg/kg的剂量施用。
[0313]
在化合物1给药28天后,分析血清脂质、血清氨基转移酶和肝脂质。肝组织的苏木精和伊红(h&e)以及天狼星红组织学染色用于定量nafld活动评分(nas)、脂肪变性、鼓胀、炎症和纤维化。测量血浆7-α-羟基-4-胆甾烯-3-酮(7ac4)作为fxr活化的生物标志物。通过rt-qpcr和rnaseq来分析rna的基因表达。
[0314]
非酒精性脂肪肝疾病活动评分(nas)是用于评估nash的综合评分。nas是根据肝脏脂肪变性、炎症和鼓胀来计算的,并通过使用h&e染色的肝组织组织学分析来确定。具体而言,根据h&e染色计算炎症评分:0分,无;1分,每200x视野《2个焦点;2分,每200x视野2-4个焦点;3分,每200x视野》4个焦点。脂肪变性评分通过h&e染色计算如下:0分,《5%;1分,5-33%;2分,》33-66%;3分,》66%)。肝细胞鼓胀是一种与细胞肿胀相关的肝细胞损伤形式,并且也可通过h&e染色的肝切片测量。鼓胀评分计算如下:0-没有肝细胞鼓胀;1-几个鼓胀肝细胞;2-许多具有明显鼓胀的肝细胞。
[0315]
如图9中所示,与未治疗的nash小鼠相比,用10、30或100mg/kg化合物1治疗的小鼠具有显著较低的nas评分。与未治疗的nash小鼠相比,用化合物1治疗也显著减轻了脂肪变性、炎症和鼓胀。图10a-c。
[0316]
通过天狼星红阳性肝切片百分比的组织学分析来定量肝纤维化。图11a显示了健康小鼠、nash小鼠和用100mg/kg化合物1治疗的nash小鼠的代表性组织学。图11b显示了用化合物1治疗的小鼠的纤维化面积的定量。与未治疗的nash对照相比,用10、30或100mg/kg化合物1治疗导致纤维化显著减轻。如图14a中所示,与对照nash小鼠相比,以10、30或100mg/kg施用的化合物1导致肝脏中1型胶原α1表达降低。
[0317]
治疗后,分析血清的丙氨酸氨基转移酶(alt)、天冬氨酸氨基转移酶(ast)、甘油三酯和总胆固醇水平。如图12a和图12b中所示,用化合物1治疗的小鼠中的血清alt和ast水平降低。图12c显示在用100mg/kg化合物1治疗的小鼠中血清甘油三酯浓度的统计显著降低。图12d显示在用10、30和100mg/kg化合物1治疗的小鼠中总胆固醇水平的统计显著降低。
[0318]
使用生化分析仪(hitachi-700)从肝组织测量肝甘油三酯。图13a显示了对照小鼠或用10、30或100mg/kg化合物1治疗的小鼠的肝脏甘油三酯浓度。用100mg/kg化合物1治疗的小鼠显示出统计显著降低的甘油三酯水平。图13b显示了代表性组织学切片。
[0319]
使用肝脏样品的rt-qpcr或rna-seq分析化合物1对基因表达的影响(图14a-c和表6)。表6显示了化合物1对肝脏中的fxr调节基因表达的影响。用化合物1治疗后每个指定基因的表达水平(如通过每百万基因计数(cpm)值定义)除以媒剂治疗的动物中所述基因的表达水平以确定化合物1相对于媒剂的活性。
[0320]
表6.fxr-靶标、炎症和纤维化基因的表达
[0321]
基因相对于媒剂的化合物1(30mg/kg)shp4.6bsep5.1ost-b135.7cyp7a10.02cyp8b10.007
[0322]
化合物1对于fxr的ec
50
浓度通过基于荧光的fxr共活化分析来确定。将化合物1或oca(奥贝胆酸,一种已知的fxr激动剂)(10μm-3nm)的半对数连续稀释液与sf9昆虫细胞中产生的人fxr配体结合域、标记的共活化因子src-1肽和tr-fret共调节缓冲液g一起在25℃下培育1小时。使用基于细胞的camp分析来测量tgr5活性。参见kawamata等人jbc 278(11)935-440(2003)。将化合物1或oca(10μm-3nm)的半对数连续稀释液添加至表达重组人tgr5的中国仓鼠卵巢细胞中。在室温下30分钟后,使用htrf读出器测量camp。使用基于细胞的rna分析来确定fxr调节基因表达的ec
50
值。将化合物1或oca(3μm-3nm)的半对数连续稀释液添加至人huh7肝癌细胞中。在37℃下11小时后,分离rna且使用以下fxr相关基因的引物通过rt-qpcr进行分析:小异二聚体伴侣(shp)、胆盐输出泵(bsep)和成纤维细胞生长因子19(fgf-19)。
[0323]
如表7中所示,化合物1是一种有效的选择性fxr激动剂。
[0324]
表7.化合物1的ec
50
[0325][0326]
总之,化合物1是一种有效的选择性fxr激动剂。在nash小鼠模型中,化合物1降低了炎症和纤维化相关基因的表达,并强烈抑制了肝脏脂肪变性、炎症、鼓胀和纤维化。
[0327]
实施例10
[0328]
背景
[0329]
氨基脲敏感性胺氧化酶(ssao)通过将伯胺(例如甲胺、mma)脱氨为醛、铵和h2o2来增加氧化应激,并通过将炎症细胞募集至肝脏,加剧肝脏炎症和损伤来促进非酒精性脂肪性肝炎(nash)。ssao水平在nash中升高并且与纤维化阶段相关。化合物2是一种选择性的共价ssao抑制剂,其减轻nash大鼠模型中的肝脏炎症和纤维化。进行了化合物2的单次递增剂量临床试验。
[0330]
本文所述的化合物可通过wo 2018/028517中所述的方法获得,该案以引用方式整体并入本文中,并且特别是关于制备本文详述的化合物的方法。
[0331]
方法
[0332]
将四组各8名健康参与者随机分至接受3:1比率的化合物2胶囊或匹配安慰剂。在给药前和给药后的不同时间点测定化合物2和pd生物标志物的血浆水平。ssao抑制是通过测量添加外源性底物(苄胺)后血浆h2o2生成的相对减少来确定的。测量了血浆中的内源性甲胺(mma)水平,预计会在ssao抑制后增加。给药后7(
±
3)天评估安全性。
[0333]
在施用单次剂量的研究药物(安慰剂或化合物)后0.25、0.5、1、2、3、4、6、8、10、12、24、48(仅ssao活性)和168(仅ssao活性)小时收集用于化合物2浓度和ssao活性测定的血浆样品。血浆pk参数通过非房室分析来确定。ssao活性通过测量来自安慰剂和活性化合物2受体的血浆样品中的过氧化氢(h2o2)生成水平来评估。相对于相应的给药前(基线)样品确定总胺氧化酶活性的百分比变化。
[0334]
血浆中的ssao特异性胺氧化酶水平使用基本上如先前(schilter等人)所述的基于动力学的分析来确定。在测量安慰剂和活性受体中的h2o2生成水平之前,通过向血浆样品中添加帕吉林(pargyline)来抑制内源性单胺氧化酶a和b。最大抑制由另外用高剂量化合物2处理的给药前(基线)样品定义,并且计算相对于基线样品的ssao特异性活性的百分比变化。
[0335]
结果
[0336]
招募了32名健康人类参与者(100%男性、63%黑人、19%亚裔、13%白人)并接受单次口服剂量的化合物2(1、3、6和10mg,各n=6)或安慰剂(n=2)。化合物2血浆pk暴露在3
与10mg剂量水平之间以大于剂量比例的方式增加。化合物2的平均半衰期在1
–
3小时范围内。在给药后4小时,在所有剂量群组中均观察到血浆ssao活性几乎完全受到抑制,并且在单次剂量的化合物2后检测到长达1周的持续抑制。最大血浆mma水平随着化合物2的增加而增加。未报告临床相关不良事件或实验室异常。
[0337]
如表8中所示,1、3、6和10mg剂量的化合物2都具有良好耐受性。
[0338]
表8治疗相关不良事件
[0339][0340]
单次剂量的化合物2的甲苯磺酸盐迅速从血浆中清除,并且表现出3与10mg之间的大于剂量比例的血浆pk。
[0341]
单次剂量的化合物2快速并且有效地降低了所有受试者的血浆胺氧化酶活性,如图15a和图15b中所示。在给药后4小时观察到ssao特异性活性接近完全抑制。图15a和图15b。血浆ssao胺氧化酶活性的抑制和血浆mma的剂量依赖性增加在单次剂量的化合物2后持续长达1周,表明有效的共价靶标参与并且支持每日一次给药,尽管血浆半衰期短。图15a和图15b。
[0342]
在所有剂量水平下,化合物2的浓度(c
最大
)比mao-a和mao
–
b的ic
50
浓度低800多倍。图15c。
[0343]
表9
–
生化活性(ic
50
μm)
[0344][0345]
观察到甲胺的剂量依赖性增加,表明在整个剂量范围内有效的血浆ssao靶标参与。图15d。
[0346]
结论
[0347]
化合物2在施用1mg至10mg范围内的单次口服剂量的健康受试者中安全且耐受性良好。化合物2在单次剂量后抑制ssao活性长达七天。这表明化合物2可通过选择性抑制ssao来有效治疗肝脏疾病或病症。它也可能在仅单次剂量后表现出持续七天的ssao活性,表明每天施用持续一周可在两周时间段内发挥治疗效果。
[0348]
实施例11
[0349]
动物处理:到达后,给大鼠2周的适应期,在此期间,它们习惯了动物设施的工作人员,并接受了经口管饲程序的训练。2周后,给动物喂食chdfd(缺乏胆碱的高脂肪饮食)并且预喂4周。接着大鼠开始用测试化合物进行治疗,并且每周3次进行腹膜内nano2注射,同时继续给它们喂食cdhfd,再持续8周。在喂食cdhfd时,在8周内每周3次(周一、周三和周五)腹膜内施用25mg/kg溶解在pbs中的nano2。
[0350]
最后处死:每个治疗组的一半动物在第84天被处死。每组的另一半动物在第二天,即第85天被处死。处死当天,动物禁食2小时,并且接受相应测试物质的最后治疗。在最后的化合物治疗后,动物在处死之前无法再获得食物。在最后一次施用后4小时,处死所有动物并且取肝样品用于进一步分析。
[0351]
结果:缺乏胆碱的高脂肪饮食(cdhfd)通常用于在啮齿类动物中诱导nash样表型。另外,在cdhfd大鼠中通过腹膜内(ip)注射亚硝酸钠(nano2)诱导肝纤维化可用于模拟晚期nash疾病。因此,大鼠cdhfd+nano
2 nash模型用于测试化合物1单独和与化合物2组合的功效。在此模型中,雄性威斯塔(wistar)大鼠被喂食cdhfd 4周以在每天口服药物和每三周ip nano2治疗之前诱发疾病。在化合物1(3mg/kg)和化合物2(25mg/kg)作为单一药剂或以组合给药8周后,肝组织经过处理用于通过rnaseq进行全转录组分析,以寻找与疾病消退相关的基因表达变化。在nash中,消退是一个复杂的过程,涉及免疫系统特化细胞(包括调节性t细胞(treg)和m2巨噬细胞)的肝脏浸润。treg和m2巨噬细胞参与免疫抑制和减轻炎症,并且似乎在包括nash在内的肝损伤动物模型中发挥有益作用。为了寻找这些细胞的存在,我们利用rnaseq表达数据来使用细胞类型特异性基因表达特征进行单样本基因集富集分析(ssgsea),以定量treg和m2巨噬细胞浸润至肝脏中的相对水平(图16)。相对于媒剂治疗的nash对照动物,化合物1(3mg/kg)和化合物2(25mg/kg)的组合显示treg和m2巨噬细胞的评
分显著更高。相反,单一药剂治疗的动物与对照没有显著差异。这些结果通过分析包括foxp3(treg)、ikzf2(treg)和cd163(m2巨噬细胞)在内的treg和m2巨噬细胞的个体标志物得到验证(图17)。只有化合物1(3mg/kg)和化合物2(25mg/kg)显示出与treg和m2巨噬细胞相关的标志物的显著更高表达。综合这些数据表明,fxr激动剂化合物1和ssao抑制剂化合物2的组合使得肝脏中与nash消退相关的免疫细胞标志物表达增加。考虑到它们不同的作用机制,化合物1和化合物2在组合使用以加速nash消退过程时可提供互补的益处。
[0352]
这些结果表明,fxr激动剂和ssao抑制剂的组合经组合而具有比单独施用的两种药物中的任一种更大的效果。
[0353]
实施例12
[0354]
将3组各8名健康参与者随机分至在7天(1mg和4mg)或14天(10mg)内接受多次每日一次(qd)剂量的3:1比率的化合物2或匹配安慰剂。在给药前和给药后的不同时间点测定化合物2和pd生物标志物的血浆水平(血浆胺氧化酶活性和甲胺水平)。在最后一次给药后至多14天内评估安全性。
[0355]
没有报告临床相关的不良事件或实验室异常。在第1天,化合物2血浆pk暴露增加大于剂量组之间的剂量比例,并且在多次qd剂量后观察到每个剂量水平的显著积累。随着剂量的增加,给药第一天与最后一天之间的积累率降低。7天后,最高剂量群组(10mg)达到稳态。化合物2的半衰期随剂量而增加,这与可饱和靶标介导的清除率一致。在所有剂量群组中,第1天观察到血浆ssao活性几乎完全受到抑制,并且在10mg群组最后一次给药后检测到长达2周的持续抑制。血浆甲胺水平以大于剂量比例的方式增加。
[0356]
当施用至多10mg qd持续14天时,化合物2在健康受试者中安全且耐受性良好。在支持qd给药方案的给药7天后达到化合物2的稳态水平。血浆ssao胺氧化酶活性的几乎完全抑制和血浆mma的剂量依赖性增加在停止给药后持续长达2周,表明在两周内每天施用化合物2可在停止给药后的两周时间段内发挥治疗效果。
[0357]
实施例13
[0358]
进行了一项研究,以显示在nash大鼠模型中组合fxr激动剂与ssao抑制剂的有益效果。
[0359]
动物处理:到达后,给大鼠2周的适应期,在此期间,它们习惯了动物设施的工作人员,并接受了经口管饲程序的训练。2周后,给动物喂食缺乏胆碱的高脂肪饮食(cdhfd)并预喂4周以诱导脂肪变性和nash样疾病表型。接着在喂食cdhfd时用测试化合物再治疗大鼠8周。在化合物治疗的同时,通过每三周一次的腹膜内(ip)注射向大鼠施用亚硝酸钠(nano2,25mg/kg溶解于pbs中)以诱导肝纤维化。
[0360]
最后处死:每个治疗组的一半动物在第84天被处死。每组的另一半动物在第二天,即第85天被处死。处死当天,动物禁食2小时,并且接受相应测试物质的最后治疗。在最后的化合物治疗后,动物在处死之前无法再获得食物。在最后一次施用后4小时,处死所有动物并且取肝样品用于进一步分析。
[0361]
取样和分析:将小肝片采集至rnalater(thermo fisher scientific dreieich germany)中,并在medgenome inc.进行rna定序(rnaseq)之前储存在-20℃下。使用标准方法通过illumina定序对肝组织进行rnaseq分析。简而言之,rnaseq文库(n=5每组)使用illumina truseq链式mrna试剂盒生成,并在novaseq 6000定序仪上进行定序。使用star
(v2.7.3a)比对器进行比对,并且在比对前移除核糖体和线粒体基因组的读段映射。原始读取计数使用htseq(v0.11.1)进行估计,并使用deseq2(v2.22.2)进行归一化。使用deseq2(r bioconductor包)确定差异表达基因(deg)。
[0362]
结果:缺乏胆碱的高脂肪饮食(cdhfd)通常用于在啮齿类动物中诱导nash样表型。另外,在cdhfd大鼠中通过腹膜内(ip)注射亚硝酸钠(nano2)诱导肝纤维化可用于模拟晚期nash疾病。因此,大鼠cdhfd+nano
2 nash模型用于测试化合物1单独和与化合物2组合的功效。在此模型中,雄性威斯塔大鼠被喂食cdhfd 4周以在每天口服药物和每三周ip nano2治疗之前诱发疾病。在化合物1(3mg/kg)和化合物2(25mg/kg)作为单一药剂或以组合给药8周后,肝组织经过处理用于通过rnaseq进行全转录组分析。表10显示了在用化合物1(3mg/mg)、化合物2(25mg/kg)或化合物1(3mg/kg)和化合物2(25mg/kg)的组合治疗的cdhfd+nano2大鼠中鉴定的差异表达基因(deg)的总数和变化方向(即相对于媒剂对照向上或向下)。使用≥1.5倍的绝对倍数变化截止值和《0.01的调节后p值,在化合物1治疗组中鉴定出309个deg,在化合物2治疗的动物中鉴定出847个deg,并且在组合治疗组中鉴定出1351个deg。这些结果表明,与单一药剂治疗组相比,组合治疗至少对deg总数产生累加效应。
[0363]
令人惊讶的是,与单独治疗组相比,在组合治疗组中观察到更多上调的deg。图18显示了使用分别为≥1.5和《0.01的绝对倍数变化和调节后p值截止值在每个治疗组中鉴定的deg的数量和重叠(相对于媒剂nash对照)。
[0364]
表10.差异表达基因(deg)
[0365][0366]
为每个治疗组鉴定的经鉴定deg数量(媒剂nash对照相对于治疗)。调节后p值《0.01且倍数变化≥1.5倍
[0367]
我们接下来检查了与先前描述的脂质代谢和甘油三酯积累相关的基因的差异表达(shepherd e,karim s,newsome p和lalor p.,inhibition of vascular adhesion protein-1modifies hepatic steatosis in vitro and in vivo.world j hepatol.2020 12(11):931-948)。化合物2治疗导致与脂质代谢和脂肪酸转运相关的基因,包括vldlr、fabp2、vegfc、ldlrap1、ldlr、ppargc1a和slc27a5(表11,用星号表示)的表达发生统计学显著变化。其中,vldlr、fabp2和slc27a5改变了≥1.5倍(以粗体显示)。只有fabp2在用化合物1治疗后显著差异表达。有趣的是,与任一单一药剂治疗组相比,化合物1和化合物2的组合使得与脂质代谢和脂肪酸转运相关的deg显著增加。此外,几个基因相对于媒剂对照的差异表达≥1.5倍,包括vldlr、fabp2、il1r2、ppara、ldlr、ppargc1a、rxra和slc27a5。
[0368]
综合这些数据表明,fxr激动剂化合物1和ssao抑制剂化合物2的组合使得参与脂质代谢和脂肪酸转运的基因的表达发生显著变化。此外,基因表达变化的模式在很大程度上与相对于单独用化合物1治疗的增强的抗脂肪变性作用一致。考虑到它们不同的作用机制,化合物1和化合物2在组合使用以加速nash消退过程时可提供互补的益处。
[0369]
表11.与脂质代谢和脂肪酸转运相关的差异表达基因
[0370]
相对于媒剂对照的差异基因表达分析(log
2-倍数变化)
[0371][0372]
cdhfd+nan02大鼠的肝脏中的基因表达分析(rnaseq)。涉及脂质代谢和脂肪酸转运的基因相对于媒剂对照的log
2-倍数变化。负变化方向(-)表示相对于媒剂减少的治疗表达;正变化方向表示相对于媒剂对照增加的基因表达。≥1.5倍(即,log
2-倍数变化≥0.6或≤-0.6)的绝对倍数变化值以粗体表示。*p值《0.05。
[0373]
实施例14:
[0374]
进行了一项随机、双盲、安慰剂对照研究,以评估组合治疗(例如化合物1和化合物2)的安全性和功效。nash受试者每天用fxr激动剂和ssao抑制剂组合治疗一次,持续12或48周。通过mri-pdff监测肝脏脂肪,且测量基于血清的非侵入性纤维化或nash标志物,例如pro-c3、timp-1、piiinp、ck-18和alt。还监测例如瘙痒和ldl-c胆固醇水平等副作用。
[0375]
本说明书中提及的所有出版物,包括专利、专利申请和科学文章均出于所有目的以引用方式整体并入本文中,其程度如同每个单独的出版物,包括专利、专利申请或科学文章均被具体和单独地指明以引用方式并入。
[0376]
尽管为了清楚理解的目的通过说明和举例的方式对前述发明进行了一些详细描述,但对于本领域技术人员来说显而易见的是,将根据上述教导进行某些微小的改变和修改。因此,描述和实施例不应被解释为限制本发明的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1